
Generation and Characterization of a New Preclinical Mouse Model of EGFR-Driven Lung Cancer with MET-Induced Osimertinib Resistance

 ,
,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
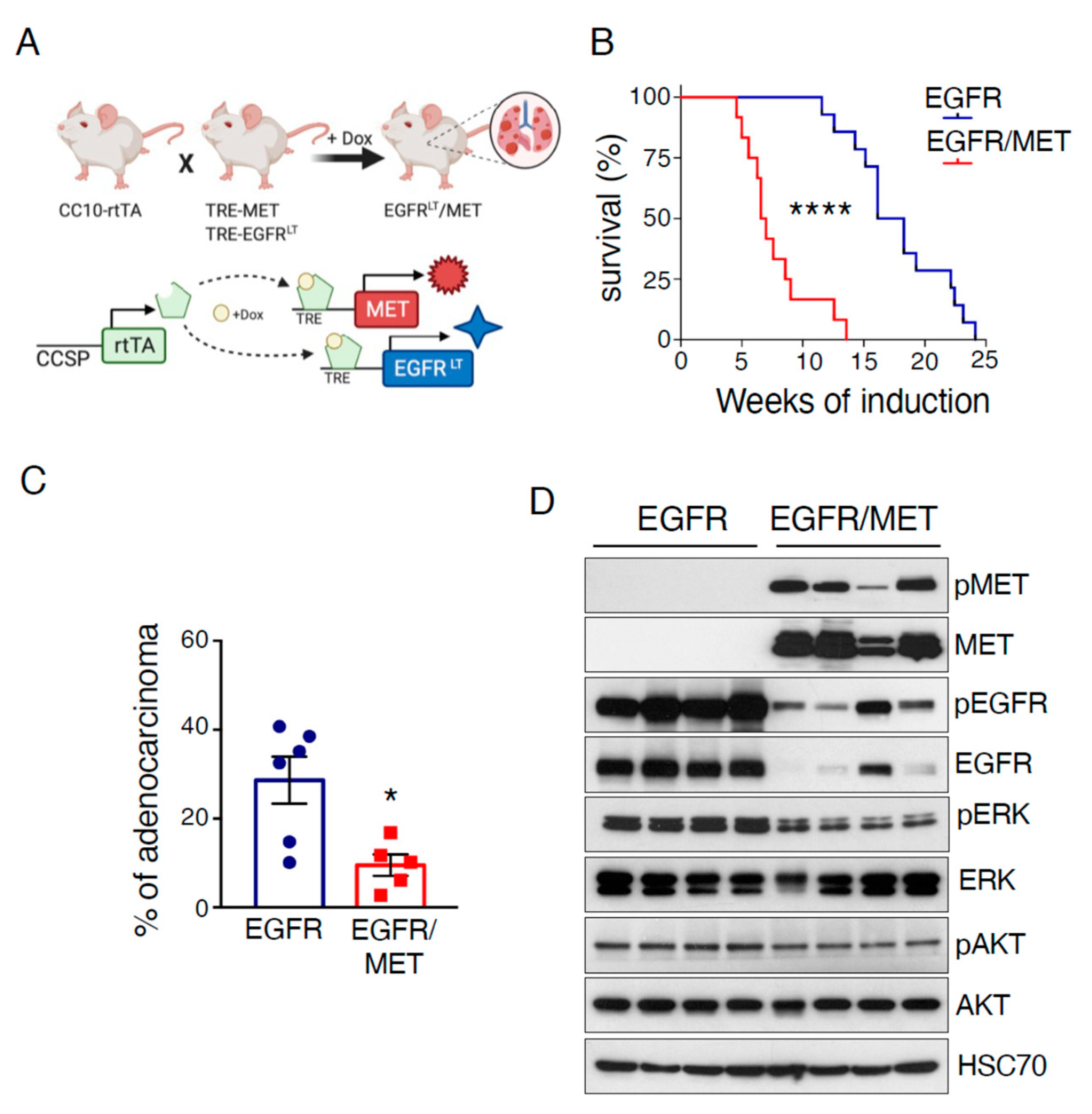
2.1. MET Overexpression Decreases Survival of Mice Harboring EGFR-Driven Lung Adenocarcinoma
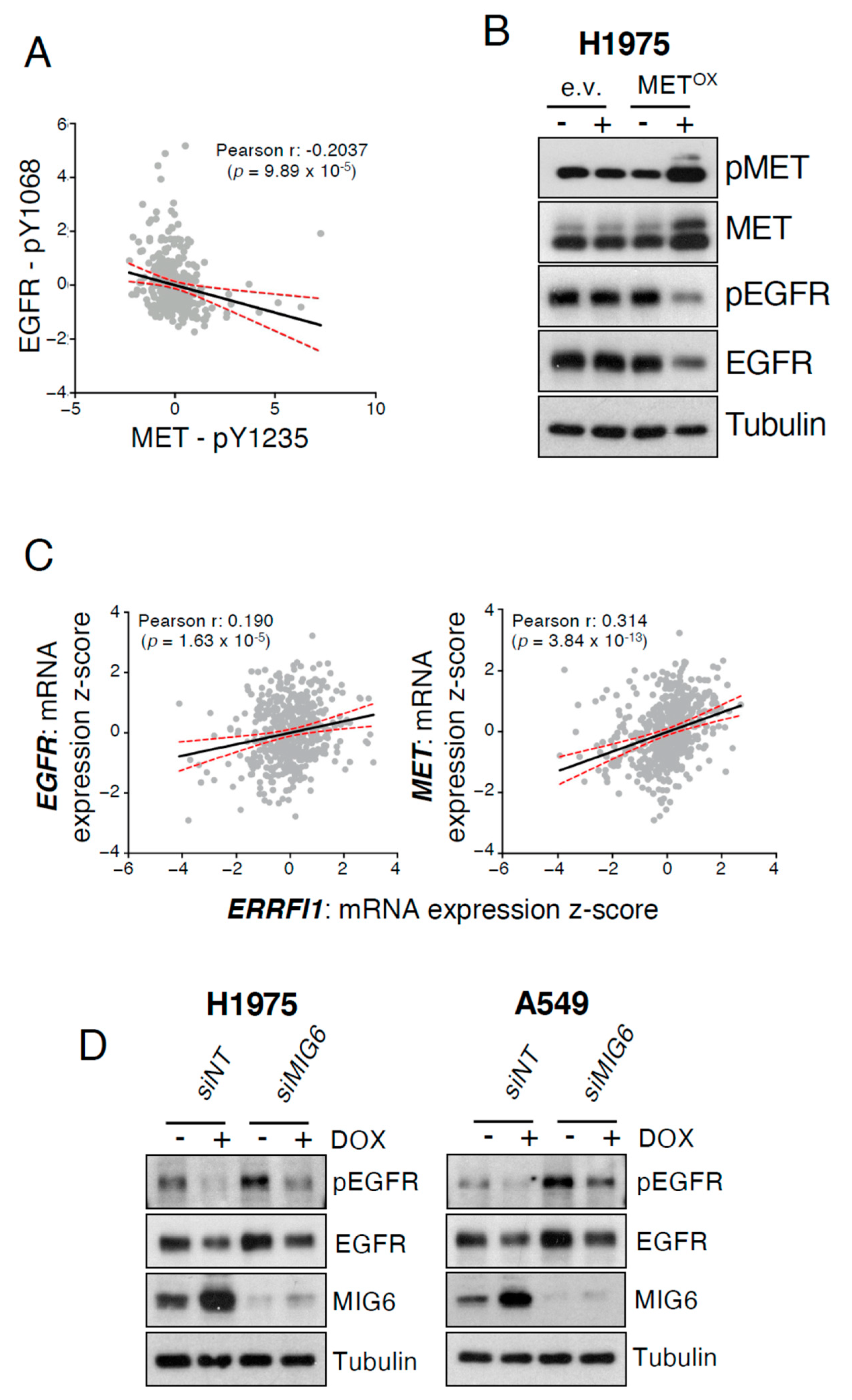
2.2. MET Overexpression Decreases EGFR Activity
2.3. EGFR/MET-Driven Tumors Are Resistant to Osimertinib
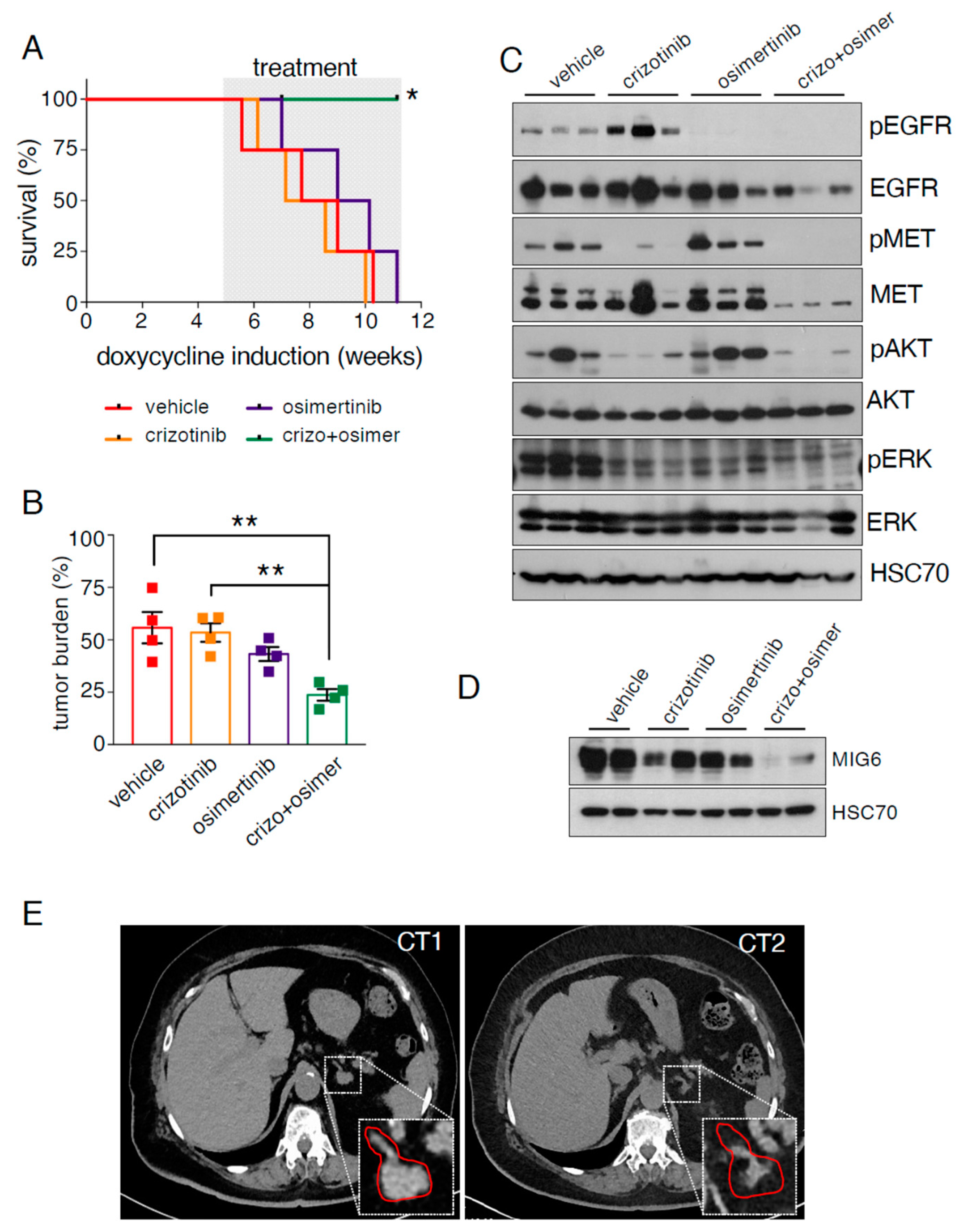
2.4. EGFR/MET-Driven Tumors Are Sensitive to the Osimertinib and Crizotinib Combination
3. Discussion
4. Material and Methods
4.1. Mice and Genotyping
4.2. Cell Culture, Western Blotting and Transfection Reagents
4.3. Generation of Cell Lines with Inducible MET Overexpression
4.4. Treatments in Mice
4.5. qPCR
4.6. Analysis of Publicly Available Datasets
4.7. Histopathology and Immunohistochemistry
4.8. Statistical Analysis
4.9. Clinical Case
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, C.S.; Gilligan, D.; Pacey, S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Bousquet Mur, E.; Bernardo, S.; Papon, L.; Mancini, M.; Fabbrizio, E.; Goussard, M.; Ferrer, I.; Giry, A.; Quantin, X.; Pujol, J.L.; et al. Notch inhibition overcomes resistance to tyrosine kinase inhibitors in EGFR-driven lung adenocarcinoma. J. Clin. Investig. 2020, 130, 612–624. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Anastasi, S.; Lamberti, D.; Alema, S.; Segatto, O. Regulation of the ErbB network by the MIG6 feedback loop in physiology, tumor suppression and responses to oncogene-targeted therapeutics. Semin. Cell Dev. Biol. 2016, 50, 115–124. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Pante, G.; Thompson, J.; Lamballe, F.; Iwata, T.; Ferby, I.; Barr, F.A.; Davies, A.M.; Maina, F.; Klein, R. Mitogen-inducible gene 6 is an endogenous inhibitor of HGF/Met-induced cell migration and neurite growth. J. Cell Biol. 2005, 171, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Landi, L.; Chiari, R.; Tiseo, M.; D’Inca, F.; Dazzi, C.; Chella, A.; Delmonte, A.; Bonanno, L.; Giannarelli, D.; Cortinovis, D.L.; et al. Crizotinib in MET-Deregulated or ROS1-Rearranged Pretreated Non-Small Cell Lung Cancer (METROS): A Phase II, Prospective, Multicenter, Two-Arms Trial. Clin. Cancer Res. 2019, 25, 7312–7319. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, H.; Lu, P.; Yu, Z.; Xu, C.; Zhuang, W.; Song, Z. Crizotinib with or without an EGFR-TKI in treating EGFR-mutant NSCLC patients with acquired MET amplification after failure of EGFR-TKI therapy: A multicenter retrospective study. J. Transl. Med. 2019, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Oh, Y.T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Eser, P.Ö.; Paranal, R.M.; Poitras, M.J.; Xu, M.; Wang, S.; Ogino, A.; Choi, J.; Missios, P.; Haikala, H.M.; Son, J.; et al. Abstract 1732: Switch to MET oncogene dependence as a novel mechanism of drug resistance in EGFR-mutant lung cancer. Cancer Res. 2019, 79, 1732. [Google Scholar] [CrossRef]
- Baldacci, S.; Mazieres, J.; Tomasini, P.; Girard, N.; Guisier, F.; Audigier-Valette, C.; Monnet, I.; Wislez, M.; Perol, M.; Do, P.; et al. Outcome of EGFR-mutated NSCLC patients with MET-driven resistance to EGFR tyrosine kinase inhibitors. Oncotarget 2017, 8, 105103–105114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrocco, I.; Romaniello, D.; Vaknin, I.; Drago-Garcia, D.; Oren, R.; Uribe, M.L.; Nataraj, B.N.; Ghosh, S.; Eilam, R.; Salame, T.M.; et al. Upfront admixing antibodies and EGFR inhibitors preempts sequential treatments in lung cancer models. EMBO Mol. Med. 2021, 13, e13144. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Lee, S.H.; Kim, S.Y.; Jeong, S.Y.; Kim, J.H.; Pyo, K.H.; Park, C.W.; Heo, S.G.; Yun, M.R.; Lim, S.; et al. Antitumor Activity of Amivantamab (JNJ-61186372), an EGFR-MET Bispecific Antibody, in Diverse Models of EGFR Exon 20 Insertion-Driven NSCLC. Cancer Discov. 2020, 10, 1194–1209. [Google Scholar] [CrossRef]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the Met Receptor by Cell Attachment Induces and Sustains Hepatocellular Carcinomas in Transgenic Mice. J. Cell Biol. 2001, 153, 1023–1034. [Google Scholar] [CrossRef]
- Tichelaar, J.W.; Lu, W.; Whitsett, J.A. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J. Biol. Chem. 2000, 275, 11858–11864. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Shimamura, T.; Ji, H.; Chen, L.; Haringsma, H.J.; McNamara, K.; Liang, M.C.; Perera, S.A.; Zaghlul, S.; Borgman, C.L.; et al. Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell 2007, 12, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Mancini, M.; Gobert, M.; Shen, S.; Furtado, G.C.; Lira, S.A.; Parkhurst, C.N.; Garambois, V.; Brengues, M.; Tadokoro, C.E.; et al. Spleen plays a major role in DLL4-driven acute T-cell lymphoblastic leukemia. Theranostics 2021, 11, 1594–1608. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancini, M.; Thomas, Q.-D.; Bourdel, S.; Papon, L.; Bousquet, E.; Jalta, P.; La Monica, S.; Travert, C.; Alfieri, R.; Quantin, X.; et al. Generation and Characterization of a New Preclinical Mouse Model of EGFR-Driven Lung Cancer with MET-Induced Osimertinib Resistance. Cancers 2021, 13, 3441. https://doi.org/10.3390/cancers13143441
Mancini M, Thomas Q-D, Bourdel S, Papon L, Bousquet E, Jalta P, La Monica S, Travert C, Alfieri R, Quantin X, et al. Generation and Characterization of a New Preclinical Mouse Model of EGFR-Driven Lung Cancer with MET-Induced Osimertinib Resistance. Cancers. 2021; 13(14):3441. https://doi.org/10.3390/cancers13143441
Chicago/Turabian StyleMancini, Maicol, Quentin-Dominique Thomas, Sylvia Bourdel, Laura Papon, Emilie Bousquet, Prisca Jalta, Silvia La Monica, Camille Travert, Roberta Alfieri, Xavier Quantin, and et al. 2021. "Generation and Characterization of a New Preclinical Mouse Model of EGFR-Driven Lung Cancer with MET-Induced Osimertinib Resistance" Cancers 13, no. 14: 3441. https://doi.org/10.3390/cancers13143441
APA StyleMancini, M., Thomas, Q.-D., Bourdel, S., Papon, L., Bousquet, E., Jalta, P., La Monica, S., Travert, C., Alfieri, R., Quantin, X., Cañamero, M., & Maraver, A. (2021). Generation and Characterization of a New Preclinical Mouse Model of EGFR-Driven Lung Cancer with MET-Induced Osimertinib Resistance. Cancers, 13(14), 3441. https://doi.org/10.3390/cancers13143441