
Integrative cBioPortal Analysis Revealed Molecular Mechanisms That Regulate EGFR-PI3K-AKT-mTOR Pathway in Diffuse Gliomas of the Brain




Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. cBioPortal Database and Bioinformatics Methods
2.2. Data Processing
2.3. Statistical Analysis
3. Results
3.1. Demographic Data of Examined Diffuse Brain Gliomas
3.2. Changes in the Copy Number (CNA) of Examined Genes
3.3. CNA of AKT1, AKT2 and AKT3—the Core of the Signaling Pathway
3.4. CNA of CHUK, GSK3β and PTEN—Regulators of PI3K-AKT-mTOR Pathway
3.5. CNA of EGFR and PIK3AP1—Activators of PI3K-AKT-mTOR Pathway
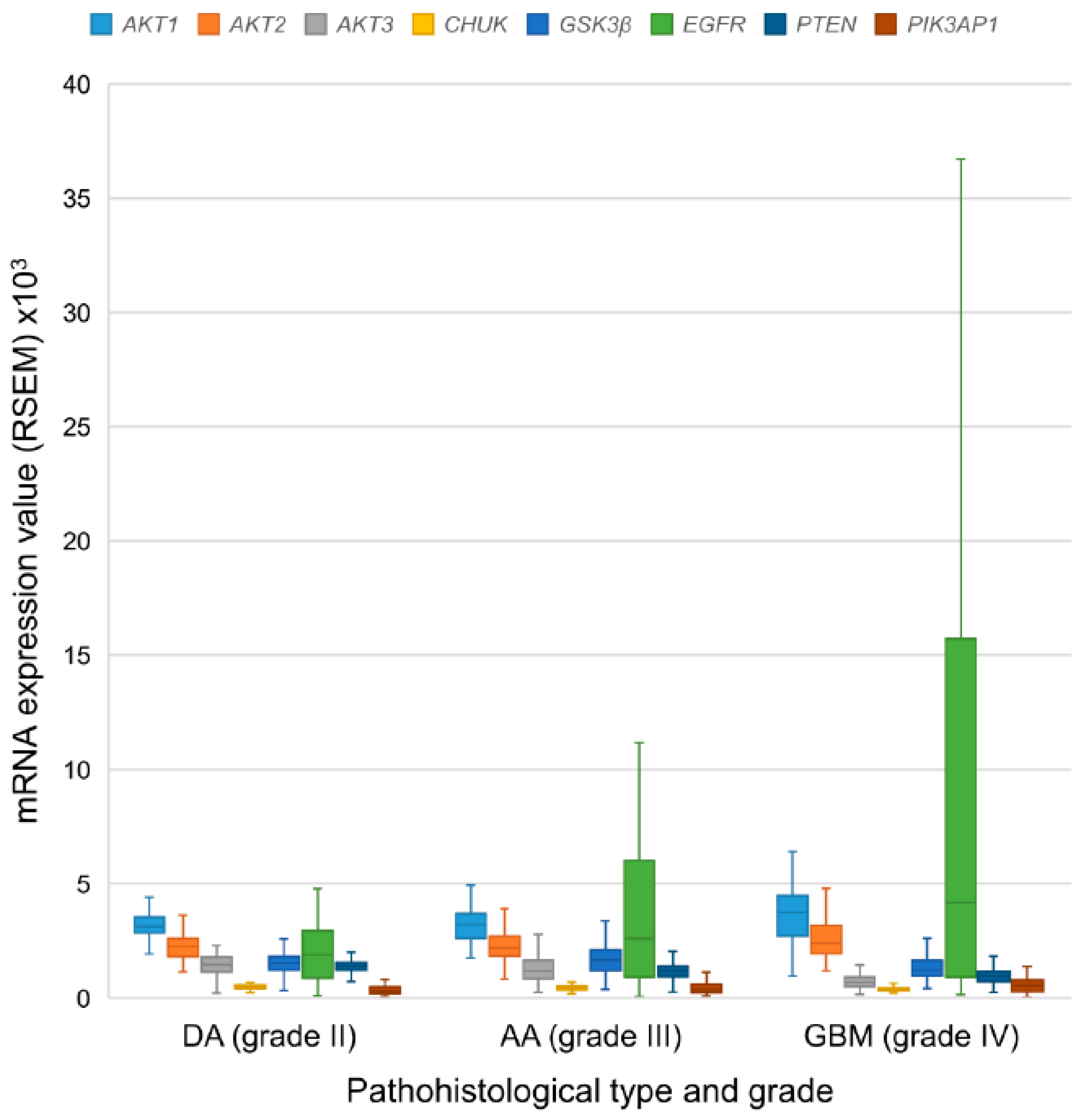
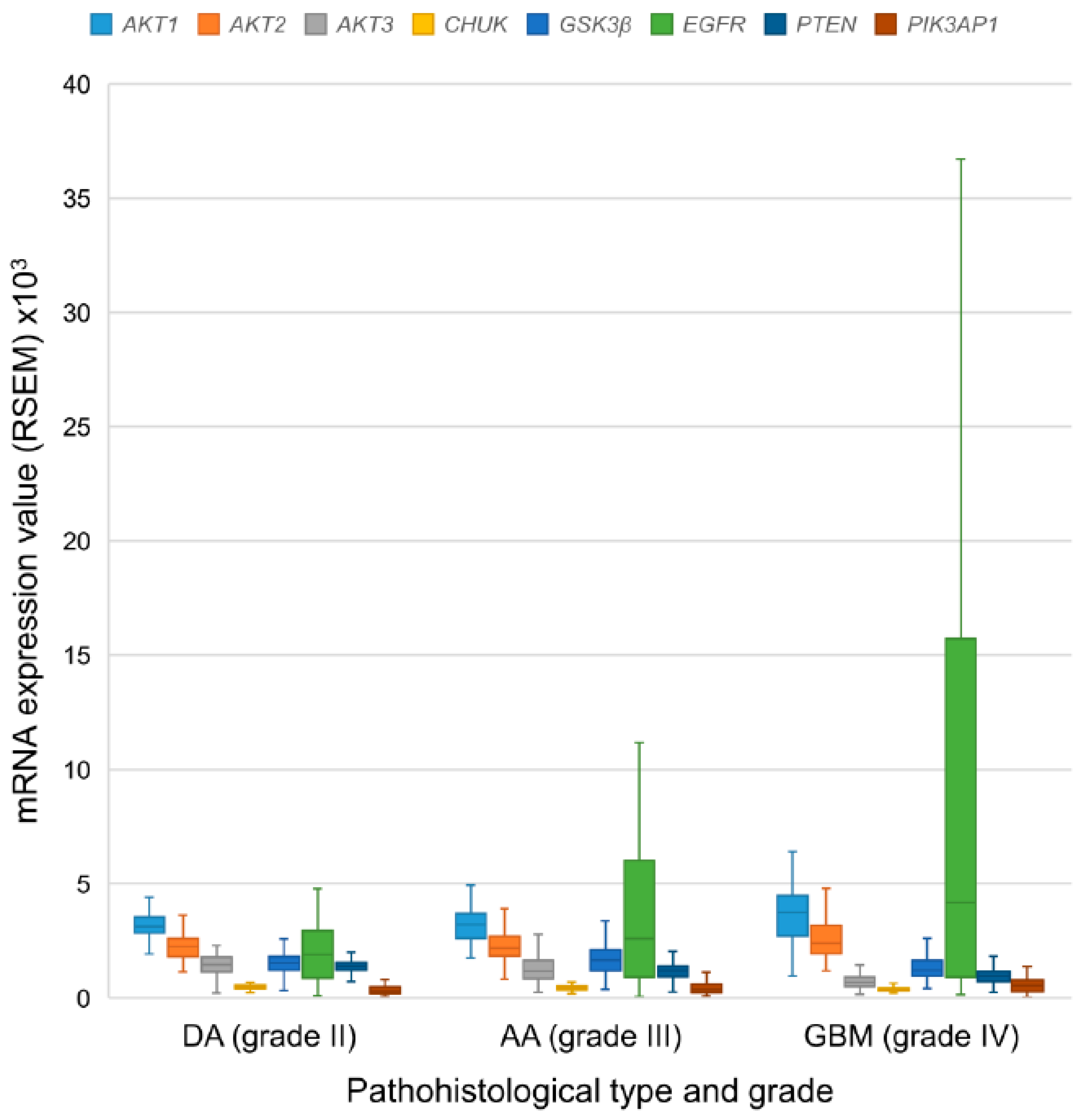
3.6. mRNA Expression Levels of the EGFR-PI3K-AKT-mTOR Pathway Participants
3.7. Protein Expression Levels of the Examined Genes
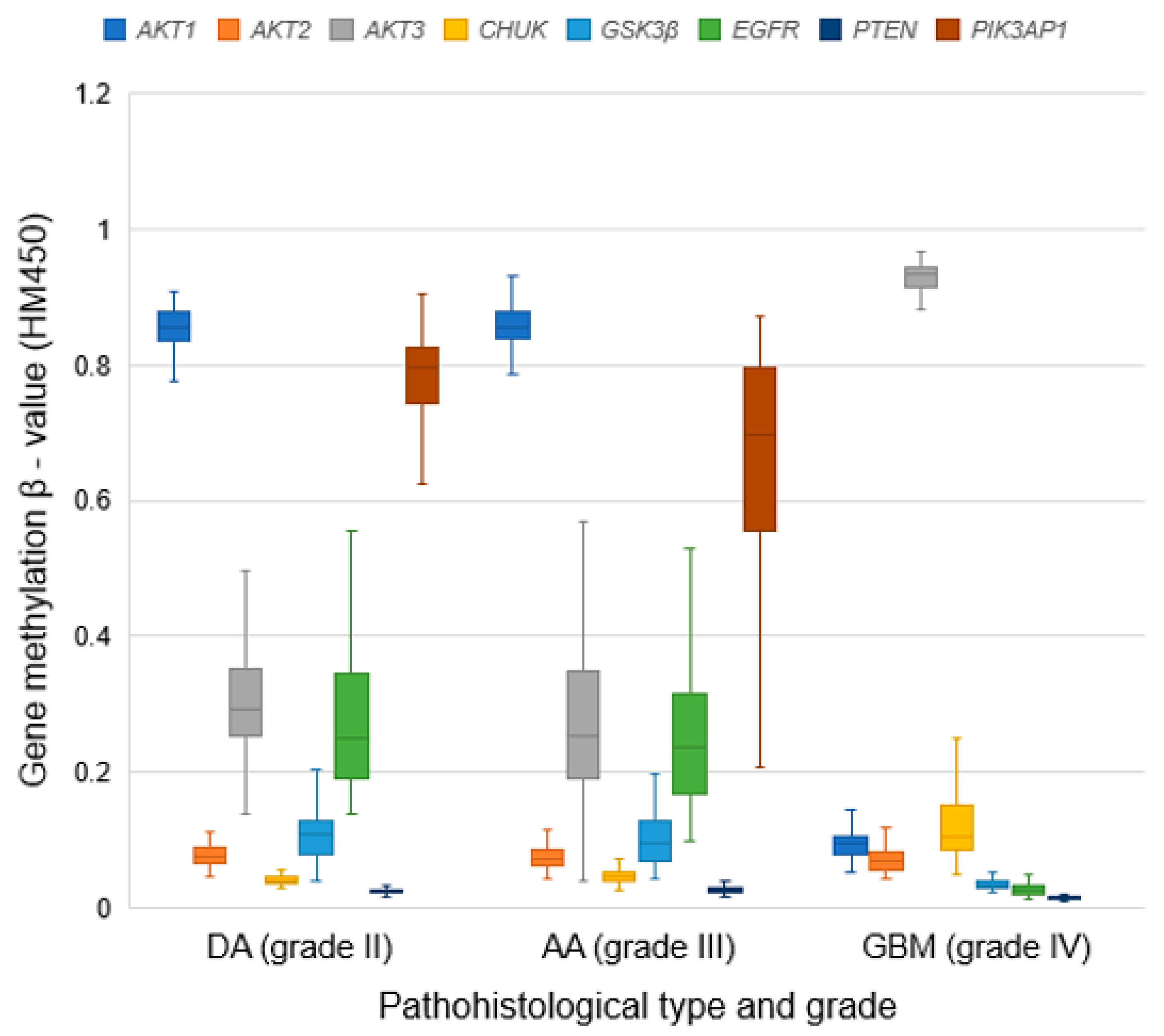
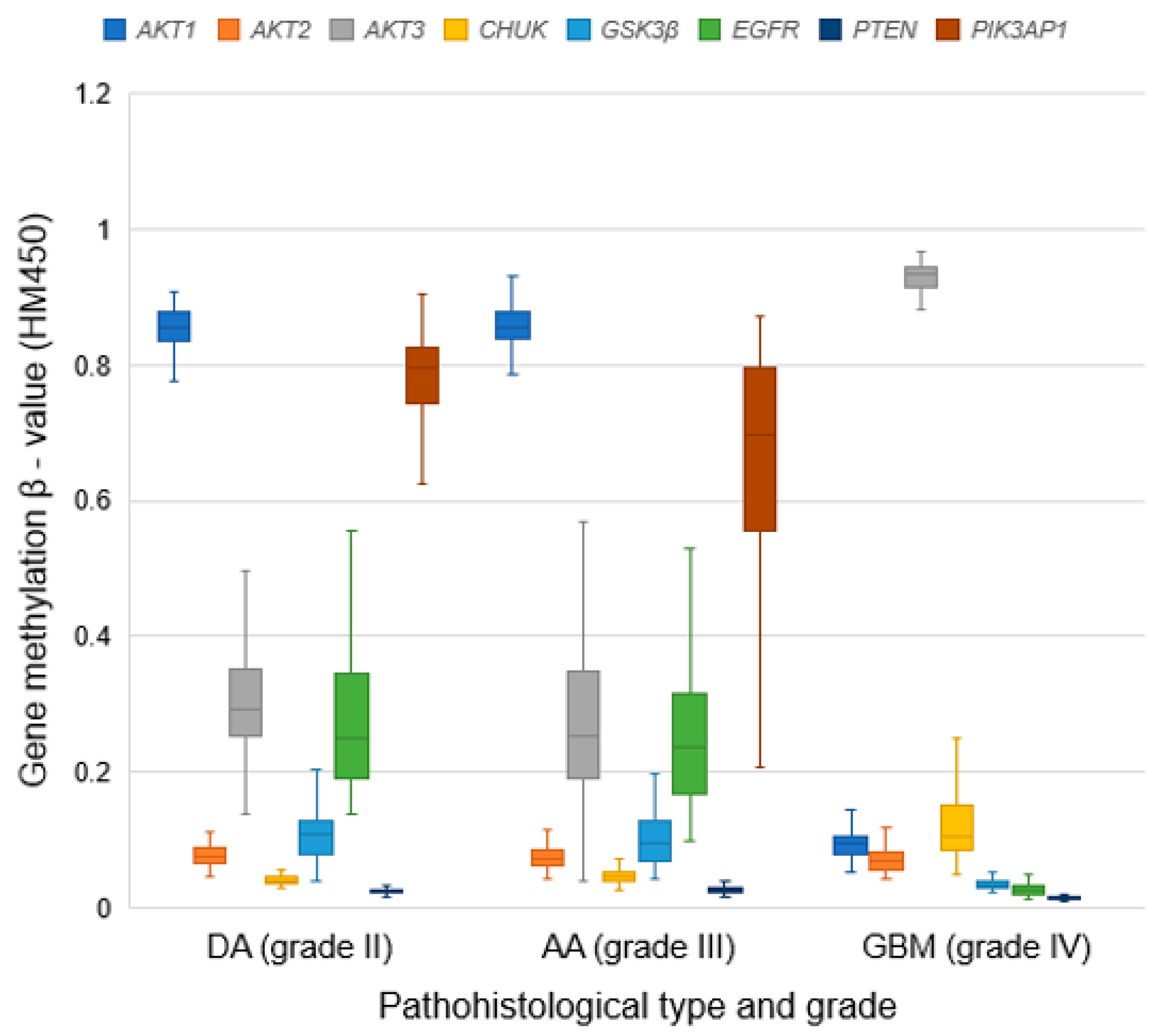
3.8. Methylation Patterns of Examined Genes
3.9. Correlation between CNA and mRNA Expression Across Glioma Types
3.10. Correlation between Mezthylation and mRNA Expression Across Glioma Types
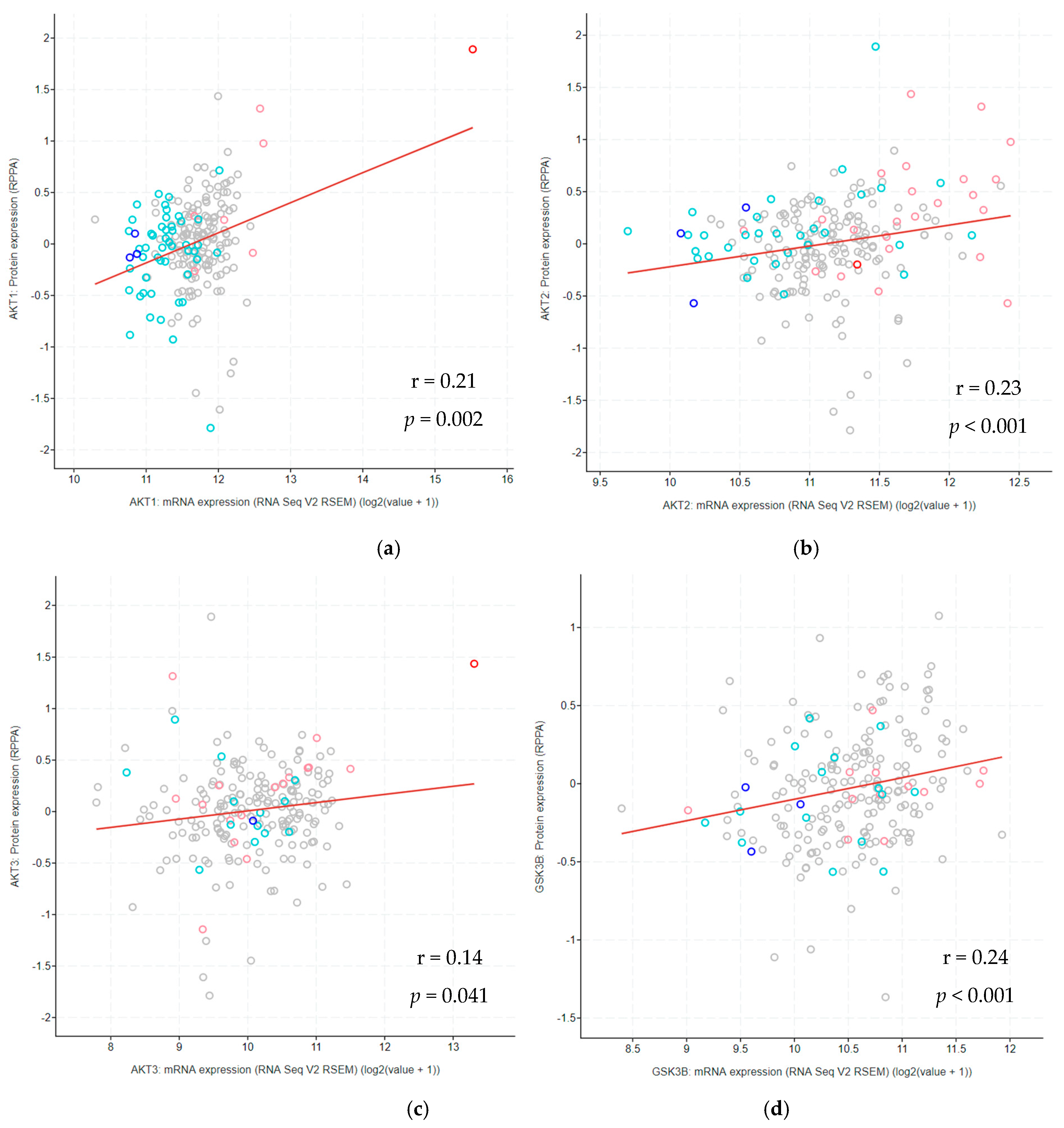
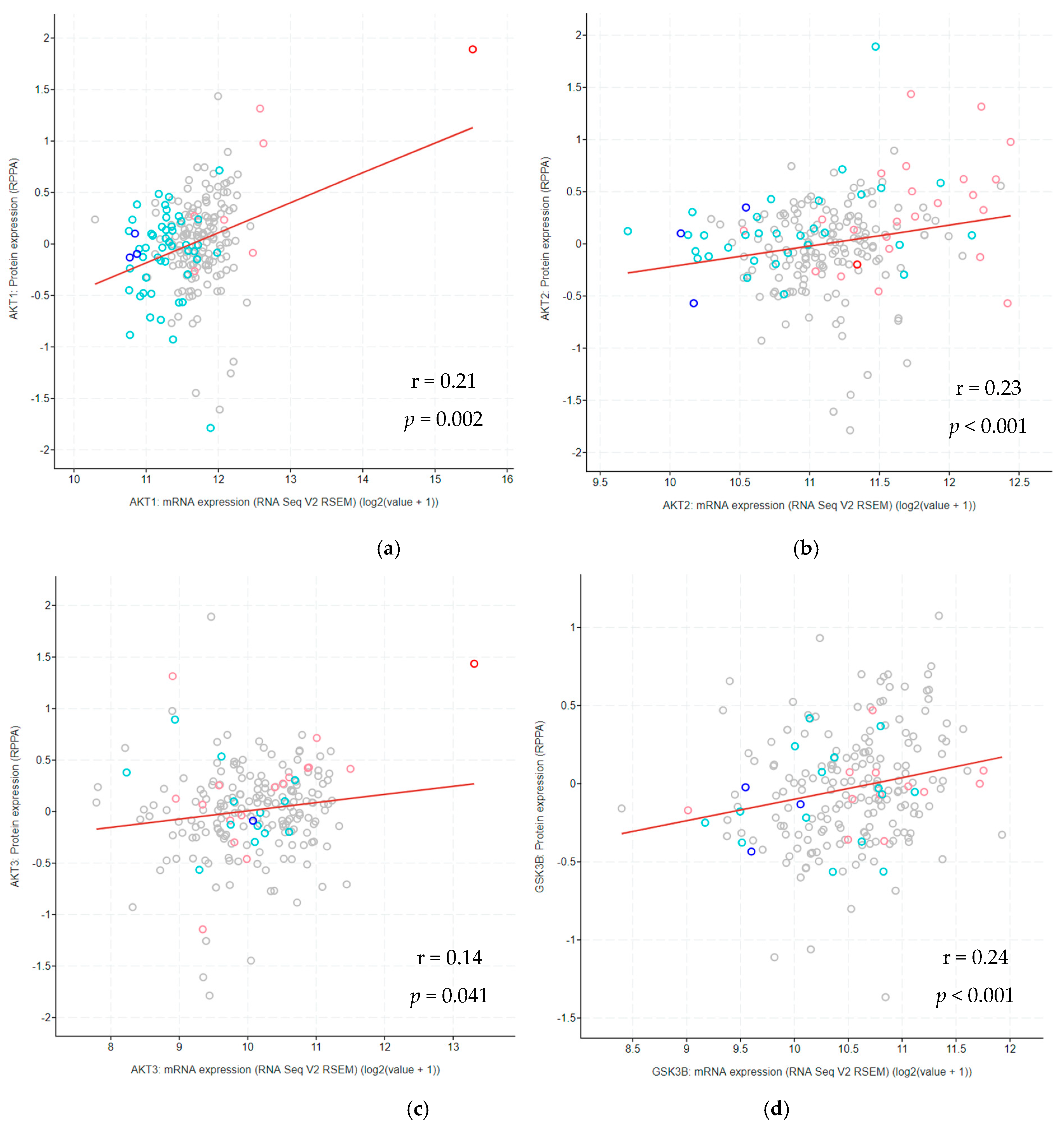
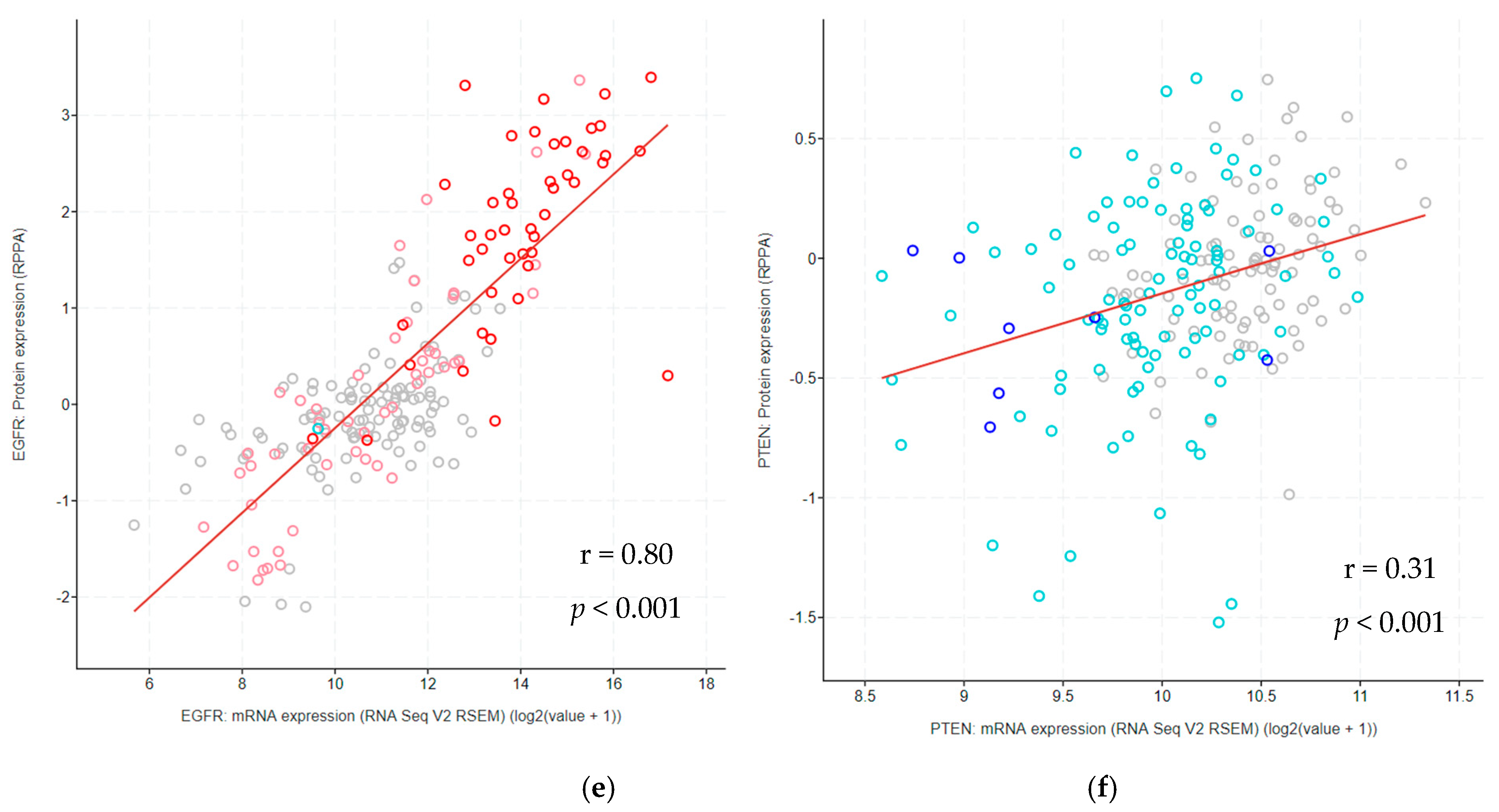
3.11. Correlation between Protein and mRNA Expression Levels Across Glioma Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maimaiti, A.; Jiang, L.; Wang, X.; Shi, X.; Pei, Y.; Hao, Y.; Paerhati, H.; Zibibula, Y.; Abudujielili, A.; Kasimu, M. Identification and validation of an individualized prognostic signature of lower-grade glioma based on nine immune related long non-coding RNA. Clin. Neurol. Neurosurg. 2021, 201, 106464. [Google Scholar] [CrossRef]
- Xia, X.; Li, X.; Li, F.; Wu, X.; Zhang, M.; Zhou, H.; Huang, N.; Yang, X.; Xiao, F.; Liu, D.; et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol. Cancer 2019, 18, 131. [Google Scholar] [CrossRef] [Green Version]
- Wolin, I.A.; Heinrich, I.A.; Nascimento, A.P.M.; Welter, P.G.; Sosa, L.D.V.; De Paul, A.L.; Zanotto-Filho, A.; Nedel, C.B.; Lima, L.D.; Osterne, V.J.S.; et al. ConBr lectin modulates MAPKs and Akt pathways and triggers autophagic glioma cell death by a mechanism dependent upon caspase-8 activation. Biochimie 2021, 180, 186–204. [Google Scholar] [CrossRef]
- Barthel, F.P.; The GLASS Consortium; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef]
- Louis, D.N.; von Deimling, A.; Cavenee, W.K. Diffuse astrocytic and oligodendroglial tumours—Introduction. In WHO Classification of Tumours of the Central Nervous System, 4th ed.; Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Eds.; The International Agency for Research on Cancer: Lyon, France, 2016; pp. 15–17. [Google Scholar]
- Lasica, A.B.; Jaunmuktane, Z.; Fersht, N.; Kirkman, M.A.; Dixon, L.; Hoskote, C.; Brandner, S.; Samandouras, G. Genomic Prognosticators and Extent of Resection in Molecularly Subtyped World Health Organization Grade II and III Gliomas–A Single-Institution, Nine-Year Data. World Neurosurg. 2021, 15. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Jerčić, K.G.; Logara, M.; Bukovac, A.; Bakarić, R.; Borovečki, F. Comparable Genomic Copy Number Aberrations Differ across Astrocytoma Malignancy Grades. Int. J. Mol. Sci. 2019, 20, 1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Yuan, X.; Jiang, Z.; Gan, D.; Ding, L.; Sun, Y.; Zhou, J.; Xu, L.; Liu, Y.; Wang, G. Regulation of AKT phosphorylation by GSK3β and PTEN to control chemoresistance in breast cancer. Breast Cancer Res. Treat. 2019, 176, 291–301. [Google Scholar] [CrossRef]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Liu, J.; Hong, W.; Fei, X.; Liu, R. Arctigenin Inhibits Glioblastoma Proliferation through the AKT/mTOR Pathway and Induces Autophagy. BioMed Res. Int. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, S.; Wu, C.; Huang, W.; Xu, B.; Lian, S.; Wang, L.; Yue, S.; Chen, N.; Zhu, Z. PD-1-Mediated PI3K/Akt/mTOR, Caspase 9/Caspase 3 and ERK Pathways Are Involved in Regulating the Apoptosis and Proliferation of CD4+ and CD8+ T Cells During BVDV Infection in vitro. Front. Immunol. 2020, 11, 467. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, Y.; Wang, Y.; Zhao, Z.; Li, T. LINC00473 rescues human bone marrow mesenchymal stem cells from apoptosis induced by dexamethasone through the PEBP1-mediated Akt/Bad/Bcl-2 signaling pathway. Int. J. Mol. Med. 2020, 47, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kan, H.; Liu, Y.; Ding, W. Plumbagin induces Ishikawa cell cycle arrest, autophagy, and apoptosis via the PI3K/Akt signaling pathway in endometrial cancer. Food Chem. Toxicol. 2021, 148, 111957. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Z.; Wang, S.; Gao, X.; Qian, M.; Qiu, W.; Zhang, Z.; Zhang, S.; Qi, W.; Sun, X.; et al. TGFβ1-induced beta-site APP-cleaving enzyme 2 upregulation promotes tumorigenesis through the NF-κB signalling pathway in human gliomas. Mol. Oncol. 2020, 14, 407–425. [Google Scholar] [CrossRef]
- Luo, L.; Lucas, R.M.; Liu, L.; Stow, J. Signalling, sorting and scaffolding adaptors for Toll-like receptors. J. Cell Sci. 2020, 133, 239194. [Google Scholar] [CrossRef] [PubMed]
- Lovšin, E.; Kovač, J.; Tesovnik, T.; Toplak, N.; Perko, D.; Rozmarič, T.; Debeljak, M.; Avčin, T. PIK3AP1 and SPON2 Genes Are Differentially Methylated in Patients With Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis (PFAPA) Syndrome. Front. Immunol. 2020, 11, 1322. [Google Scholar] [CrossRef]
- Shahcheraghi, S.H.; Tchokonte-Nana, V.; Lotfi, M.; Lotfi, M.; Ghorbani, A.; Sadeghnia, H.R. Wnt/beta-catenin and PI3K/Akt/mTOR Signaling Pathways in Glioblastoma: Two Main Targets for Drug Design: A Review. Curr. Pharm. Des. 2020, 26, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Liang, W.-W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.-W.; Reynolds, S.M.; Wyczalkowski, M.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; LaLonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, J.; Li, Y.; Wang, Z.; Kang, S. Genome-wide analysis of DNA methylation in endometriosis using Illumina Human Methylation 450 K BeadChips. Mol. Reprod. Dev. 2019, 86, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, Y.-W.; Zhang, M.Q.; Gazdar, A.F. DNA methylation data analysis and its application to cancer research. Epigenomics 2013, 5, 301–316. [Google Scholar] [CrossRef] [Green Version]
- Mu, M.; Niu, W.; Zhang, X.; Hu, S.; Niu, C. LncRNA BCYRN1 inhibits glioma tumorigenesis by competitively binding with miR-619-5p to regulate CUEDC2 expression and the PTEN/AKT/p21 pathway. Oncogene 2020, 39, 6879–6892. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Minna, E.; Romeo, P.; Dugo, M.; De Cecco, L.; Todoerti, K.; Pilotti, S.; Perrone, F.; Seregni, E.; Agnelli, L.; Neri, A.; et al. miR-451a is underexpressed and targets AKT/mTOR pathway in papillary thyroid carcinoma. Oncotarget 2016, 7, 12731–12747. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Li, X.; Lin, Z.; Li, H.; Mo, Y.; Su, F.; Mo, W.; Yang, Z. EBV-LMP1 regulating AKT/mTOR signaling pathway and WWOX in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol 2017, 10, 8619–8625. [Google Scholar] [PubMed]
- Song, M.; Liu, X.; Liu, K.; Zhao, R.; Huang, H.; Shi, Y.; Zhang, M.; Zhou, S.; Xie, H.; Chen, H.; et al. Targeting AKT with Oridonin Inhibits Growth of Esophageal Squamous Cell Carcinoma In Vitro and Patient-Derived Xenografts In Vivo. Mol. Cancer Ther. 2018, 17, 1540–1553. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Park, D.; Magis, A.T.; Behera, M.; Ramalingam, S.S.; Owonikoko, T.K.; Sica, G.L.; Ye, K.; Zhang, C.; Chen, Z.; et al. Mcl-1 Interacts with Akt to Promote Lung Cancer Progression. Cancer Res. 2019, 79, 6126–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Majewska, E.; Szeliga, M. AKT/GSK3β Signaling in Glioblastoma. Neurochem. Res. 2017, 42, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Joy, A.; Kapoor, M.; Georges, J.; Butler, L.; Chang, Y.; Li, C.; Crouch, A.; Smirnov, I.; Nakada, M.; Hepler, J.; et al. The role of AKT isoforms in glioblastoma: AKT3 delays tumor progression. J. Neuro-Oncology 2016, 130, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Paul-Samojedny, M.; Suchanek, R.; Borkowska, P.; Pudełko, A.; Owczarek, A.; Kowalczyk, M.; Machnik, G.; Fila-Daniłow, A.; Kowalski, J. Knockdown of AKT3 (PKBγ) and PI3KCA suppresses cell viability and proliferation and induces the apoptosis of glioblastoma multiforme T98G cells. Biomed. Res. Int. 2014, 2014, 768181. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Jia, J.; Shi, Y.; Fu, C.; Chen, L.; Jiang, Y.; Zhou, L.; Liu, S.; Tao, Y. Opposed expression of IKKα: Loss in keratinizing carcinomas and gain in non-keratinizing carcinomas. Oncotarget 2015, 6, 25499–25505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Li, Q.; Shi, Z.; Li, C.; Wang, L.; Liu, X.; Jiang, C.; Qian, X.; You, Y.; Liu, N.; et al. GSK-3β regulates tumor growth and angiogenesis in human glioma cells. Oncotarget 2015, 6, 31901–31915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ji, F.; Jiang, Y.; Zhao, T.; Xu, C. Correlation analysis of expressions of PTEN and p53 with the value obtained by magnetic resonance spectroscopy and apparent diffusion coefficient in the tumor and the tumor-adjacent area in magnetic resonance imaging for glioblastoma. J. BUON Off. J. Balk. Union Oncol. 2018, 23, 391–397. [Google Scholar]
- Yang, Q.; Jiang, W.; Hou, P. Emerging role of PI3K/AKT in tumor-related epigenetic regulation. Semin. Cancer Biol. 2019, 59, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Lauenstein, J.U.; Udgata, A.; Bartram, A.; De Sutter, D.; Fisher, D.I.; Halabi, S.; Eyckerman, S.; Gay, N.J. Phosphorylation of the multifunctional signal transducer B-cell adaptor protein (BCAP) promotes recruitment of multiple SH2/SH3 proteins including GRB2. J. Biol. Chem. 2019, 294, 19852–19861. [Google Scholar] [CrossRef] [Green Version]
- Bukovac, A.; Kafka, A.; Raguž, M.; Brlek, P.; Dragičević, K.; Müller, D.; Pećina-Šlaus, N. Are We Benign? What Can Wnt Signaling Pathway and Epithelial to Mesenchymal Transition Tell Us about Intracranial Meningioma Progression. Cancers 2021, 13, 1633. [Google Scholar] [CrossRef] [PubMed]
- Kafka, A.; Bačić, M.; Tomas, D.; Žarković, K.; Bukovac, A.; Njirić, N.; Mrak, G.; Krsnik, Ž.; Pećina-Šlaus, N. Different behaviour of DVL1, DVL2, DVL3 in astrocytoma malignancy grades and their association to TCF1 and LEF1 upregulation. J. Cell. Mol. Med. 2018, 23, 641–655. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PHT and Grade | Age/Years | Sex | ||
|---|---|---|---|---|
| Median | IQR | M | F | |
| DA (grade II) | 34.5 *,*** | 12 | 36 | 26 |
| AA (grade III) | 44.0 *,** | 21 | 70 | 59 |
| GBM (grade IV) | 59.0 **,*** | 18 | 340 | 220 |
| Total | 55.0 | 22 | 446 | 305 |
| GENE | PHT | N | CNA/% | ||||
|---|---|---|---|---|---|---|---|
| HOMDEL | HETLOSS | DIPLOID | GAIN | AMP | |||
| AKT1 | DA | 62 | 0 | 15 | 85 | 0 | 0 |
| AA | 129 | 2 | 26 | 70 | 2 | 0 | |
| GBM | 560 | 0 | 27 | 66 | 6 | 1 | |
| AKT2 **,*** | DA | 62 | 3 | 6 | 89 | 0 | 2 |
| AA | 129 | 2 | 20 | 67 | 10 | 1 | |
| GBM | 560 | 0 | 10 | 57 | 33 | 1 | |
| AKT3 **,*** | DA | 62 | 0 | 0 | 98 | 2 | 0 |
| AA | 129 | 1 | 7 | 81 | 10 | 1 | |
| GBM | 560 | 0 | 4 | 78 | 15 | 2 | |
| CHUK *,**,*** | DA | 62 | 0 | 8 | 92 | 0 | 0 |
| AA | 129 | 1 | 47 | 51 | 1 | 0 | |
| GBM | 560 | 0 | 88 | 12 | 0 | 0 | |
| GSK3β | DA | 62 | 5 | 5 | 89 | 2 | 0 |
| AA | 129 | 0 | 6 | 90 | 4 | 0 | |
| GBM | 560 | 0 | 9 | 79 | 11 | 0 | |
| EGFR *,**,*** | DA | 62 | 0 | 2 | 92 | 5 | 2 |
| AA | 129 | 0 | 1 | 59 | 22 | 19 | |
| GBM | 560 | 0 | 1 | 11 | 44 | 44 | |
| PTEN *,**,*** | DA | 62 | 0 | 8 | 92 | 0 | 0 |
| AA | 129 | 4 | 64 | 50 | 0 | 0 | |
| GBM | 560 | 10 | 79 | 11 | 0 | 0 | |
| PIK3AP1 *,**,*** | DA | 62 | 0 | 8 | 92 | 0 | 0 |
| AA | 129 | 2 | 47 | 51 | 0 | 1 | |
| GBM | 560 | 1 | 88 | 11 | 0 | 0 | |
| PHT | N | mRNA Expression | GENE | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AKT1 | AKT2 | AKT3 | CHUK | GSK3β | EGFR | PTEN | PIK3AP1 | |||
| DA grade II | 62 | M | 3123.58 * | 2245.96 * | 1450.79 * | 501.64 * | 1530.27 * | 1901.81 * | 1403.18 * | 311.42 * |
| IQR | 712.24 | 785.86 | 632.17 | 140.73 | 594.58 | 2082.12 | 337.54 | 289.02 | ||
| AA grade III | 129 | M | 3213.98 * | 2179.75 * | 1174.06 * | 442.53 * | 1659.60 * | 2612.05 * | 1212.54 * | 364.66 * |
| IQR | 1076.78 | 853.40 | 796.81 | 147.41 | 905.05 | 5112.99 | 464.74 | 362.59 | ||
| GBM grade IV | 140 | M | 3737.21 * | 2394.95 * | 693.48 * | 383.62* | 1215.72 * | 4179.58 * | 952.07* | 536.57 * |
| IQR | 1778.33 | 1223.51 | 414.06 | 128.15 | 693.82 | 14,797.33 | 448.76 | 517.91 | ||
| PHT | N | Protein Expression | PROTEIN | |||
|---|---|---|---|---|---|---|
| AKT1/AKT2/AKT3 | GSK3β | EGFR | PTEN | |||
| DA grade II | 46 | M | −0.024 | −0.160 | 0.042 | −0.055 |
| IQR | 0.388 | 0.531 | 0.468 | 0.467 | ||
| AA grade III | 99 | M | 0.082 | −0.051 | 0.023 | −0.162 |
| IQR | 0.432 | 0.435 | 1.276 | 0.421 | ||
| GBM grade IV | 213 | M | 0.073 | 0.0004 | −0.116 | 0.018 |
| IQR | 0.605 | 0.499 | 2.691 | 0.456 | ||
| PHT | N | Methylation | GENE | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AKT1 | AKT2 | AKT3 | CHUK | GSK3β | EGFR | PTEN | PIK3AP1 | |||
| DA grade II | 62 | M | 0.856 * | 0.075 * | 0.294 * | 0.040 * | 0.108 * | 0.250 * | 0.025 * | 0.796 * |
| IQR | 0.044 | 0.024 | 0.099 | 0.010 | 0.049 | 0.155 | 0.005 | 0.083 | ||
| AA grade III | 129 | M | 0.856 * | 0.073 * | 0.252 * | 0.045 * | 0.095 * | 0.235 * | 0.026 * | 0.707 * |
| IQR | 0.039 | 0.022 | 0.154 | 0.013 | 0.060 | 0.147 | 0.006 | 0.238 | ||
| GBM grade IV | 123 | M | 0.096 * | 0.068 * | 0.934 * | 0.105 * | 0.032 * | 0.025 * | 0.014 * | - |
| IQR | 0.027 | 0.024 | 0.029 | 0.067 | 0.010 | 0.012 | 0.003 | - | ||
| PHT | GENE | CNA vs. mRNA Expression | Methylation vs. mRNA Expression | Protein Expression vs. mRNA Expression |
|---|---|---|---|---|
| r | r | r | ||
| DA grade II | AKT1 | 0.10 | −0.15 | 0.10 |
| AKT2 | 0.21 | −0.10 | 0.12 | |
| AKT3 | −0.06 | −0.22 | 0.27 | |
| CHUK | 0.22 | −0.16 | - | |
| GSK3β | 0.17 | −0.29 * | 0.16 | |
| EGFR | 0.25 * | −0.32 * | 0.55 ** | |
| PTEN | 0.28 * | −0.02 | 0.46 * | |
| PIK3AP1 | 0.11 | −0.28 * | - | |
| AA grade III | AKT1 | 0.50 ** | −0.26 * | 0.32 * |
| AKT2 | 0.49 ** | −0.42 ** | 0.29 * | |
| AKT3 | −0.03 | −0.45 ** | 0.01 | |
| CHUK | 0.69 ** | −0.45 ** | - | |
| GSK3β | 0.15 | −0.36 ** | 0.23 * | |
| EGFR | 0.48 ** | −0.49 ** | 0.73 ** | |
| PTEN | 0.59 ** | −0.32 ** | 0.37 ** | |
| PIK3AP1 | 0.14 | −0.12 | - | |
| GBM grade IV | AKT1 | 0.61 ** | −0.06 | 0.24 |
| AKT2 | 0.47 ** | −0.30 | 0.27 * | |
| AKT3 | 0.05 | 0.34 * | 0.15 | |
| CHUK | 0.56 ** | 0.05 | - | |
| GSK3β | 0.30 ** | 0.09 | 0.28 * | |
| EGFR | 0.79 ** | −0.44 * | 0.89 ** | |
| PTEN | 0.56 ** | −0.40 ** | 0.19 | |
| PIK3AP1 | 0.49 ** | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brlek, P.; Kafka, A.; Bukovac, A.; Pećina-Šlaus, N. Integrative cBioPortal Analysis Revealed Molecular Mechanisms That Regulate EGFR-PI3K-AKT-mTOR Pathway in Diffuse Gliomas of the Brain. Cancers 2021, 13, 3247. https://doi.org/10.3390/cancers13133247
Brlek P, Kafka A, Bukovac A, Pećina-Šlaus N. Integrative cBioPortal Analysis Revealed Molecular Mechanisms That Regulate EGFR-PI3K-AKT-mTOR Pathway in Diffuse Gliomas of the Brain. Cancers. 2021; 13(13):3247. https://doi.org/10.3390/cancers13133247
Chicago/Turabian StyleBrlek, Petar, Anja Kafka, Anja Bukovac, and Nives Pećina-Šlaus. 2021. "Integrative cBioPortal Analysis Revealed Molecular Mechanisms That Regulate EGFR-PI3K-AKT-mTOR Pathway in Diffuse Gliomas of the Brain" Cancers 13, no. 13: 3247. https://doi.org/10.3390/cancers13133247
APA StyleBrlek, P., Kafka, A., Bukovac, A., & Pećina-Šlaus, N. (2021). Integrative cBioPortal Analysis Revealed Molecular Mechanisms That Regulate EGFR-PI3K-AKT-mTOR Pathway in Diffuse Gliomas of the Brain. Cancers, 13(13), 3247. https://doi.org/10.3390/cancers13133247