
Androgen Receptor Signaling in Prostate Cancer Genomic Subtypes

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
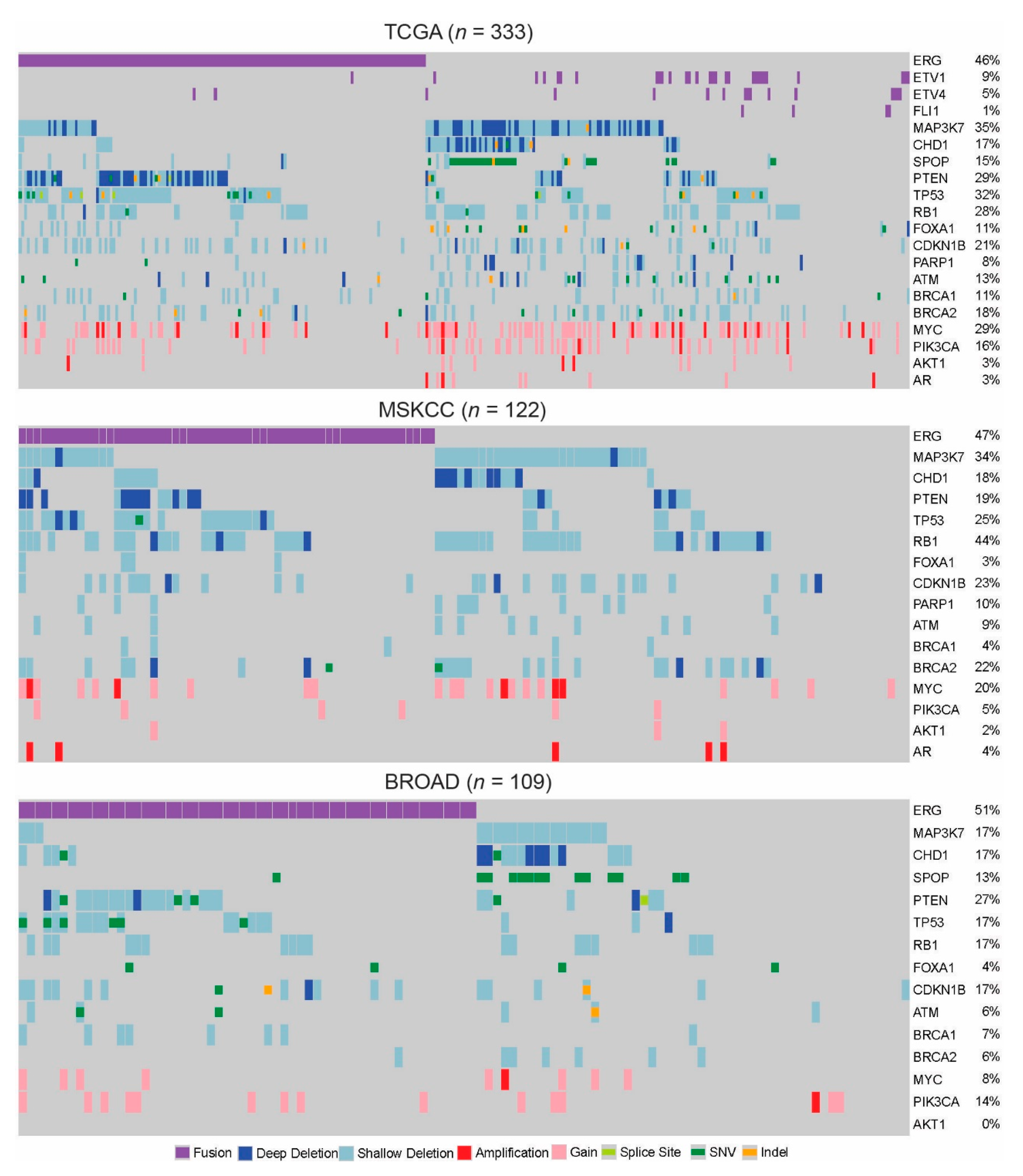
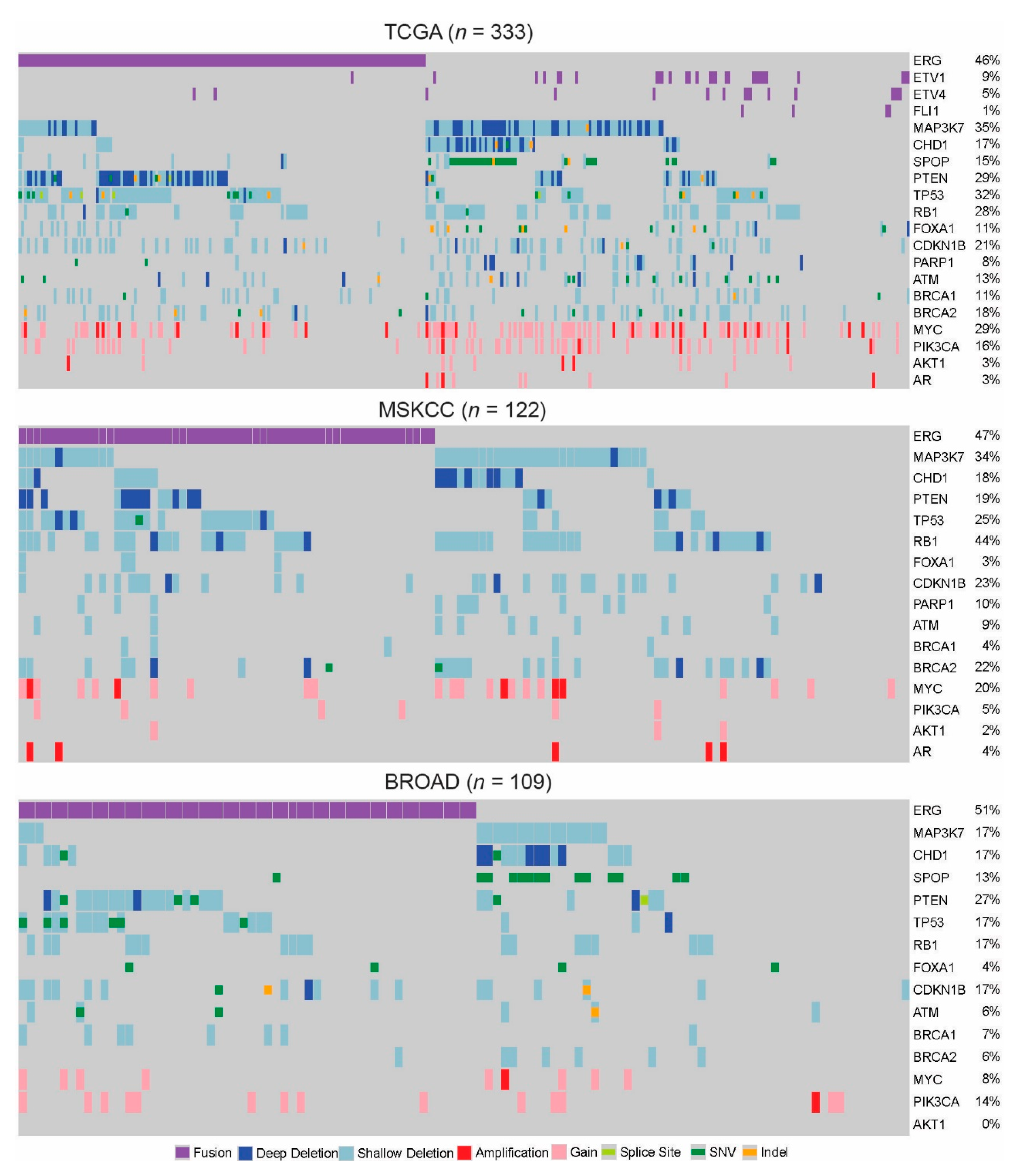
2. Individual Alterations
2.1. ETS-Factors
2.2. MAP3K7
2.3. CHD1
2.4. SPOP
2.5. PI3K Pathway
2.6. TP53
2.7. Cell Cycle: RB1 and CDKN1B
2.8. FOXA1
2.9. DNA Repair: ATM, PARP1, BRCA1, BRCA2
2.10. MYC
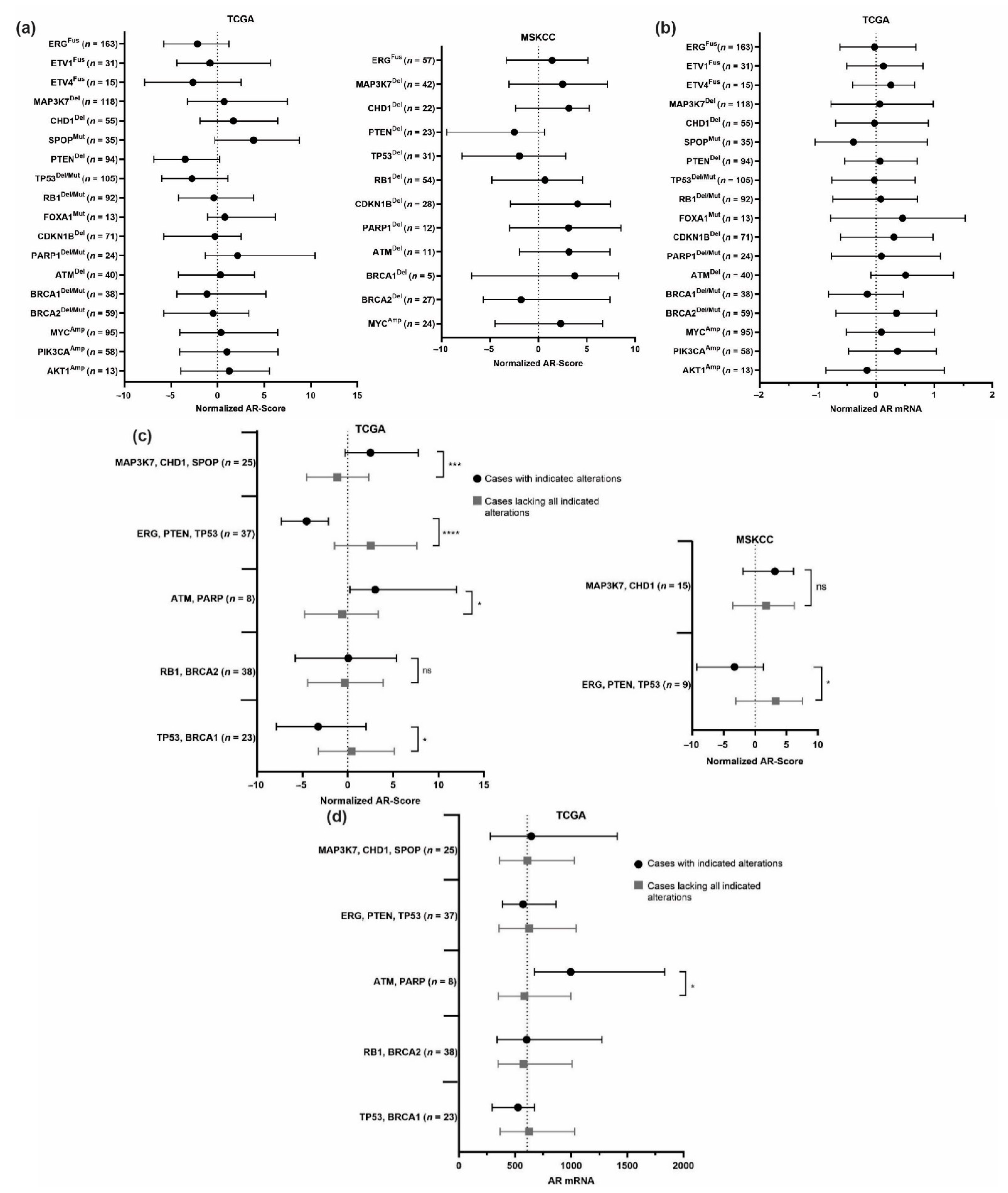
3. Genomic Subtypes
3.1. MAP3K7, CHD1, SPOP
3.2. Alterations Mutually Exclusive from ERG/ETS Fusions
3.3. ERG, PTEN, TP53
3.4. ATM and PARP1
3.5. BRCA1/TP53 and BRCA2/RB1
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.A.-O.; Miller, K.A.-O.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Nelson, W.G.; Meeker, A.K.; Coffey, D.S. Stem cell features of benign and malignant prostate epithelial cells. J. Urol. 1998, 160, 2381–2392. [Google Scholar] [CrossRef]
- Cunha, G.R.; Donjacour, A.A.; Cooke, P.S.; Mee, S.; Bigsby, R.M.; Higgins, S.J.; Sugimura, Y. The endocrinology and developmental biology of the prostate. Endocr. Rev. 1987, 8, 338–362. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, G.; Irvine, R.A.; Coetzee, G.A.; Tilley, W.D. Contribution of the androgen receptor to prostate cancer predisposition and progression. Cancer Metastasis Rev. 2001, 20, 207–223. [Google Scholar] [CrossRef] [PubMed]
- van der Kwast, T.H.; Schalken, J.; Ruizeveld de Winter, J.A.; van Vroonhoven, C.C.; Mulder, E.; Boersma, W.; Trapman, J. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int. J. Cancer 1991, 48, 189–193. [Google Scholar] [CrossRef]
- Denis, L.J.; Griffiths, K. Endocrine treatment in prostate cancer. Semin. Surg. Oncol. 2000, 18, 52–74. [Google Scholar] [CrossRef]
- Zhu, Y.; Wen, J.; Huang, G.; Mittlesteadt, J.; Wen, X.; Lu, X.A.-O. CHD1 and SPOP synergistically protect prostate epithelial cells from DNA damage. Prostate 2021, 81, 81–88. [Google Scholar] [CrossRef]
- Pienta, K.J.; Bradley, D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res. 2006, 12, 1665–1671. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Goldstein, A.S. Adaptation or selection--mechanisms of castration-resistant prostate cancer. Nat. Rev. Urol. 2013, 10, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Grivas, P.D.; Robins, D.M.; Hussain, M. Predicting response to hormonal therapy and survival in men with hormone sensitive metastatic prostate cancer. Crit. Rev. Oncol. Hematol. 2013, 85, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, M.; Oberg, A.L.; Mahoney, D.W.; Riska, S.M.; Sherwood, R.; Zhang, Y.; Zenka, R.M.; Sahasrabudhe, D.; Qin, R.; Zhang, S. Serum Proteomics on the Basis of Discovery of Predictive Biomarkers of Response to Androgen Deprivation Therapy in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2019, 17, 248–253.e7. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cancer Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cancer Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claessens, F.; Helsen, C.; Prekovic, S.; Van den Broeck, T.; Spans, L.; Van Poppel, H.; Joniau, S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol. 2014, 11, 712–716. [Google Scholar] [CrossRef]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef]
- Bubendorf, L.; Kononen, J.; Koivisto, P.; Schraml, P.; Moch, H.; Gasser, T.C.; Willi, N.; Mihatsch, M.J.; Sauter, G.; Kallioniemi, O.P. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999, 59, 803–806. [Google Scholar]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hieronymus, H.; Lamb, J.; Ross, K.N.; Peng, X.P.; Clement, C.; Rodina, A.; Nieto, M.; Du, J.; Stegmaier, K.; Raj, S.M.; et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006, 10, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Smith, L.R.; Roulston, D.; Helgeson, B.E.; Cao, X.; Wei, J.T.; Rubin, M.A.; Shah, R.B.; et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006, 66, 3396–3400. [Google Scholar] [CrossRef] [Green Version]
- Helgeson, B.E.; Tomlins, S.A.; Shah, N.; Laxman, B.; Cao, Q.; Prensner, J.R.; Cao, X.; Singla, N.; Montie, J.E.; Varambally, S.; et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008, 68, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.-M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006, 66, 8337–8341. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostwick, D.G.; Liu, L.; Brawer, M.K.; Qian, J. High-grade prostatic intraepithelial neoplasia. Rev. Urol. 2004, 6, 171–179. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.-G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.-D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef]
- Mani, R.S.; Tomlins, S.A.; Callahan, K.; Ghosh, A.; Nyati, M.K.; Varambally, S.; Palanisamy, N.; Chinnaiyan, A.M. Induced chromosomal proximity and gene fusions in prostate cancer. Science 2009, 326, 1230. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Dobi, A.; Mohamed, A.; Li, H.; Thangapazham, R.L.; Furusato, B.; Shaheduzzaman, S.; Tan, S.H.; Vaidyanathan, G.; Whitman, E.; et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 2008, 27, 5348–5353. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.E.; Tsai, M.J.; Aebersold, R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Mol. Endocrinol. 2003, 17, 1726–1737. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Mani, R.-S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; Gong, Y.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–554. [Google Scholar] [CrossRef] [Green Version]
- Kunderfranco, P.; Mello-Grand, M.; Cangemi, R.; Pellini, S.; Mensah, A.; Albertini, V.; Malek, A.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS ONE 2010, 5, e10547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Mei, Q.; Zielinska-Kwiatkowska, A.; Matsui, Y.; Blackburn, M.L.; Benedetti, D.; Krumm, A.A.; Taborsky, G.J.; Chansky, H.A. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem. J. 2003, 369, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Graff, R.E.; Pettersson, A.; Lis, R.T.; DuPre, N.; Jordahl, K.M.; Nuttall, E.; Rider, J.R.; Fiorentino, M.; Sesso, H.D.; Kenfield, S.A.; et al. The TMPRSS2:ERG fusion and response to androgen deprivation therapy for prostate cancer. Prostate 2015, 75, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Bernasocchi, T.; El Tekle, G.A.-O.; Bolis, M.; Mutti, A.; Vallerga, A.; Brandt, L.P.; Spriano, F.; Svinkina, T.; Zoma, M.; Ceserani, V.; et al. Dual functions of SPOP and ERG dictate androgen therapy responses in prostate cancer. Nat. Commun. 2021, 12, 734. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Shi, L.; Cimic, A.; Romero, L.; Sui, G.; Lees, C.J.; Cline, J.M.; Seals, D.F.; Sirintrapun, J.S.; McCoy, T.P.; et al. Suppression of Tak1 promotes prostate tumorigenesis. Cancer Res. 2012, 72, 2833–2843. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.U.; Rider, L.; Nieto, C.; Romero, L.; Karimpour-Fard, A.; Loda, M.; Lucia, M.S.; Wu, M.; Shi, L.; Cimic, A.; et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res. 2015, 75, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chang, B.L.; Cramer, S.; Koty, P.P.; Li, T.; Sun, J.; Turner, A.R.; Von Kap-Herr, C.; Bobby, P.; Rao, J.; et al. Deletion of a small consensus region at 6q15, including the MAP3K7 gene, is significantly associated with high-grade prostate cancers. Clin. Cancer Res. 2007, 13, 5028–5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluth, M.; Hesse, J.; Heinl, A.; Krohn, A.; Steurer, S.; Sirma, H.; Simon, R.; Mayer, P.-S.; Schumacher, U.; Grupp, K.; et al. Genomic deletion of MAP3K7 at 6q12-22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod. Pathol. 2013, 26, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Jillson, L.K.; Rider, L.C.; Rodrigues, L.U.; Romero, L.; Karimpour-Fard, A.; Nieto, C.; Gillette, C.A.-O.; Torkko, K.; Danis, E.; Smith, E.E.; et al. MAP3K7 Loss Drives Enhanced Androgen Signaling and Independently Confers Risk of Recurrence in Prostate Cancer with Joint Loss of CHD1. Mol. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ormond, D.A.-O.; Kleinschmidt-DeMasters, B.K.; Cavalcante, D.; Smith, E.E.; Cramer, S.D.; Lucia, M.S. Prostatic adenocarcinoma CNS parenchymal and dural metastases: Alterations in ERG, CHD1 and MAP3K7 expression. J. Neurooncol. 2019, 142, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, Y.; Ta, H.Q.; Gioeli, D. Androgen receptor phosphorylation: Biological context and functional consequences. Endocr. Relat. Cancer 2014, 21, T131–T145. [Google Scholar] [CrossRef]
- Gioeli, D.; Black, B.E.; Gordon, V.; Spencer, A.; Kesler, C.T.; Eblen, S.T.; Paschal, B.M.; Weber, M.J. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol. Endocrinol. 2006, 20, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washino, S.; Rider, L.C.; Romero, L.; Jillson, L.K.; Affandi, T.A.-O.; Ohm, A.A.-O.; Lam, E.T.; Reyland, M.E.; Costello, J.A.-O.; Cramer, S.D. Loss of MAP3K7 Sensitizes Prostate Cancer Cells to CDK1/2 Inhibition and DNA Damage by Disrupting Homologous Recombination. Mol. Cancer Res. 2019, 17, 1985–1998. [Google Scholar] [CrossRef] [Green Version]
- Simic, R.; Lindstrom, D.L.; Tran, H.G.; Roinick, K.L.; Costa, P.J.; Johnson, A.D.; Hartzog, G.A.; Arndt, K.M. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J. 2003, 22, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Lusser, A.; Urwin, D.L.; Kadonaga, J.T. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat. Struct. Mol. Biol. 2005, 12, 160–166. [Google Scholar] [CrossRef]
- Augello, M.A.; Liu, D.; Deonarine, L.D.; Robinson, B.D.; Huang, D.; Stelloo, S.; Blattner, M.; Doane, A.S.; Wong, E.W.P.; Chen, Y.; et al. CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell 2019, 35, 603–617.e8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.D.; Deng, S.; Hoover, E.; Chen, C.C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e11. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2018, 19, e46783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef]
- Mani, R.S. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov. Today 2014, 19, 1498–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-substrate complexes: Insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurillat, J.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015, 4, e09207. [Google Scholar] [CrossRef]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.E.; Eedunuri, V.K.; Chew, S.A.; Zimmermann, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Wang, C.; Deng, Y.; Yu, L.; Huang, H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014, 6, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Batra, J. Speckle-type POZ protein mutations interrupt tumor suppressor function of speckle-type POZ protein in prostate cancer by affecting androgen receptor degradation. Asian J. Androl. 2014, 16, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Rajapakshe, K.; Shah, S.S.; Shou, J.; Eedunuri, V.K.; Foley, C.; Fiskus, W.; Rajendran, M.; Chew, S.A.; Zimmermann, M.; et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2019, 79, 4552. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; An, J.; Yang, Y.; Wu, D.; Bai, Y.; Cao, W.; Ma, L.; Chen, J.; Yu, Z.; He, Y.; et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol. Med. 2018, 10, e8478. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.A.-O.X.; Guo, J.; Zhang, J.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [Green Version]
- Saal, L.H.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.-B.; Maurer, M.; Koujak, S.; Ferrando, A.; Malmström, P.; Memeo, L.; et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef] [Green Version]
- Krohn, A.; Diedler, T.; Burkhardt, L.; Mayer, P.-S.; De Silva, C.; Meyer-Kornblum, M.; Kötschau, D.; Tennstedt, P.; Huang, J.; Gerhäuser, C.; et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am. J. Pathol. 2012, 18, 401–412. [Google Scholar] [CrossRef]
- Kaarbø, M.; Mikkelsen, O.L.; Malerød, L.; Qu, S.; Lobert, V.H.; Akgul, G.; Halvorsen, T.; Maelandsmo, G.M.; Saatcioglu, F. PI3K-AKT-mTOR pathway is dominant over androgen receptor signaling in prostate cancer cells. Cell Oncol. 2010, 32, 11–27. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Johnson, D.; Luong, R.; Sun, Z. Crosstalking between androgen and PI3K/AKT signaling pathways in prostate cancer cells. J. Biol. Chem. 2015, 290, 2759–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.K.; Yeh, S.; Kang, H.Y.; Chang, C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7200–7205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Klempner, S.J.; Myers, A.P.; Cantley, L.C. What a tangled web we weave: Emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013, 3, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Flesken-Nikitin, A.; Corney, D.C.; Wang, W.; Goodrich, D.W.; Roy-Burman, P.; Nikitin, A.Y. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006, 66, 7889–7898. [Google Scholar] [CrossRef] [Green Version]
- Burchardt, M.; Burchardt, T.; Shabsigh, A.; Ghafar, M.; Chen, M.W.; Anastasiadis, A.; de la Taille, A.; Kiss, A.; Buttyan, R. Reduction of wild type p53 function confers a hormone resistant phenotype on LNCaP prostate cancer cells. Prostate 2001, 48, 225–230. [Google Scholar] [CrossRef]
- Navone, N.M.; Troncoso, P.; Pisters, L.L.; Goodrow, T.L.; Palmer, J.L.; Nichols, W.W.; von Eschenbach, A.C.; Conti, C.J. p53 protein accumulation and gene mutation in the progression of human prostate carcinoma. J. Natl. Cancer Inst. 1993, 85, 1657–1669. [Google Scholar] [CrossRef]
- Cronauer, M.V.; Troncoso, P.; Pisters, L.L.; Goodrow, T.L.; Palmer, J.L.; Nichols, W.W.; von Eschenbach, A.C.; Conti., C.J. Inhibition of p53 function diminishes androgen receptor-mediated signaling in prostate cancer cell lines. Oncogene 2004, 23, 3541–3549. [Google Scholar] [CrossRef] [Green Version]
- Chappell, W.H.; Lehmann, B.D.; Terrian, D.M.; Abrams, S.L.; Steelman, L.S.; McCubrey, J.A. p53 expression controls prostate cancer sensitivity to chemotherapy and the MDM2 inhibitor Nutlin-3. Cell Cycle 2012, 11, 4579–4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddison, L.A.; Sutherland, B.W.; Barrios, R.J.; Greenberg, N.M. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004, 64, 6018–6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.S.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faisal, F.A.; Murali, S.; Kaur, H.; Vidotto, T.A.-O.; Guedes, L.B.; Salles, D.C.; Kothari, V.; Tosoian, J.A.-O.; Han, S.; Hovelson, D.H.; et al. CDKN1B Deletions are Associated with Metastasis in African American Men with Clinically Localized, Surgically Treated Prostate Cancer. Clin. Cancer Res. 2020, 26, 2595–2602. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Cirillo, L.A.; McPherson, C.E.; Bossard, P.; Stevens, K.; Cherian, S.; Shim, E.Y.; Clark, K.L.; Burley, S.K.; Zaret, K.S. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998, 17, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Parolia, A.; Cieslik, M.; Chu, S.C.; Xiao, L.; Ouchi, T.; Zhang, Y.; Wang, X.; Vats, P.; Cao, X.; Pitchiaya, S.; et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019, 571, 413–418. [Google Scholar] [CrossRef]
- Xu, B.; Song, B.; Lu, X.; Kim, J.; Hu, M.; Zhao, J.C.; Yu, J. Altered chromatin recruitment by FOXA1 mutations promotes androgen independence and prostate cancer progression. Cell Res. 2019, 29, 773–775. [Google Scholar] [CrossRef]
- Gao, N.; Zhang, J.; Rao, M.A.; Case, T.C.; Mirosevich, J.; Wang, Y.; Jin, R.; Gupta, A.; Rennie, P.S.; Matusik, R.J. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003, 17, 1484–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.J.; Zhao, J.C.; Wu, L.; Kim, J.; Yu, J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014, 5, 3972. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.J.; Karthaus, W.R.; Hoover, E.; Liu, D.; Gruet, A.; Zhang, Z.; Cho, H.; DiLoreto, R.; Chhangawala, S.; Liu, Y.; et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature 2019, 571, 408–412. [Google Scholar] [CrossRef]
- Gao, S.A.-O.; Chen, S.; Han, D.; Barrett, D.; Han, W.A.-O.; Ahmed, M.; Patalano, S.; Macoska, J.A.; He, H.H.; Cai, C.A.-O. Forkhead domain mutations in FOXA1 drive prostate cancer progression. Cell Res. 2019, 29, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Mehta, R.J.; Nakshatri, H.; Idrees, M.T.; Badve, S.S. High-level expression of forkhead-box protein A1 in metastatic prostate cancer. Histopathology 2011, 58, 766–772. [Google Scholar] [CrossRef]
- Messina, C.; Cattrini, C.; Soldato, D.; Vallome, G.; Caffo, O.; Castro, E.; Olmos, D.; Boccardo, F.; Zanardi, E.A.-O. BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications. J. Oncol. 2020, 2020, 4986365. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.A.-O.; Ma, H.T.; Poon, R.A.-O. Synergism between ATM and PARP1 Inhibition Involves DNA Damage and Abrogating the G(2) DNA Damage Checkpoint. Mol. Cancer Ther. 2020, 19, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [Green Version]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [Green Version]
- Spratt, D.E.; Evans, M.J.; Davis, B.J.; Doran, M.G.; Lee, M.X.; Shah, N.; Wongvipat, J.; Carnazza, K.E.; Klee, G.G.; Polkinghorn, W.; et al. Androgen Receptor Upregulation Mediates Radioresistance after Ionizing Radiation. Cancer Res. 2015, 75, 4688–4696. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, Z.A.; Krauss, D.J. Adjuvant androgen deprivation therapy for prostate cancer treated with radiation therapy. Transl. Androl. Urol. 2018, 7, 378–389. [Google Scholar] [CrossRef]
- Spratt, D.E.; Malone, S.; Roy, S.; Grimes, S.; Eapen, L.; Morgan, S.C.; Malone, J.; Craig, J.; Dess, R.T.; Jackson, W.C.; et al. Prostate Radiotherapy with Adjuvant Androgen Deprivation Therapy (ADT) Improves Metastasis-Free Survival Compared to Neoadjuvant ADT: An Individual Patient Meta-Analysis. J. Clin. Oncol. 2021, 39, 136–144. [Google Scholar] [CrossRef]
- Jenkins, R.B.; Qian, J.; Lieber, M.M.; Bostwick, D.G. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 1997, 57, 524–531. [Google Scholar]
- Fromont, G.; Godet, J.; Peyret, A.; Irani, J.; Celhay, O.; Rozet, F.; Cathelineau, X.; Cussenot, O. 8q24 amplification is associated with Myc expression and prostate cancer progression and is an independent predictor of recurrence after radical prostatectomy. Hum. Pathol. 2013, 44, 1617–1623. [Google Scholar] [CrossRef]
- Barfeld, S.J.; Fazli, L.; Persson, M.; Marjavaara, L.; Urbanucci, A.; Kaukoniemi, K.M.; Rennie, P.S.; Ceder, Y.; Chabes, A.; Visakorpi, T.; et al. Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer. Oncotarget 2015, 6, 12587–12602. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Gurel, B.; Sutcliffe, S.; Aryee, M.J.; Schultz, D.; Iwata, T.; Uemura, M.; Zeller, K.I.; Anele, U.; Zheng, Q.; et al. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am. J. Pathol. 2011, 178, 1824–1834. [Google Scholar] [CrossRef]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003, 4, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Iwata, T.; Schultz, D.; Hicks, J.; Hubbard, G.K.; Mutton, L.N.; Lotan, T.L.; Bethel, C.; Lotz, M.T.; Yegnasubramanian, S.; Nelson, W.G.; et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS ONE 2010, 5, e9427. [Google Scholar] [CrossRef]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Vander Griend, D.J.; Litvinov, I.V.; Isaacs, J.T. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int. J. Biol. Sci. 2014, 10, 627–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barfeld, S.J.; Urbanucci, A.; Itkonen, H.M.; Fazli, L.; Hicks, J.L.; Thiede, B.; Rennie, P.S.; Yegnasubramanian, S.; DeMarzo, A.M.; Mills, I.G. c-Myc Antagonises the Transcriptional Activity of the Androgen Receptor in Prostate Cancer Affecting Key Gene Networks. EBioMedicine 2017, 18, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Bernard, D.; Pourtier-Manzanedo, A.; Gil, J.; Beach, D.H. Myc confers androgen-independent prostate cancer cell growth. J. Clin. Investig. 2003, 112, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Cao, S.; Jin, L.; Kobelski, M.; Schouest, B.; Wang, X.; Ungerleider, N.; Baddoo, M.; Zhang, W.; Corey, E.; et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019, 38, 4977–4989. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef]
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The Dual Inhibition of RNA Pol I Transcription and PIM Kinase as a New Therapeutic Approach to Treat Advanced Prostate Cancer. Clin. Cancer Res. 2016, 22, 5539–5552. [Google Scholar] [CrossRef] [Green Version]
- Boysen, G.; Rodrigues, D.N.; Rescigno, P.; Seed, G.; Dolling, D.; Riisnaes, R.; Crespo, M.; Zafeiriou, Z.; Sumanasuriya, S.; Bianchini, D.; et al. SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin. Cancer Res. 2018, 24, 5585–5593. [Google Scholar] [CrossRef] [Green Version]
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell 2011, 19, 664–678. [Google Scholar] [CrossRef] [Green Version]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 2020, 52, 984. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chi, P.; Rockowitz, S.; Iaquinta, P.J.; Shamu, T.; Shukla, S.; Gao, D.; Sirota, I.; Carver, B.S.; Wongvipat, J.; et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat. Med. 2013, 19, 1023–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, N.; Gao, D.; Hu, W.; Hieronymus, H.; Wang, S.; Lee, Y.A.-O.; Lee, C.A.-O.; Choi, D.; Gopalan, A.; Chen, Y.; et al. Aberrant Expression of ERG Promotes Resistance to Combined PI3K and AR Pathway Inhibition through Maintenance of AR Target Genes. Mol. Cancer Ther. 2019, 18, 1577–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Liu, Y.N.; Pierce, R.; Abou-Kheir, W.; Casey, O.; Seng, V.; Camacho, D.; Simpson, R.M.; Kelly, K. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am. J. Pathol. 2011, 179, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Blee, A.M.; He, Y.; Yang, Y.; Ye, Z.; Yan, Y.; Pan, Y.; Ma, T.; Dugdale, J.; Kuehn, E.; Kohli, M.; et al. TMPRSS2-ERG Controls Luminal Epithelial Lineage and Antiandrogen Sensitivity in PTEN and TP53-Mutated Prostate Cancer. Clin. Cancer Res. 2018, 24, 4551–4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Pezaro, C. PARP inhibitor combinations in prostate cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835919897537. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Antonarakis, E.S. PARP inhibition - not all gene mutations are created equal. Nat. Rev. Urol. 2019, 16, 4–6. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers (Basel) 2020, 12, 687. [Google Scholar] [CrossRef] [Green Version]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.A.-O.; Cai, M.Y.; Elmarakeby, H.A.-O.; Park, J.A.-O.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.A.-O.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 2019, 76, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Armenia, J.; Mazzu, Y.Z.; Nandakumar, S.; Stopsack, K.A.-O.; Atiq, M.A.-O.; Komura, K.A.-O.; Jehane, L.; Hirani, R.; Chadalavada, K.; et al. Significance of BRCA2 and RB1 Co-loss in Aggressive Prostate Cancer Progression. Clin. Cancer Res. 2020, 26, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Schweizer, M.T.; Lucas, J.M.; Coleman, I.; Nyquist, M.D.; Frank, S.B.; Tharakan, R.; Mostaghel, E.; Luo, J.; Pritchard, C.C.; et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. Clin. Investig. 2019, 129, 4245–4260. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, O.S.; Nyquist, M.D.; Schweizer, M.T.; Balk, S.P.; Corey, E.; Plymate, S.; Nelson, P.S.; Mostaghel, E.A. Supraphysiologic Testosterone Therapy in the Treatment of Prostate Cancer: Models, Mechanisms and Questions. Cancers 2017, 9, 166. [Google Scholar] [CrossRef] [Green Version]
- Nyquist, M.D.; Ang, L.S.; Corella, A.; Coleman, I.M.; Meers, M.P.; Christiani, A.J.; Pierce, C.; Janssens, D.H.; Meade, H.E.; Bose, A.; et al. Selective androgen receptor modulators activate the canonical prostate cancer androgen receptor program and repress cancer growth. J. Clin. Investig. 2021, 131, e146777. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Subtype | A | B | Neither | A Not B | B Not A | Both | Log2 Odds Ratio | p-Value | q-Value | Tendency |
|---|---|---|---|---|---|---|---|---|---|---|
| MAP3K7, CHD1, SPOP | MAP3K7: HOMDEL HETLOSS | CHD1: HOMDEL HETLOSS | 203 | 76 | 12 | 42 | >3 | <0.001 | <0.001 | Co-occurrence |
| MAP3K7: HOMDEL HETLOSS | SPOP: MUT = MISSENSE | 210 | 88 | 5 | 30 | >3 | <0.001 | <0.001 | Co-occurrence | |
| CHD1: HOMDEL HETLOSS | SPOP: MUT = MISSENSE | 272 | 26 | 7 | 28 | >3 | <0.001 | <0.001 | Co-occurrence | |
| CDKN1B: HOMDEL HETLOSS | MAP3K7: HOMDEL HETLOSS | 180 | 35 | 83 | 35 | 1.117 | 0.004 | 0.017 | Co-occurrence | |
| CHD1: HOMDEL HETLOSS | PARP1: HOMDEL HETLOSS | 264 | 45 | 15 | 9 | 1.816 | 0.007 | 0.033 | Co-occurrence | |
| MAP3K7: HOMDEL HETLOSS | PARP1: HOMDEL HETLOSS | 209 | 100 | 6 | 18 | 2.648 | <0.001 | <0.001 | Co-occurrence | |
| Exclusive from ERG | ERG: FUSION | MAP3K7: HOMDEL HETLOSS | 92 | 123 | 89 | 29 | −2.037 | <0.001 | <0.001 | Mutual exclusivity |
| ERG: FUSION | ETV1: FUSION | 153 | 151 | 28 | 1 | <−3 | <0.001 | <0.001 | Mutual exclusivity | |
| ERG: FUSION | CHD1: HOMDEL HETLOSS | 135 | 144 | 46 | 8 | −2.617 | <0.001 | <0.001 | Mutual exclusivity | |
| ERG: FUSION | PARP1: HOMDEL HETLOSS | 158 | 151 | 23 | 1 | <−3 | <0.001 | <0.001 | Mutual exclusivity | |
| ERG: FUSION | FOXA1: MUT | 168 | 152 | 13 | 0 | <−3 | <0.001 | 0.002 | Mutual exclusivity | |
| ERG: FUSION | MYC: AMP GAIN MUT = MISSENSE | 115 | 123 | 66 | 29 | −1.283 | <0.001 | 0.002 | Mutual exclusivity | |
| ERG: FUSION | ETV4: FUSION | 167 | 150 | 14 | 2 | −2.652 | 0.005 | 0.022 | Mutual exclusivity | |
| ERG: FUSION | SPOP: MUT = MISSENSE | 146 | 152 | 35 | 0 | <−3 | <0.001 | <0.001 | Mutual exclusivity | |
| Exclusive from SPOP | TP53: HOMDEL HETLOSS MUT | SPOP: MUT = MISSENSE | 194 | 104 | 34 | 1 | <−3 | <0.001 | <0.001 | Mutual exclusivity |
| PTEN: HOMDEL HETLOSS | SPOP: MUT = MISSENSE | 207 | 91 | 33 | 2 | −2.859 | <0.001 | 0.004 | Mutual exclusivity | |
| SPOP: MUT = MISSENSE | BRCA1: HOMDEL HETLOSS MUT | 260 | 35 | 38 | 0 | <−3 | 0.011 | 0.045 | Mutual exclusivity | |
| ERG, PTEN, TP53 | ERG: FUSION | PTEN: HOMDEL HETLOSS | 151 | 89 | 30 | 63 | 1.833 | <0.001 | <0.001 | Co-occurrence |
| PTEN: HOMDEL HETLOSS | TP53: HOMDEL HETLOSS MUT | 181 | 47 | 59 | 46 | 1.586 | <0.001 | <0.001 | Co-occurrence | |
| ERG: FUSION | TP53: HOMDEL HETLOSS MUT | 141 | 87 | 40 | 65 | 1.397 | <0.001 | <0.001 | Co-occurrence | |
| PTEN: HOMDEL HETLOSS | BRCA2: HOMDEL HETLOSS MUT | 206 | 68 | 34 | 25 | 1.155 | 0.006 | 0.028 | Co-occurrence | |
| ATM, PARP1 | PARP1: HOMDEL HETLOSS | ATM: HOMDEL HETLOSS | 285 | 17 | 24 | 7 | 2.29 | 0.003 | 0.017 | Co-occurrence |
| RB1, BRCA2 | RB1: HOMDEL HETLOSS MUT | BRCA2: HOMDEL HETLOSS MUT | 220 | 54 | 21 | 38 | 2.882 | <0.001 | <0.001 | Co-occurrence |
| RB1: HOMDEL HETLOSS MUT | CHD1: HOMDEL HETLOSS | 218 | 61 | 23 | 31 | 2.268 | <0.001 | <0.001 | Co-occurrence | |
| RB1: HOMDEL HETLOSS MUT | SPOP: MUT = MISSENSE | 224 | 74 | 17 | 18 | 1.68 | 0.001 | 0.008 | Co-occurrence | |
| PIK3CA | PIK3CA: AMP GAIN MUT = MISSENSE | TP53: HOMDEL HETLOSS MUT | 201 | 27 | 74 | 31 | 1.641 | <0.001 | <0.001 | Co-occurrence |
| PIK3CA: AMP GAIN MUT = MISSENSE | AKT1: AMP GAIN MUT = MISSENSE | 270 | 50 | 5 | 8 | >3 | <0.001 | 0.002 | Co-occurrence | |
| PIK3CA: AMP GAIN MUT = MISSENSE | MAP3K7: HOMDEL HETLOSS | 188 | 27 | 87 | 31 | 1.311 | 0.002 | 0.009 | Co-occurrence | |
| PIK3CA: AMP GAIN MUT = MISSENSE | SPOP: MUT = MISSENSE | 252 | 46 | 23 | 12 | 1.515 | 0.008 | 0.036 | Co-occurrence | |
| PIK3CA: AMP GAIN MUT = MISSENSE | CHD1: HOMDEL HETLOSS | 237 | 42 | 38 | 16 | 1.248 | 0.011 | 0.045 | Co-occurrence | |
| FOXA1 | FOXA1: MUT | ATM: HOMDEL HETLOSS | 294 | 8 | 26 | 5 | 2.821 | 0.004 | 0.018 | Co-occurrence |
| FOXA1: MUT | PARP1: HOMDEL HETLOSS | 300 | 9 | 20 | 4 | 2.737 | 0.01 | 0.04 | Co-occurrence | |
| CHD1: HOMDEL HETLOSS | FOXA1: MUT | 274 | 46 | 5 | 8 | >3 | <0.001 | 0.002 | Co-occurrence | |
| MAP3K7: HOMDEL HETLOSS | FOXA1: MUT | 211 | 109 | 4 | 9 | 2.123 | 0.012 | 0.047 | Co-occurrence | |
| RB1: HOMDEL HETLOSS MUT | FOXA1: MUT | 237 | 83 | 4 | 9 | 2.684 | 0.002 | 0.01 | Co-occurrence | |
| MYC | MYC: AMP GAIN MUT = MISSENSE | BRCA1: HOMDEL HETLOSS MUT | 219 | 76 | 19 | 19 | 1.527 | 0.002 | 0.013 | Co-occurrence |
| MYC: AMP GAIN MUT = MISSENSE | BRCA2: HOMDEL HETLOSS MUT | 212 | 62 | 26 | 33 | 2.118 | <0.001 | <0.001 | Co-occurrence | |
| MYC: AMP GAIN MUT = MISSENSE | ATM: HOMDEL HETLOSS | 224 | 78 | 14 | 17 | 1.802 | 0.001 | 0.007 | Co-occurrence | |
| CHD1: HOMDEL HETLOSS | MYC: AMP GAIN MUT = MISSENSE | 209 | 29 | 70 | 25 | 1.364 | 0.002 | 0.01 | Co-occurrence | |
| MAP3K7: HOMDEL HETLOSS | MYC: AMP GAIN MUT = MISSENSE | 169 | 69 | 46 | 49 | 1.384 | <0.001 | <0.001 | Co-occurrence | |
| PIK3CA: AMP GAIN MUT = MISSENSE | MYC: AMP GAIN MUT = MISSENSE | 213 | 25 | 62 | 33 | 2.181 | <0.001 | <0.001 | Co-occurrence | |
| RB1: HOMDEL HETLOSS MUT | MYC: AMP GAIN MUT = MISSENSE | 185 | 53 | 56 | 39 | 1.282 | <0.001 | 0.004 | Co-occurrence | |
| TP53: HOMDEL HETLOSS MUT | MYC: AMP GAIN MUT = MISSENSE | 178 | 60 | 50 | 45 | 1.417 | <0.001 | <0.001 | Co-occurrence |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jillson, L.K.; Yette, G.A.; Laajala, T.D.; Tilley, W.D.; Costello, J.C.; Cramer, S.D. Androgen Receptor Signaling in Prostate Cancer Genomic Subtypes. Cancers 2021, 13, 3272. https://doi.org/10.3390/cancers13133272
Jillson LK, Yette GA, Laajala TD, Tilley WD, Costello JC, Cramer SD. Androgen Receptor Signaling in Prostate Cancer Genomic Subtypes. Cancers. 2021; 13(13):3272. https://doi.org/10.3390/cancers13133272
Chicago/Turabian StyleJillson, Lauren K., Gabriel A. Yette, Teemu D. Laajala, Wayne D. Tilley, James C. Costello, and Scott D. Cramer. 2021. "Androgen Receptor Signaling in Prostate Cancer Genomic Subtypes" Cancers 13, no. 13: 3272. https://doi.org/10.3390/cancers13133272
APA StyleJillson, L. K., Yette, G. A., Laajala, T. D., Tilley, W. D., Costello, J. C., & Cramer, S. D. (2021). Androgen Receptor Signaling in Prostate Cancer Genomic Subtypes. Cancers, 13(13), 3272. https://doi.org/10.3390/cancers13133272