
Novel Insights into YB-1 Signaling and Cell Death Decisions

 , , and
, , and
Abstract
:Simple Summary
Abstract
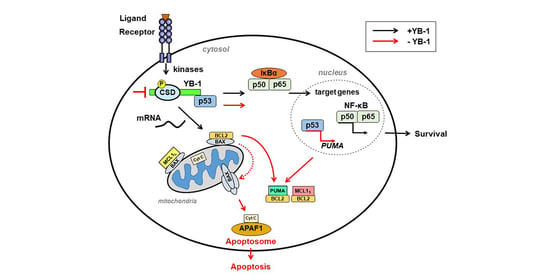
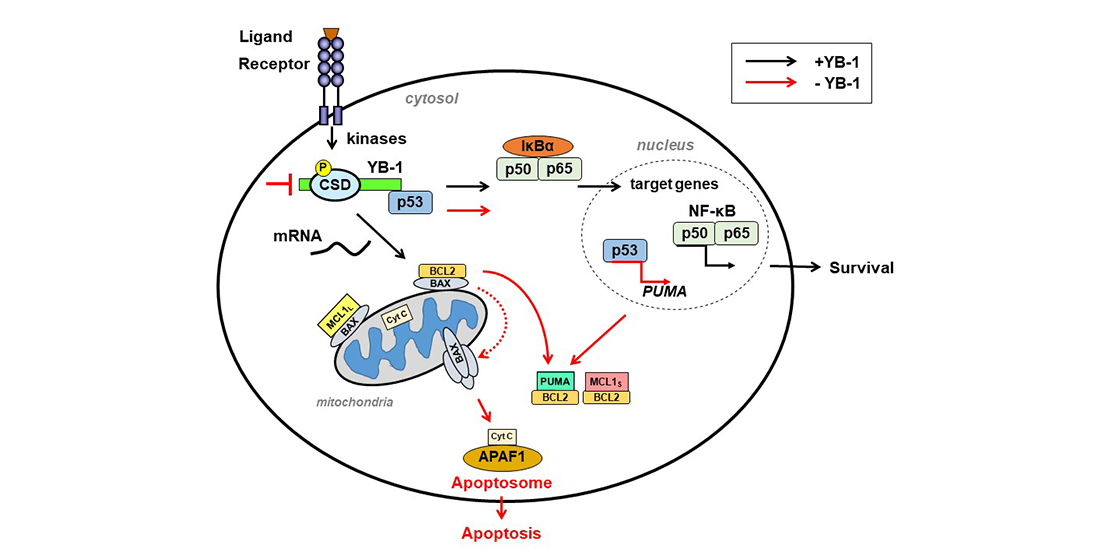
1. Introduction
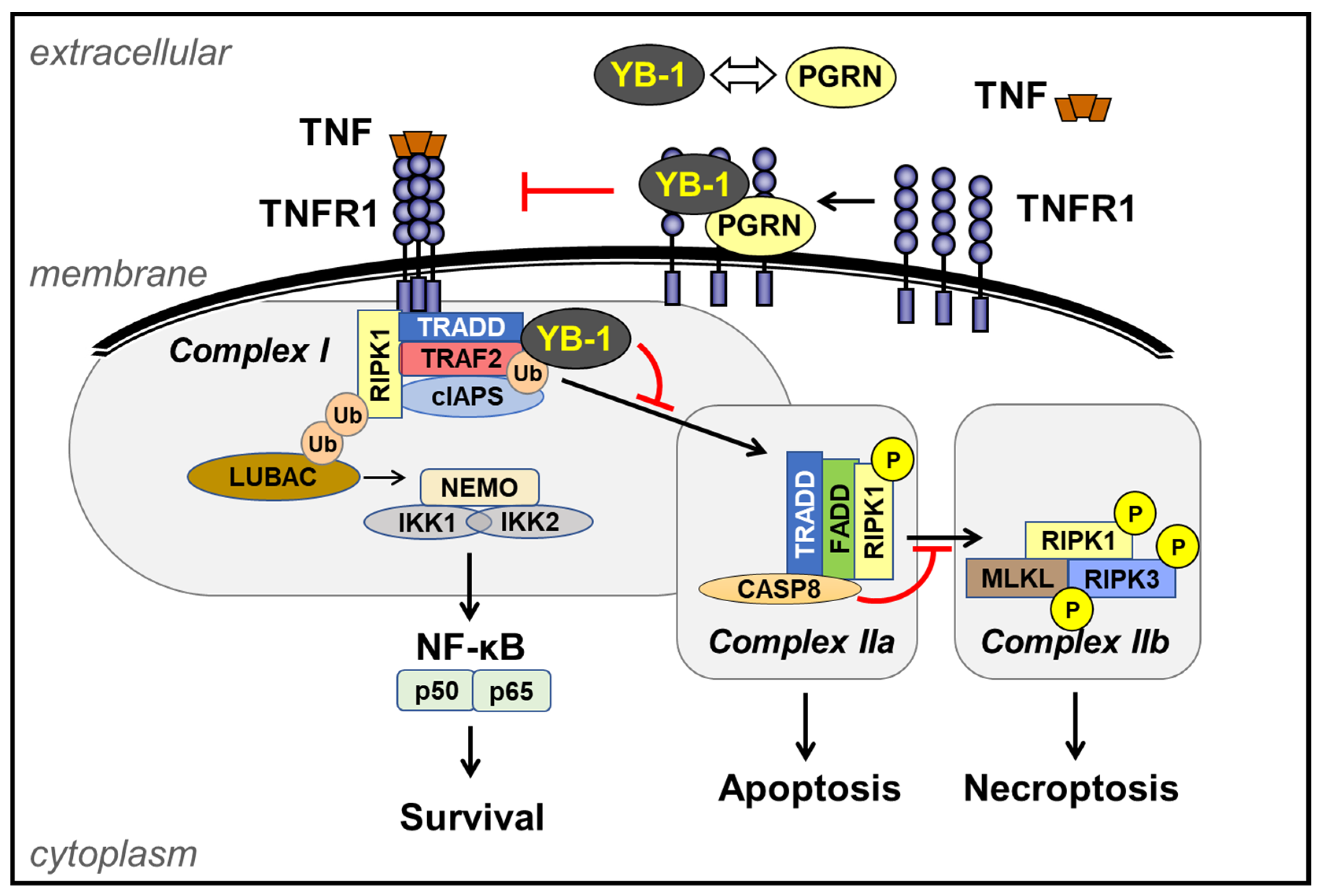
2. Extracellular YB-1
3. TNF Receptor-1 (TNFR1) Signaling Pathway
4. Targeting YB-1 Influences Cell Death Decisions
4.1. Myeloproliferative Neoplasia (MPN)
4.2. Chronic Myeloid Leukemia
4.3. Systemic Lupus Erythematosus (SLE)
4.4. T Cell Leukemia
4.5. Ovarian Cancer
5. Outlook
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
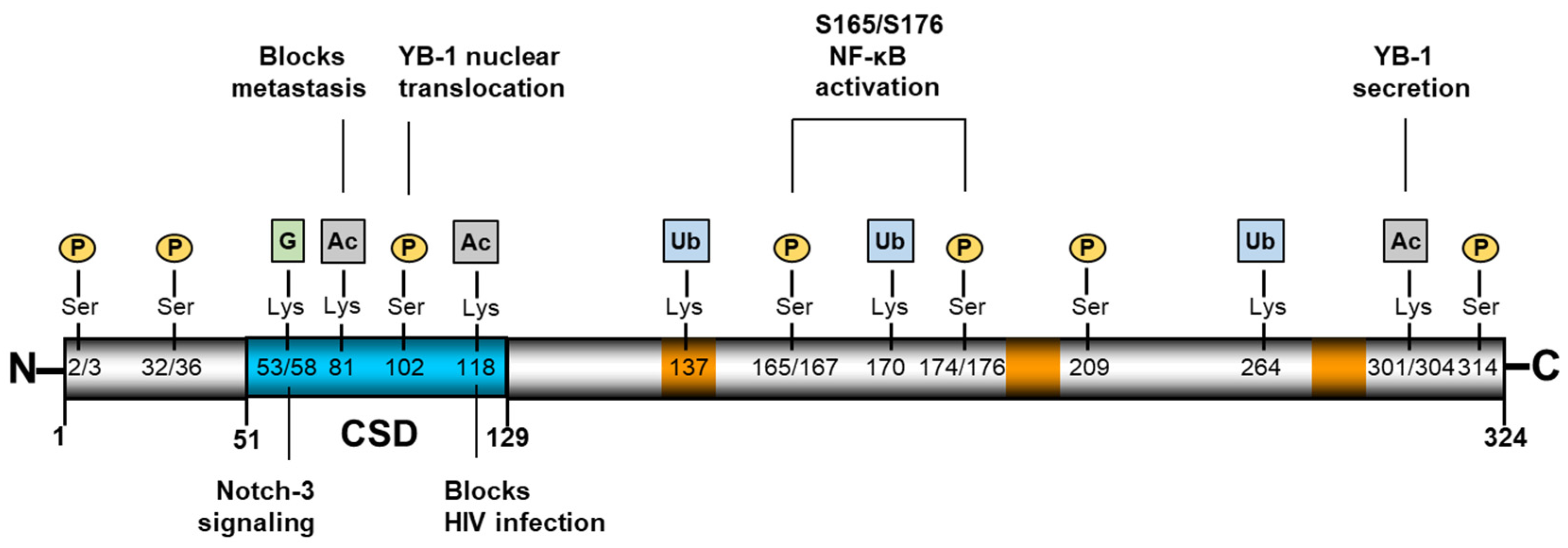
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Site on YB-1 | Type of PTM | Ref. |
|---|---|---|
| S32 | Phosphorylation | [79] |
| S36 | Phosphorylation | [79] |
| K53 | Guanidinylation | [26] |
| K58 | Guanidinylation | [26] |
| K81 | Acetylation | [7] |
| S102 | Phosphorylation | [93] |
| K118 | Ubiquitination | [81] |
| K137 | Ubiquitination | [24] |
| K170 | Ubiquitination | [24] |
| S165 | Phosphorylation | [81] |
| S167 | Phosphorylation | [24] |
| S174 | Phosphorylation | [24] |
| S176 | Phosphorylation | [80] |
| S209 | Phosphorylation | [24] |
| K264 | Ubiquitination | [24] |
| K301 | Acetylation | [31] |
| K304 | Acetylation | [31] |
| S314 | Phosphorylation | [79] |
References
- Budkina, K.S.; Zlobin, N.E.; Kononova, S.V.; Ovchinnikov, L.P.; Babakov, A.V. Cold Shock Domain Proteins: Structure and Interaction with Nucleic Acids. Biochemistry 2020, 85, S1–S19. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, U.; Roske, Y. Cold-Shock Domains-Abundance, Structure, Properties, and Nucleic-Acid Binding. Cancers 2021, 13, 190. [Google Scholar] [CrossRef] [PubMed]
- Mordovkina, D.; Lyabin, D.N.; Smolin, E.A.; Sogorina, E.M.; Ovchinnikov, L.P.; Eliseeva, I. Y-Box Binding Proteins in mRNP Assembly, Translation, and Stability Control. Biomolecules 2020, 10, 591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangermano, F.; Delicato, A.; Calabro, V. Y Box Binding Protein 1 (YB-1) Oncoprotein at the Hub of DNA Proliferation, Damage and Cancer Progression. Biochimie 2020, 179, 205–216. [Google Scholar] [CrossRef]
- Eliseeva, I.A.; Kim, E.R.; Guryanov, S.G.; Ovchinnikov, L.P.; Lyabin, D.N. Y-Box-Binding Protein 1 (YB-1) and Its Functions. Biochemistry 2011, 76, 1402–1433. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Pratt, G.; Yeo, G.W.; Moore, M.J. The Clothes Make the mRNA: Past and Present Trends in mRNP Fashion. Annu. Rev. Biochem. 2015, 84, 325–354. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, A.M.; Veinotte, C.J.; Cheng, H.; Grunewald, T.G.; Negri, G.L.; Somasekharan, S.P.; Corkery, D.P.; Tirode, F.; Mathers, J.; Khan, D.; et al. Translational Activation of HIF1alpha by YB-1 Promotes Sarcoma Metastasis. Cancer Cell 2015, 27, 682–697. [Google Scholar] [CrossRef] [Green Version]
- Evdokimova, V.; Tognon, C.; Ng, T.; Ruzanov, P.; Melnyk, N.; Fink, D.; Sorokin, A.; Ovchinnikov, L.P.; Davicioni, E.; Triche, T.J.; et al. Translational Activation of Snail1 and Other Developmentally Regulated Transcription Factors by YB-1 Promotes an Epithelial-Mesenchymal Transition. Cancer Cell 2009, 15, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Kretov, D.A.; Curmi, P.A.; Hamon, L.; Abrakhi, S.; Desforges, B.; Ovchinnikov, L.P.; Pastre, D. mRNA and DNA Selection via Protein Multimerization: YB-1 as a Case Study. Nucleic Acids Res. 2015, 43, 9457–9473. [Google Scholar] [CrossRef] [Green Version]
- Kretov, D.A.; Clement, M.J.; Lambert, G.; Durand, D.; Lyabin, D.N.; Bollot, G.; Bauvais, C.; Samsonova, A.; Budkina, K.; Maroun, R.C.; et al. YB-1, an Abundant Core mRNA-Binding Protein, Has the Capacity to Form an RNA Nucleoprotein Filament: A Structural Analysis. Nucleic Acids Res. 2019, 47, 3127–3141. [Google Scholar] [CrossRef]
- Naumenko, K.N.; Sukhanova, M.V.; Hamon, L.; Kurgina, T.A.; Alemasova, E.E.; Kutuzov, M.M.; Pastre, D.; Lavrik, O.I. Regulation of Poly(ADP-Ribose) Polymerase 1 Activity by Y-Box-Binding Protein 1. Biomolecules 2020, 10, 1325. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.S.; Li, S.; Yang, Y.; Sun, P.; Zhu, Q.; Wang, J.; Jiang, B.; Yang, D.; Liu, M. Structural Basis of DNA Binding to Human YB-1 Cold Shock Domain Regulated by Phosphorylation. Nucleic Acids Res. 2020, 48, 9361–9371. [Google Scholar] [CrossRef]
- Evdokimova, V.; Ruzanov, P.; Anglesio, M.S.; Sorokin, A.V.; Ovchinnikov, L.P.; Buckley, J.; Triche, T.J.; Sonenberg, N.; Sorensen, P.H. Akt-Mediated YB-1 Phosphorylation Activates Translation of Silent mRNA Species. Mol. Cell Biol. 2006, 26, 277–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyabin, D.N.; Eliseeva, I.A.; Ovchinnikov, L.P. YB-1 Protein: Functions and Regulation. Wiley Interdiscip. Rev. RNA 2014, 5, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Yokoyama, G.; Takahashi, H.; Namoto, R.; Nakagawa, S.; Toh, U.; Kage, M.; Shirouzu, K.; Kuwano, M. Preclinical Studies of Molecular-Targeting Diagnostic and Therapeutic Strategies against Breast Cancer. Breast Cancer 2008, 15, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Kashihara, M.; Azuma, K.; Kawahara, A.; Basaki, Y.; Hattori, S.; Yanagawa, T.; Terazaki, Y.; Takamori, S.; Shirouzu, K.; Aizawa, H.; et al. Nuclear Y-box Binding Protein-1, a Predictive Marker of Prognosis, is Correlated with Expression of HER2/ErbB2 and HER3/ErbB3 in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2009, 4, 1066–1074. [Google Scholar] [CrossRef] [Green Version]
- Hohlfeld, R.; Brandt, S.; Bernhardt, A.; Gorny, X.; Schindele, D.; Jandrig, B.; Schostak, M.; Isermann, B.; Lindquist, J.A.; Mertens, P.R. Crosstalk between Akt Signaling and Cold Shock Proteins in Mediating Invasive cell Phenotypes. Oncotarget 2018, 9, 19039–19049. [Google Scholar] [CrossRef] [Green Version]
- Dolfini, D.; Mantovani, R. Targeting the Y/CCAAT Box in Cancer: YB-1 (YBX1) or NF-Y? Cell Death Differ. 2013, 20, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Wang, H.; Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Kohli, S.; Hoffmann, J.; Dhople, V.M.; et al. Activated Protein C Ameliorates Renal Ischemia-Reperfusion Injury by Restricting Y-Box Binding Protein-1 Ubiquitination. J. Am. Soc. Nephrol. 2015, 26, 2789–2799. [Google Scholar] [CrossRef] [Green Version]
- Palicharla, V.R.; Maddika, S. HACE1 Mediated K27 Ubiquitin Linkage Leads to YB-1 Protein Secretion. Cell Signal. 2015, 27, 2355–2362. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Chan, D.W.; Jung, S.Y.; Malovannaya, A.; Wang, Y.; Qin, J. A Data Set of Human Endogenous Protein Ubiquitination Sites. Mol. Cell Proteom. 2011, 10, M110.002089. [Google Scholar] [CrossRef] [Green Version]
- Hansen, F.M.; Tanzer, M.C.; Bruning, F.; Bludau, I.; Stafford, C.; Schulman, B.A.; Robles, M.S.; Karayel, O.; Mann, M. Data-Independent Acquisition Method for Ubiquitinome Analysis Reveals Regulation of Circadian Biology. Nat. Commun. 2021, 12, 254. [Google Scholar] [CrossRef]
- Wagner, S.A.; Satpathy, S.; Beli, P.; Choudhary, C. SPATA2 Links CYLD to the TNF-Alpha Receptor Signaling Complex and Modulates the Receptor Signaling Outcomes. EMBO J. 2016, 35, 1868–1884. [Google Scholar] [CrossRef] [Green Version]
- Chibi, M.; Meyer, M.; Skepu, A.; DJ, G.R.; Moolman-Smook, J.C.; Pugh, D.J. RBBP6 Interacts with Multifunctional Protein YB-1 through its RING Finger Domain, Leading to Ubiquitination and Proteosomal Degradation of YB-1. J. Mol. Biol. 2008, 384, 908–916. [Google Scholar] [CrossRef]
- Breitkopf, D.M.; Jankowski, V.; Ohl, K.; Hermann, J.; Hermert, D.; Tenbrock, K.; Liu, X.; Martin, I.V.; Wang, J.; Groll, F.; et al. The YB-1:Notch-3 Axis Modulates Immune Cell Responses and Organ Damage in Systemic Lupus Erythematosus. Kidney Int. 2020, 97, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Hessman, C.L.; Hildebrandt, J.; Shah, A.; Brandt, S.; Bock, A.; Frye, B.C.; Raffetseder, U.; Geffers, R.; Brunner-Weinzierl, M.C.; Isermann, B.; et al. YB-1 Interferes with TNFalpha-TNFR Binding and Modulates Progranulin-Mediated Inhibition of TNFalpha Signaling. Int. J. Mol. Sci. 2020, 21, 7076. [Google Scholar] [CrossRef]
- Rauen, T.; Raffetseder, U.; Frye, B.C.; Djudjaj, S.; Muhlenberg, P.J.; Eitner, F.; Lendahl, U.; Bernhagen, J.; Dooley, S.; Mertens, P.R. YB-1 Acts as a Ligand for Notch-3 Receptors and Modulates Receptor Activation. J. Biol. Chem. 2009, 284, 26928–26940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Kanig, N.; En-Nia, A.; Kaehne, T.; Eberhardt, C.S.; Shpacovitch, V.; Trautwein, C.; Mertens, P.R. Y-Box Protein-1/p18 Fragment Identifies Malignancies in Patients with Chronic Liver Disease. BMC Cancer 2011, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Galm, O.; Kanig, N.; Yagmur, E.; Brandt, S.; Lindquist, J.A.; Eberhardt, C.S.; Raffetseder, U.; Mertens, P.R. High Prevalence of Y-box Protein-1/p18 Fragment in Plasma of Patients with Malignancies of Different Origin. BMC Cancer 2014, 14, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, B.C.; Halfter, S.; Djudjaj, S.; Muehlenberg, P.; Weber, S.; Raffetseder, U.; En-Nia, A.; Knott, H.; Baron, J.M.; Dooley, S.; et al. Y-Box Protein-1 is Actively Secreted through a Non-Classical Pathway and Acts as an Extracellular Mitogen. EMBO Rep. 2009, 10, 783–789. [Google Scholar] [CrossRef]
- Hanssen, L.; Alidousty, C.; Djudjaj, S.; Frye, B.C.; Rauen, T.; Boor, P.; Mertens, P.R.; van Roeyen, C.R.; Tacke, F.; Heymann, F.; et al. YB-1 is an Early and Central Mediator of Bacterial and Sterile Inflammation In Vivo. J. Immunol. 2013, 191, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- van Roeyen, C.R.; Eitner, F.; Martinkus, S.; Thieltges, S.R.; Ostendorf, T.; Bokemeyer, D.; Luscher, B.; Luscher-Firzlaff, J.M.; Floege, J.; Mertens, P.R. Y-Box Protein 1 Mediates PDGF-B Effects in Mesangioproliferative Glomerular Disease. J. Am. Soc. Nephrol. 2005, 16, 2985–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarino, A.M.; Troiano, A.; Pizzo, E.; Bosso, A.; Vivo, M.; Pinto, G.; Amoresano, A.; Pollice, A.; La Mantia, G.; Calabro, V. Oxidative Stress Causes Enhanced Secretion of YB-1 Protein that Restrains Proliferation of Receiving Cells. Genes 2018, 9, 513. [Google Scholar] [CrossRef] [Green Version]
- Raffetseder, U.; Liehn, E.A.; Weber, C.; Mertens, P.R. Role of Cold Shock Y-Box Protein-1 in Inflammation, Atherosclerosis and Organ Transplant Rejection. Eur. J. Cell Biol. 2012, 91, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Aburjania, Z.; Jang, S.; Whitt, J.; Jaskula-Stzul, R.; Chen, H.; Rose, J.B. The Role of Notch3 in Cancer. Oncologist 2018, 23, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Djudjaj, S.; Chatziantoniou, C.; Raffetseder, U.; Guerrot, D.; Dussaule, J.C.; Boor, P.; Kerroch, M.; Hanssen, L.; Brandt, S.; Dittrich, A.; et al. Notch-3 Receptor Activation Drives Inflammation and Fibrosis Following Tubulointerstitial Kidney Injury. J. Pathol. 2012, 228, 286–299. [Google Scholar] [CrossRef]
- Ou, Y.; Zhao, Z.; Zhang, W.; Wu, Q.; Wu, C.; Liu, X.; Fu, M.; Ji, N.; Wang, D.; Qiu, J.; et al. Kindlin-2 Interacts with Beta-Catenin and YB-1 to Enhance EGFR Transcription during Glioma Progression. Oncotarget 2016, 7, 74872–74885. [Google Scholar] [CrossRef] [Green Version]
- Karathanasis, C.; Medler, J.; Fricke, F.; Smith, S.; Malkusch, S.; Widera, D.; Fulda, S.; Wajant, H.; van Wijk, S.J.L.; Dikic, I.; et al. Single-Molecule Imaging Reveals the Oligomeric State of Functional TNFalpha-Induced Plasma Membrane TNFR1 Clusters in Cells. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Shah, A.; Plaza-Sirvent, C.; Weinert, S.; Buchbinder, J.H.; Lavrik, I.N.; Mertens, P.R.; Schmitz, I.; Lindquist, J.A. YB-1 Mediates TNF-Induced Pro-Survival Signaling by Regulating NF-kappaB Activation. Cancers 2020, 12, 2188. [Google Scholar] [CrossRef]
- Black, R.A. Tumor Necrosis Factor-Alpha Converting Enzyme. Int. J. Biochem. Cell Biol. 2002, 34, 726–729. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-Mediated Inflammatory Disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Laha, D.; Grant, R.; Mishra, P.; Nilubol, N. The Role of Tumor Necrosis Factor in Manipulating the Immunological Response of Tumor Microenvironment. Front. Immunol. 2021, 12, 6908. [Google Scholar] [CrossRef]
- Chen, A.Y.; Wolchok, J.D.; Bass, A.R. TNF in the Era of Immune Checkpoint Inhibitors: Friend or Foe? Nat. Rev. Rheumatol. 2021, 17, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, I.; Coornaert, B.; Beyaert, R. Function and Regulation of Tumor Necrosis Factor Receptor Type 2. Curr. Med. Chem. 2004, 11, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.; Davis, M. TNF Receptor 2 Pathway: Drug Target for Autoimmune Diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The Molecular Architecture of the TNF Superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.Y.; Lu, M.; Yin, Q.; Wu, H. Structural Revelations of TRAF2 Function in TNF Receptor Signaling Pathway. Adv. Exp. Med. Biol. 2007, 597, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF Receptor 1-Associated Protein TRADD Signals Cell Death and NF-Kappa B Activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 Facilitate Cancer Cell Survival by Functioning as E3 Ligases that Promote RIP1 Ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. c-IAP1 and c-IAP2 are Critical Mediators of Tumor Necrosis Factor Alpha (TNFalpha)-Induced NF-kappaB Activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tailor, D.; Resendez, A.; Garcia-Marques, F.J.; Pandrala, M.; Going, C.C.; Bermudez, A.; Kumar, V.; Rafat, M.; Nambiar, D.K.; Honkala, A.; et al. Y Box Binding Protein 1 Inhibition as a Targeted Therapy for Ovarian Cancer. Cell Chem. Biol. 2021. [Google Scholar] [CrossRef]
- Iwai, K.; Fujita, H.; Sasaki, Y. Linear Ubiquitin Chains: NF-kappaB Signalling, Cell Death and Beyond. Nat. Rev. Mol. Cell Biol. 2014, 15, 503–508. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the Linear Ubiquitin Chain Assembly Complex Stabilizes the TNF-R1 Signaling Complex and is Required for TNF-Mediated Gene Induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Schmukle, A.C.; Haas, T.L.; Gerlach, B.; Cordier, S.M.; Rieser, E.; Walczak, H. The Linear Ubiquitin Chain Assembly Complex Forms Part of the TNF-R1 Signalling Complex and is Required for Effective TNF-Induced Gene Induction and Prevents TNF-Induced Apoptosis. Adv. Exp. Med. Biol. 2011, 691, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-Alpha-Induced Apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in Apoptosis: A Post-Translational Modification at the Edge of Life and Death. Nat. Rev. Mol. Cell Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and Its Role in Inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Mayo, M.W.; Baldwin, A.S., Jr. TNF- and Cancer Therapy-Induced Apoptosis: Potentiation by Inhibition of NF-kappaB. Science 1996, 274, 784–787. [Google Scholar] [CrossRef]
- Khraishi, M. Comparative Overview of Safety of the Biologics in Rheumatoid Arthritis. J. Rheumatol. Suppl. 2009, 82, 25–32. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-Receptors: From Mediators of Cell Death and Inflammation to Therapeutic Giants—Past, Present and Future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Lee, C.K.; Cha, H.S.; Choe, J.Y.; Park, E.J.; Kim, J. Effect of Anti-Tumor Necrosis Factor Alpha Treatment of Rheumatoid Arthritis and Chronic Kidney Disease. Rheumatol. Int. 2015, 35, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The Pathogenesis of Atherosclerosis: A Perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Schreyer, S.A.; Peschon, J.J.; LeBoeuf, R.C. Accelerated Atherosclerosis in Mice Lacking Tumor Necrosis Factor Receptor p55. J. Biol. Chem. 1996, 271, 26174–26178. [Google Scholar] [CrossRef] [Green Version]
- Tipping, P.G.; Hancock, W.W. Production of Tumor Necrosis Factor and Interleukin-1 by Macrophages from Human Atheromatous Plaques. Am. J. Pathol. 1993, 142, 1721–1728. [Google Scholar] [PubMed]
- Askling, J.; Bongartz, T. Malignancy and Biologic Therapy in Rheumatoid Arthritis. Curr. Opin. Rheumatol. 2008, 20, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Dixon, W. The Safety of Anti-Tumour Necrosis Factor Therapy in Rheumatoid Arthritis. Curr. Opin. Rheumatol. 2008, 20, 138–144. [Google Scholar] [CrossRef]
- Matteson, E.L.; Bongartz, T. Tumor Necrosis Factor Antagonists and Cancer in Patients with Rheumatoid Arthritis. Nat. Clin. Pract. Rheumatol. 2007, 3, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional Regulation of Endothelial Cell Adhesion Molecules: NF-Kappa B and Cytokine-Inducible Enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudeck, J.; Kotrba, J.; Immler, R.; Hoffmann, A.; Voss, M.; Alexaki, V.I.; Morton, L.; Jahn, S.R.; Katsoulis-Dimitriou, K.; Winzer, S.; et al. Directional Mast Cell Degranulation of Tumor Necrosis Factor into Blood Vessels Primes Neutrophil Extravasation. Immunity 2021, 54, 468–483.e465. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Peter, M.E. Programmed Cell Death: Apoptosis Meets Necrosis. Nature 2011, 471, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Virag, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) Signaling in Cell Death. Mol. Asp. Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Schnoder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing Factor YBX1 Mediates Persistence of JAK2-Mutated Neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Hua, L.; Wang, B.; Wei, H.; Prabhu, L.; Hartley, A.V.; Jiang, G.; Liu, Y.; Lu, T. Novel Serine 176 Phosphorylation of YBX1 Activates NF-kappaB in Colon Cancer. J. Biol. Chem. 2017, 292, 3433–3444. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, L.; Mundade, R.; Wang, B.; Wei, H.; Hartley, A.V.; Martin, M.; McElyea, K.; Temm, C.J.; Sandusky, G.; Liu, Y.; et al. Critical Role of Phosphorylation of Serine 165 of YBX1 on the Activation of NF-kappaB in Colon Cancer. Oncotarget 2015, 6, 29396–29412. [Google Scholar] [CrossRef] [Green Version]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High Expression of Bcl-2 Protein in Acute Myeloid Leukemia Cells is Associated with Poor Response to Chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients with Acute Myeloid Leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Xie, X.; Han, G.; Zhang, T.; Li, Y.; Li, Y.; Yin, R.; Wang, Q.; Zhang, T.; Wang, P.; et al. YBX1 is Required for Maintaining Myeloid Leukemia Cell Survival by Regulating BCL2 Stability in an m6A-Dependent Manner. Blood 2021. [Google Scholar] [CrossRef]
- Ban, Y.; Tan, P.; Cai, J.; Li, J.; Hu, M.; Zhou, Y.; Mei, Y.; Tan, Y.; Li, X.; Zeng, Z.; et al. LNCAROD is Stabilized by m6A Methylation and Promotes Cancer Progression via Forming a Ternary Complex with HSPA1A and YBX1 in Head and Neck Squamous Cell Carcinoma. Mol. Oncol. 2020, 14, 1282–1296. [Google Scholar] [CrossRef]
- Li, N.; Zhan, X. Identification of Pathology-Specific Regulators of m(6)A RNA Modification to Optimize Lung Cancer Management in the Context of Predictive, Preventive, and Personalized Medicine. EPMA J. 2020, 11, 485–504. [Google Scholar] [CrossRef]
- Lasham, A.; Print, C.G.; Woolley, A.G.; Dunn, S.E.; Braithwaite, A.W. YB-1: Oncoprotein, Prognostic Marker and Therapeutic Target? Biochem. J. 2013, 449, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Gieseler-Halbach, S.; Meltendorf, S.; Pierau, M.; Weinert, S.; Heidel, F.H.; Fischer, T.; Handschuh, J.; Braun-Dullaeus, R.C.; Schrappe, M.; Lindquist, J.A.; et al. RSK-Mediated Nuclear Accumulation of the Cold-Shock Y-Box Protein-1 Controls Proliferation of T Cells and T-ALL Blasts. Cell Death Differ. 2017, 24, 371–383. [Google Scholar] [CrossRef]
- Meltendorf, S.; Fu, H.; Pierau, M.; Lindquist, J.A.; Finzel, S.; Mertens, P.R.; Gieseler-Halbach, S.; Ambach, A.; Thomas, U.; Lingel, H.; et al. Cell Survival Failure in Effector T Cells from Patients With Systemic Lupus Erythematosus Following Insufficient Up-Regulation of Cold-Shock Y-Box Binding Protein 1. Arthritis Rheumatol. 2020, 72, 1721–1733. [Google Scholar] [CrossRef]
- Billen, L.P.; Kokoski, C.L.; Lovell, J.F.; Leber, B.; Andrews, D.W. Bcl-XL Inhibits Membrane Permeabilization by Competing with Bax. PLoS Biol. 2008, 6, e147. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Gartner, D.; Soezeri, O.; Radbruch, A.; Schulze-Osthoff, K.; Brunner-Weinzierl, M.C. CD152 (CTLA-4) Determines the Unequal Resistance of Th1 and Th2 Cells against Activation-Induced Cell Death by a Mechanism Requiring PI3 Kinase Function. J. Exp. Med. 2004, 199, 831–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, B.W.; Kucab, J.; Wu, J.; Lee, C.; Cheang, M.C.; Yorida, E.; Turbin, D.; Dedhar, S.; Nelson, C.; Pollak, M.; et al. Akt Phosphorylates the Y-Box Binding Protein 1 at Ser102 Located in the Cold Shock Domain and Affects the Anchorage-Independent Growth of Breast Cancer Cells. Oncogene 2005, 24, 4281–4292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poudyal, D.; Yang, J.; Chen, Q.; Goswami, S.; Adelsberger, J.W.; Das, S.; Herman, A.; Hornung, R.L.; Andresson, T.; Imamichi, T. IL-27 Posttranslationally Regulates Y-Box Binding Protein-1 to Inhibit HIV-1 Replication in Human CD4+ T Cells. Aids 2019, 33, 1819–1830. [Google Scholar] [CrossRef]
- Ben-Mustapha, I.; Agrebi, N.; Barbouche, M.R. Novel Insights into FAS Defects Underlying Autoimmune Lymphoproliferative Syndrome Revealed by Studies in Consanguineous Patients. J. Leukoc. Biol. 2018, 103, 501–508. [Google Scholar] [CrossRef]
- Ito, K.; Tsutsumi, K.; Kuzumaki, T.; Gomez, P.F.; Otsu, K.; Ishikawa, K. A Novel Growth-Inducible Gene that Encodes a Protein with a Conserved Cold-Shock Domain. Nucleic. Acids Res. 1994, 22, 2036–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keilhoff, G.; Titze, M.; Esser, T.; Langnaese, K.; Ebmeyer, U. Constitutive and Functional Expression of YB-1 in Microglial Cells. Neuroscience 2015, 301, 439–453. [Google Scholar] [CrossRef]
- Raffetseder, U.; Rauen, T.; Djudjaj, S.; Kretzler, M.; En-Nia, A.; Tacke, F.; Zimmermann, H.W.; Nelson, P.J.; Frye, B.C.; Floege, J.; et al. Differential Regulation of Chemokine CCL5 Expression in Monocytes/Macrophages and Renal Cells by Y-Box Protein-1. Kidney Int. 2009, 75, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Stenina, O.I.; Poptic, E.J.; DiCorleto, P.E. Thrombin Activates a Y Box-Binding Protein (DNA-Binding Protein B) in Endothelial Cells. J. Clin. Invest. 2000, 106, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Sprent, J.; Surh, C.D. Normal T Cell Homeostasis: The Conversion of Naive Cells into Memory-Phenotype Cells. Nat. Immunol. 2011, 12, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Surh, C.D.; Sprent, J. Homeostasis of Naive and Memory T Cells. Immunity 2008, 29, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Gherzi, R.; Andersen, J.S.; Gaietta, G.; Jurchott, K.; Royer, H.D.; Mann, M.; Karin, M. Nucleolin and YB-1 are Required for JNK-Mediated Interleukin-2 mRNA Stabilization during T-cell Activation. Genes Dev. 2000, 14, 1236–1248. [Google Scholar] [PubMed]
- Johnson, T.G.; Schelch, K.; Mehta, S.; Burgess, A.; Reid, G. Why Be One Protein When You Can Affect Many? The Multiple Roles of YB-1 in Lung Cancer and Mesothelioma. Front. Cell Dev. Biol. 2019, 7, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurya, P.K.; Mishra, A.; Yadav, B.S.; Singh, S.; Kumar, P.; Chaudhary, A.; Srivastava, S.; Murugesan, S.N.; Mani, A. Role of Y Box Protein-1 in Cancer: As Potential Biomarker and Novel Therapeutic Target. J. Cancer 2017, 8, 1900–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, S.; Mertens, P.R. A Remedy for Kidney Disease Successfully Alters the Cold Shock Protein Response during Inflammation. Kidney Int. 2016, 90, 1148–1150. [Google Scholar] [CrossRef]
- Khan, M.I.; Adhami, V.M.; Lall, R.K.; Sechi, M.; Joshi, D.C.; Haidar, O.M.; Syed, D.N.; Siddiqui, I.A.; Chiu, S.Y.; Mukhtar, H. YB-1 Expression Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer that is Inhibited by a Small Molecule Fisetin. Oncotarget 2014, 5, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Sechi, M.; Lall, R.K.; Afolabi, S.O.; Singh, A.; Joshi, D.C.; Chiu, S.Y.; Mukhtar, H.; Syed, D.N. Fisetin Targets YB-1/RSK Axis Independent of its Effect on ERK Signaling: Insights from In Vitro and In Vivo Melanoma models. Sci. Rep. 2018, 8, 15726. [Google Scholar] [CrossRef]
- Xiao, Y.Z.; Yang, M.; Xiao, Y.; Guo, Q.; Huang, Y.; Li, C.J.; Cai, D.; Luo, X.H. Reducing Hypothalamic Stem Cell Senescence Protects against Aging-Associated Physiological Decline. Cell Metab 2020, 31, 534–548.e535. [Google Scholar] [CrossRef]
- Kuwano, M.; Shibata, T.; Watari, K.; Ono, M. Oncogenic Y-Box Binding Protein-1 as an Effective Therapeutic Target in Drug-Resistant Cancer. Cancer Sci. 2019, 110, 1536–1543. [Google Scholar] [CrossRef]
- Morgenroth, R.; Reichardt, C.; Steffen, J.; Busse, S.; Frank, R.; Heidecke, H.; Mertens, P.R. Autoantibody Formation and Mapping of Immunogenic Epitopes against Cold-Shock-Protein YB-1 in Cancer Patients and Healthy Controls. Cancers 2020, 12, 3507. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Shaheen, A.A.; Baeza, N.; Lytvyak, E.; Urbanski, S.J.; Mason, A.L.; Norman, G.L.; Fritzler, M.J.; Swain, M.G. Evaluation of Classical and Novel Autoantibodies for the Diagnosis of Primary Biliary Cholangitis-Autoimmune Hepatitis Overlap Syndrome (PBC-AIH OS). PLoS ONE 2018, 13, e0193960. [Google Scholar] [CrossRef] [PubMed]
- Bloch, D.B.; Nobre, R.A.; Yang, W.H. GW/P-Bodies and Autoimmune Disease. Adv. Exp. Med. Biol. 2013, 768, 61–70. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, A.; Lindquist, J.A.; Rosendahl, L.; Schmitz, I.; Mertens, P.R. Novel Insights into YB-1 Signaling and Cell Death Decisions. Cancers 2021, 13, 3306. https://doi.org/10.3390/cancers13133306
Shah A, Lindquist JA, Rosendahl L, Schmitz I, Mertens PR. Novel Insights into YB-1 Signaling and Cell Death Decisions. Cancers. 2021; 13(13):3306. https://doi.org/10.3390/cancers13133306
Chicago/Turabian StyleShah, Aneri, Jonathan A. Lindquist, Lars Rosendahl, Ingo Schmitz, and Peter R. Mertens. 2021. "Novel Insights into YB-1 Signaling and Cell Death Decisions" Cancers 13, no. 13: 3306. https://doi.org/10.3390/cancers13133306
APA StyleShah, A., Lindquist, J. A., Rosendahl, L., Schmitz, I., & Mertens, P. R. (2021). Novel Insights into YB-1 Signaling and Cell Death Decisions. Cancers, 13(13), 3306. https://doi.org/10.3390/cancers13133306