
Knockout of Putative Tumor Suppressor Aldh1l1 in Mice Reprograms Metabolism to Accelerate Growth of Tumors in a Diethylnitrosamine (DEN) Model of Liver Carcinogenesis

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Genotyping
2.3. DEN Administration and Sample Collection
2.4. Liver Tumor Analysis
2.5. Immunohistochemical (IHC) Staining and Review
2.6. Western Blot Assays
2.7. Real-Time PCR
2.8. RT-PCR Data Pretreatment/Transformation
2.9. Metabolome Analysis
2.10. Statistical Analysis
3. Results
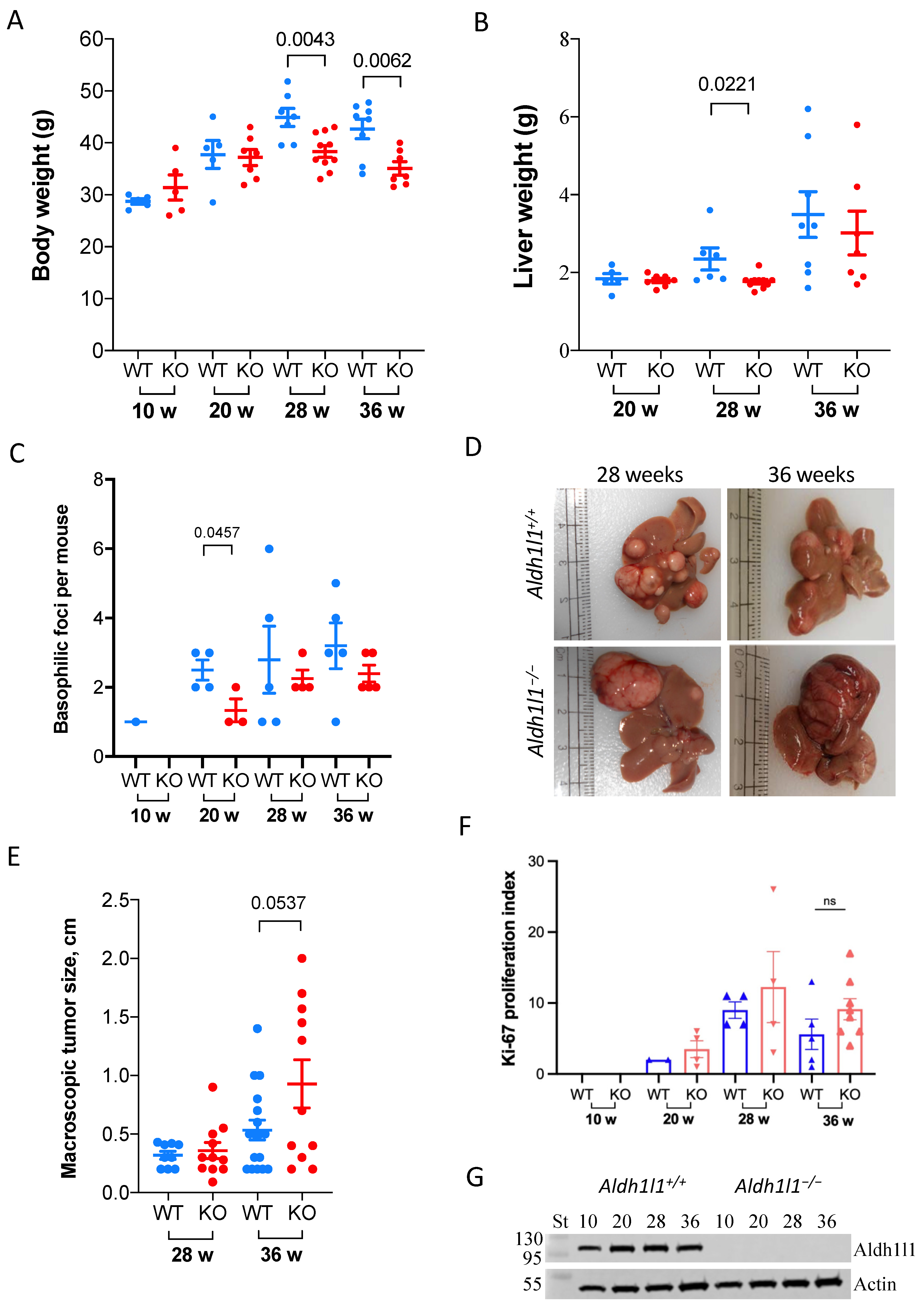
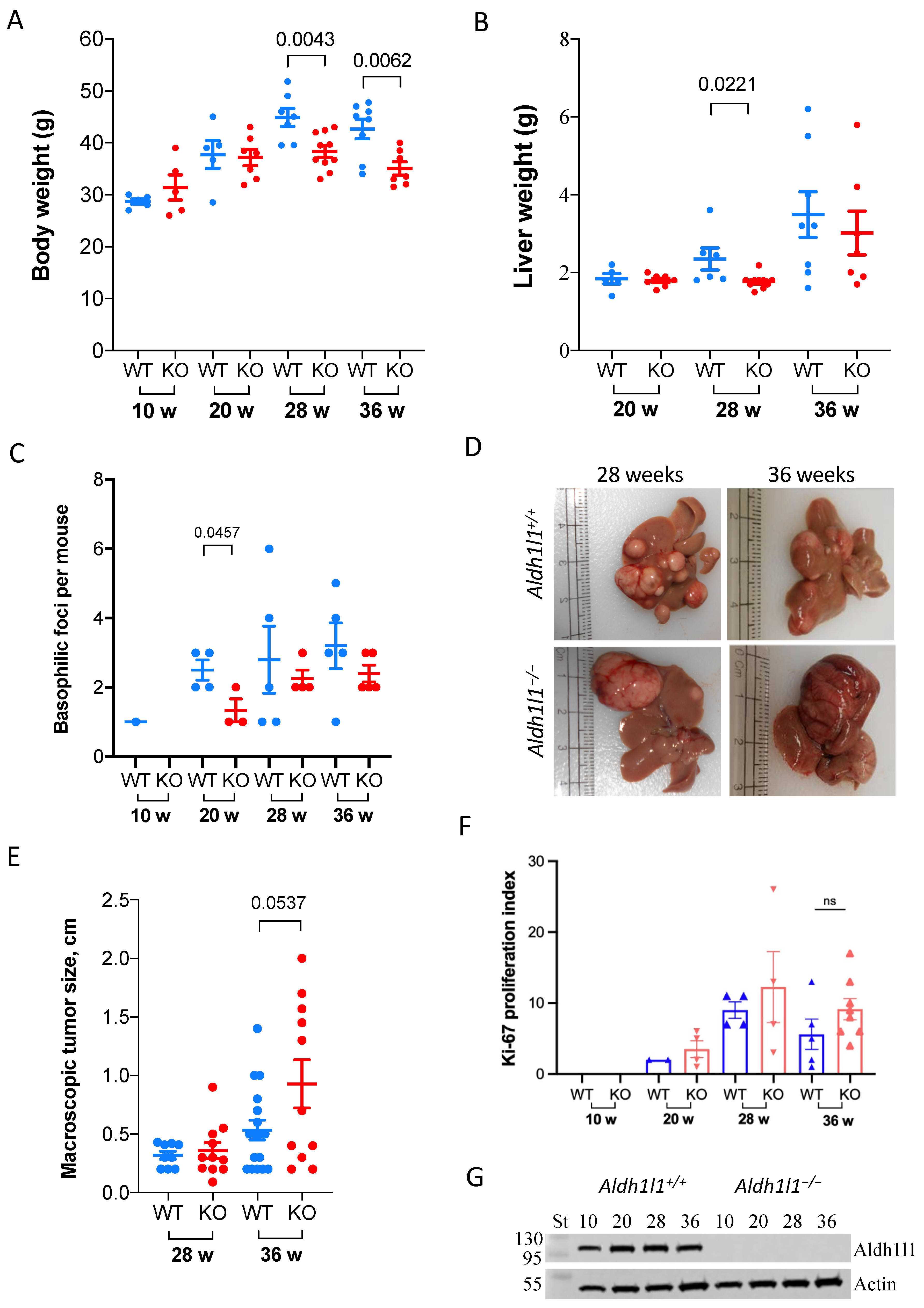
3.1. Aldh1l1 Knockout Alters Tumor Growth Dynamics in a DEN Model Of Carcinogenesis
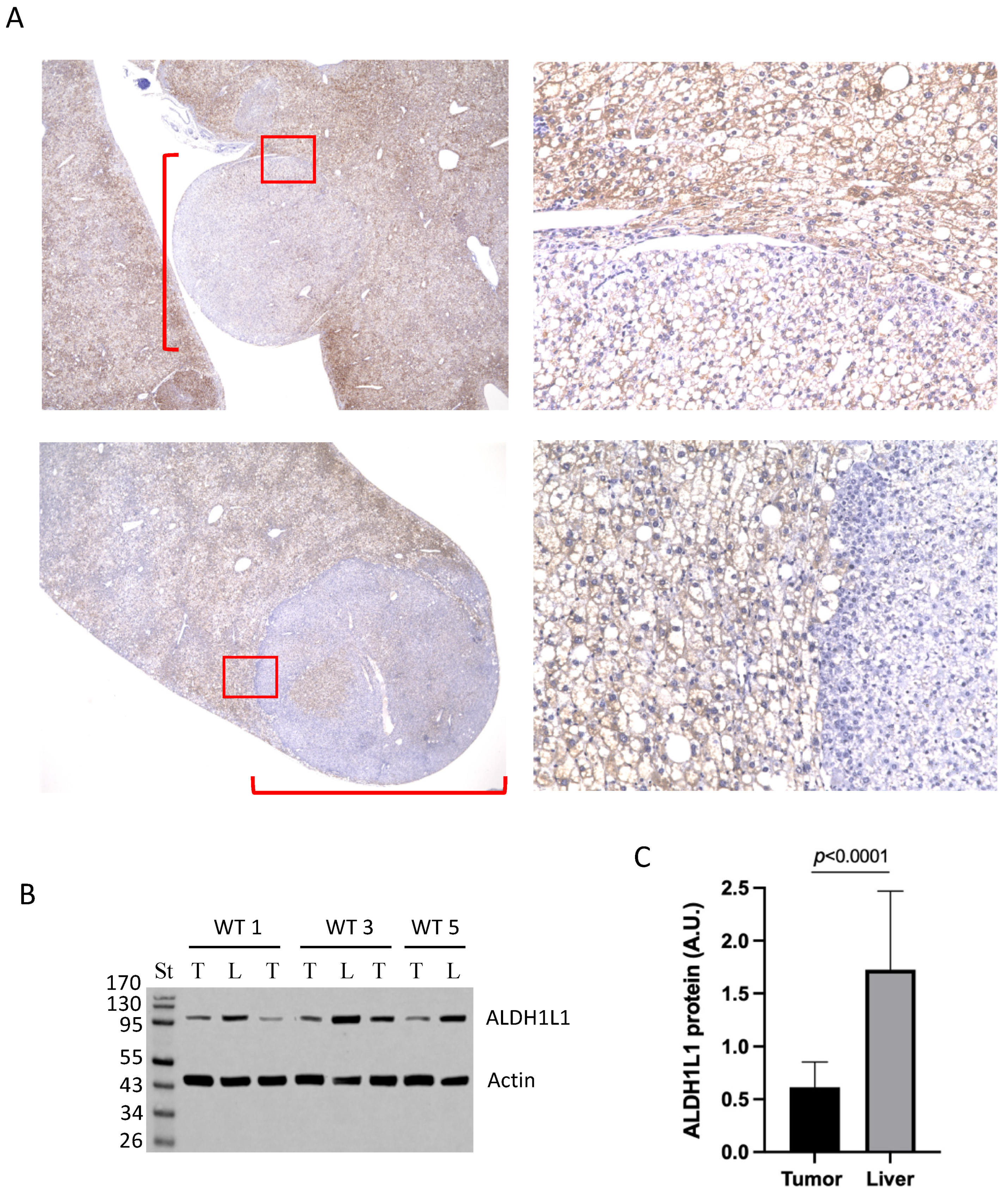
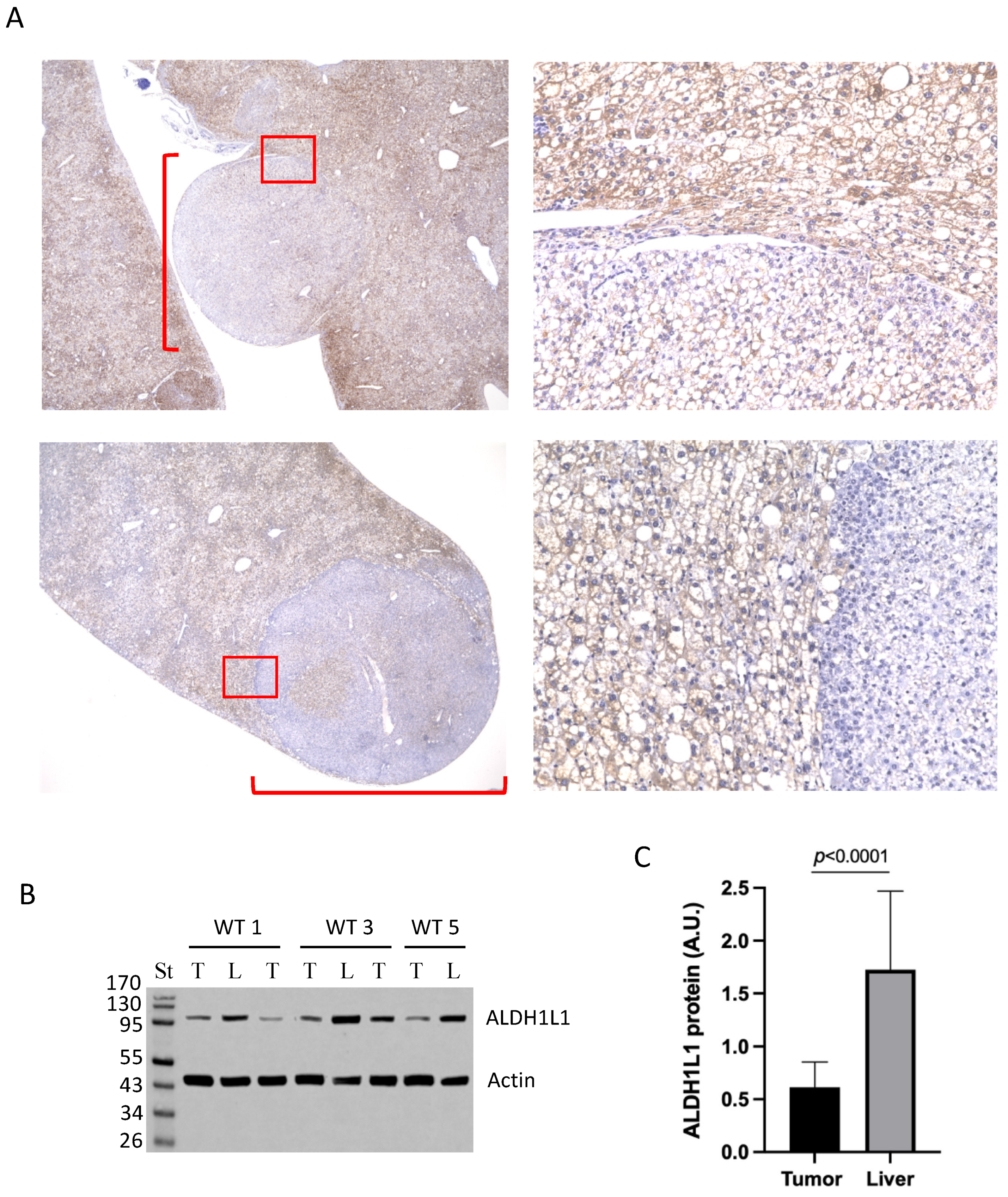
3.2. Large Tumors in DEN-Treated Wild-Type Mice Lost ALDH1L1 Protein
3.3. Vascular Density in Livers of Aldh1l1−/− and Aldh1l1+/+ Mice
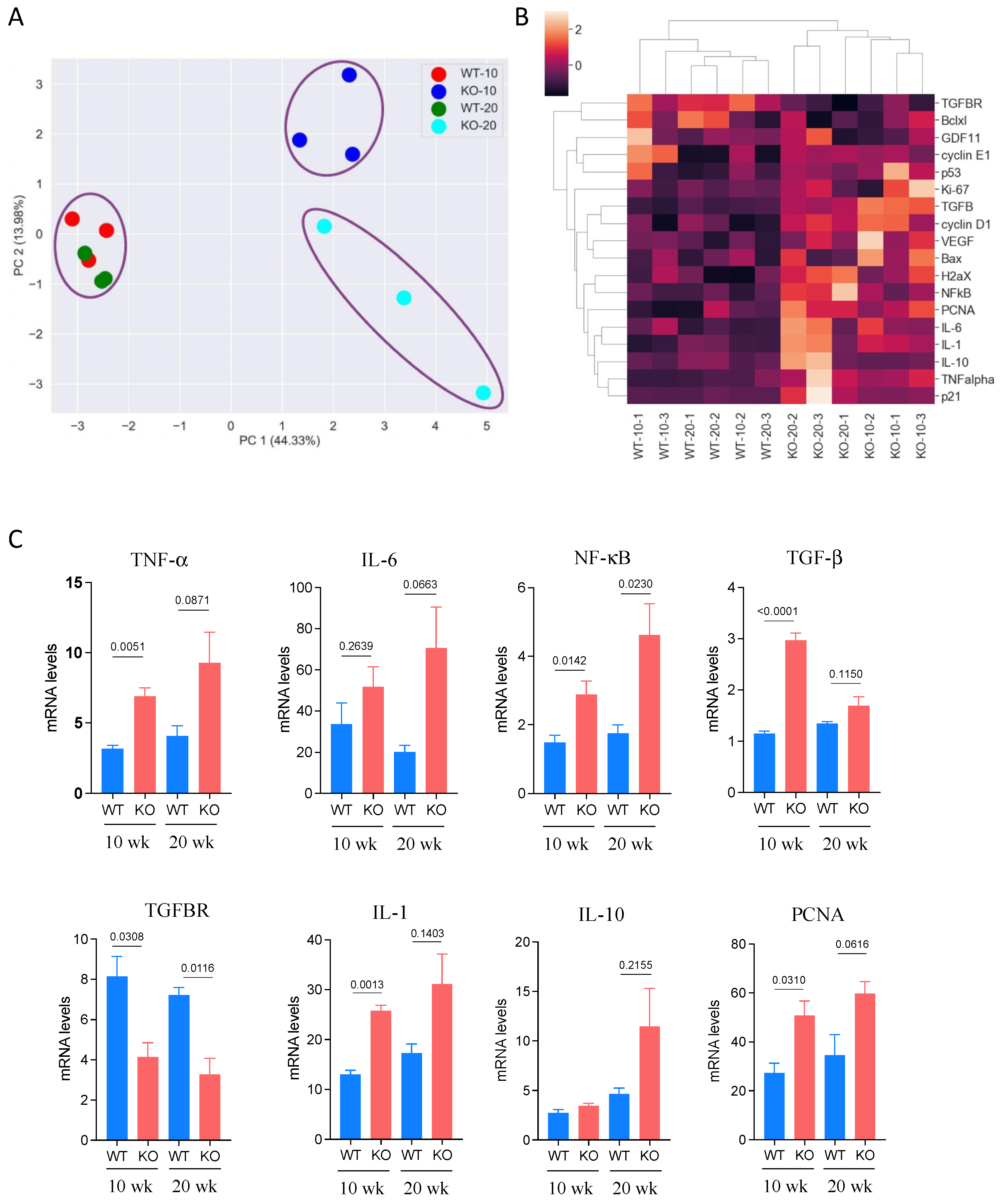
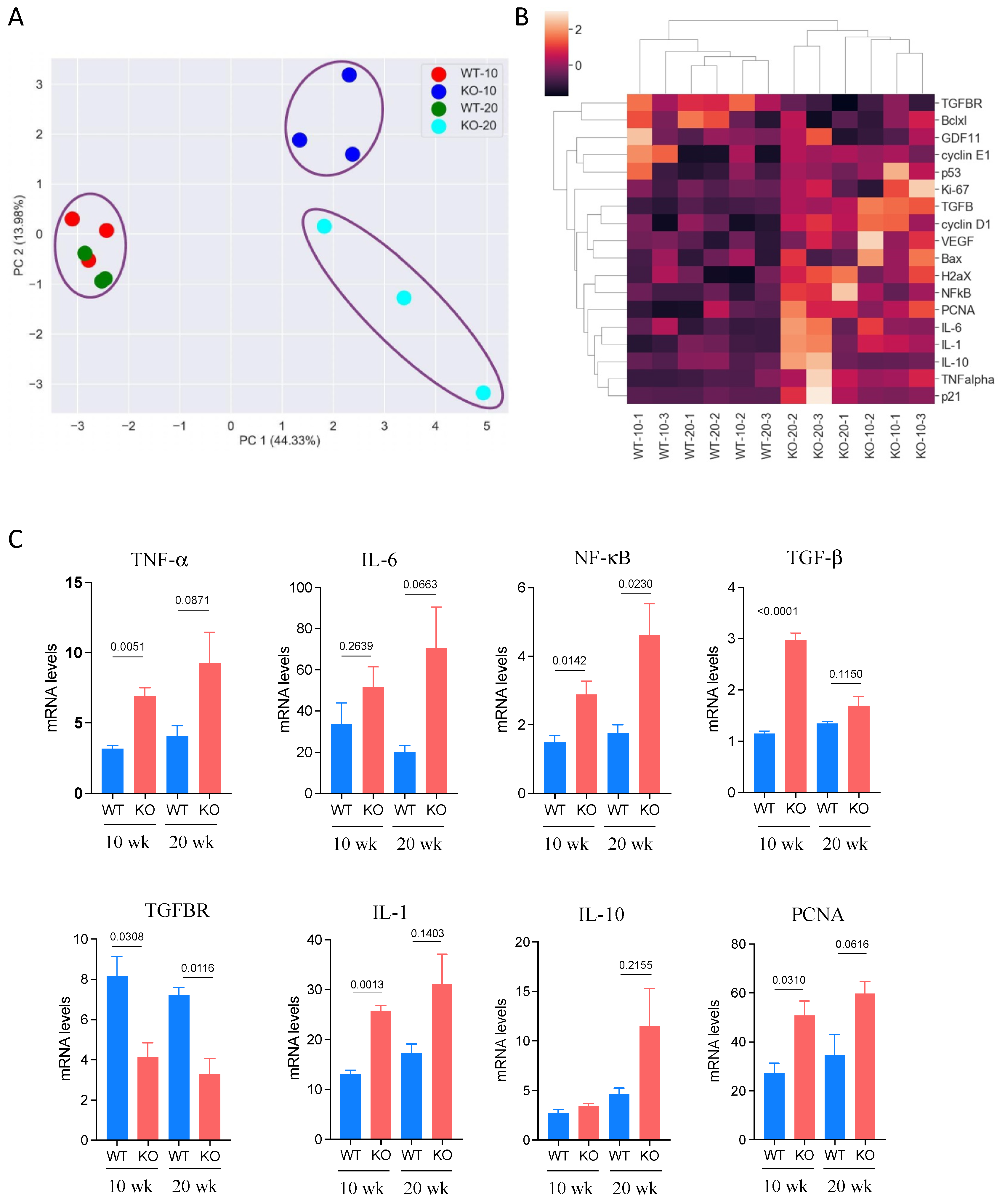
3.4. Expression of Cancer-Related Genes in DEN-Treated Mice
3.5. Aldh1l1−/− Mice Demonstrate an Altered Metabolic Response to DEN Compared To Wild-Type Mice
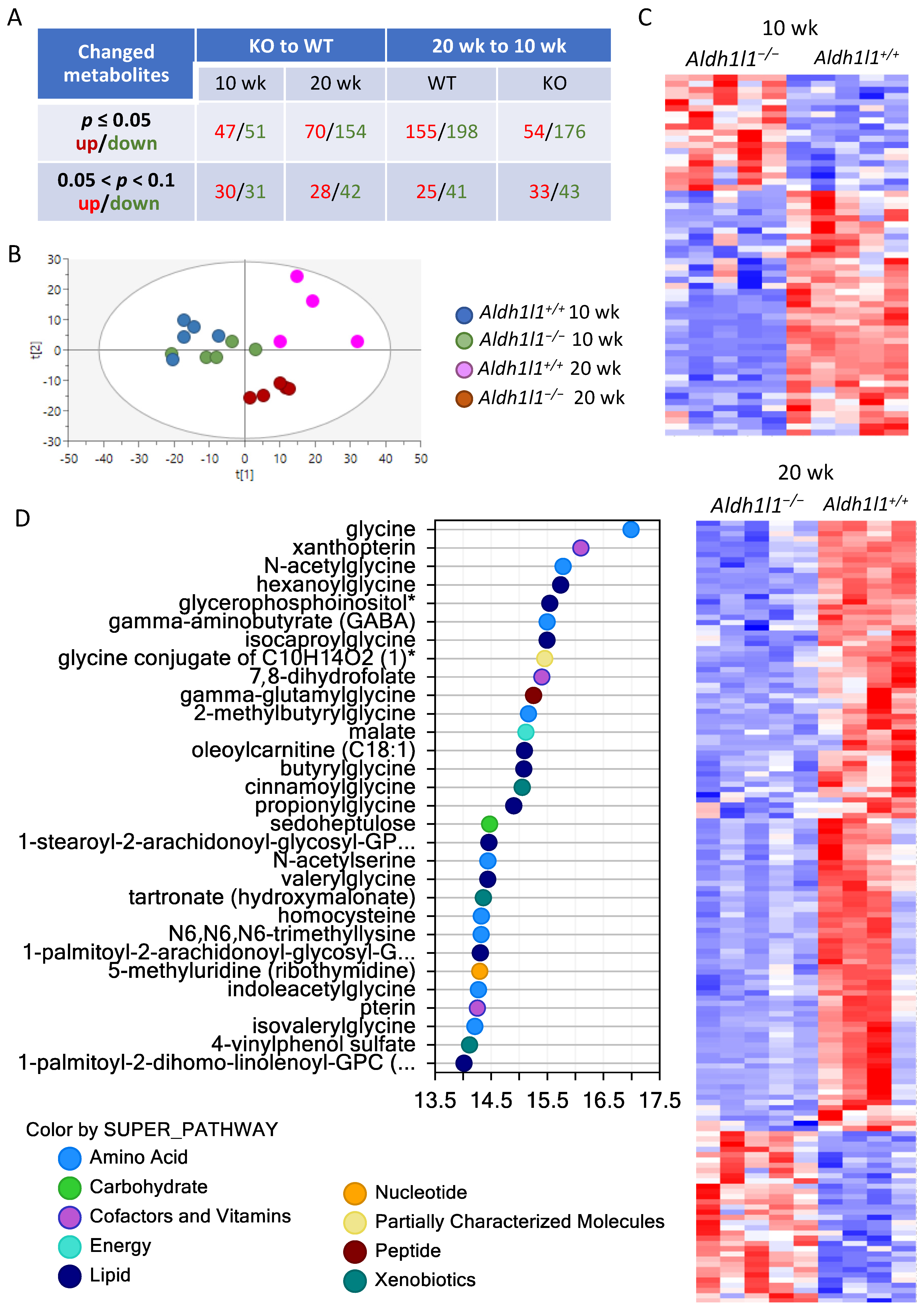
3.5.1. Overall Metabolic Changes
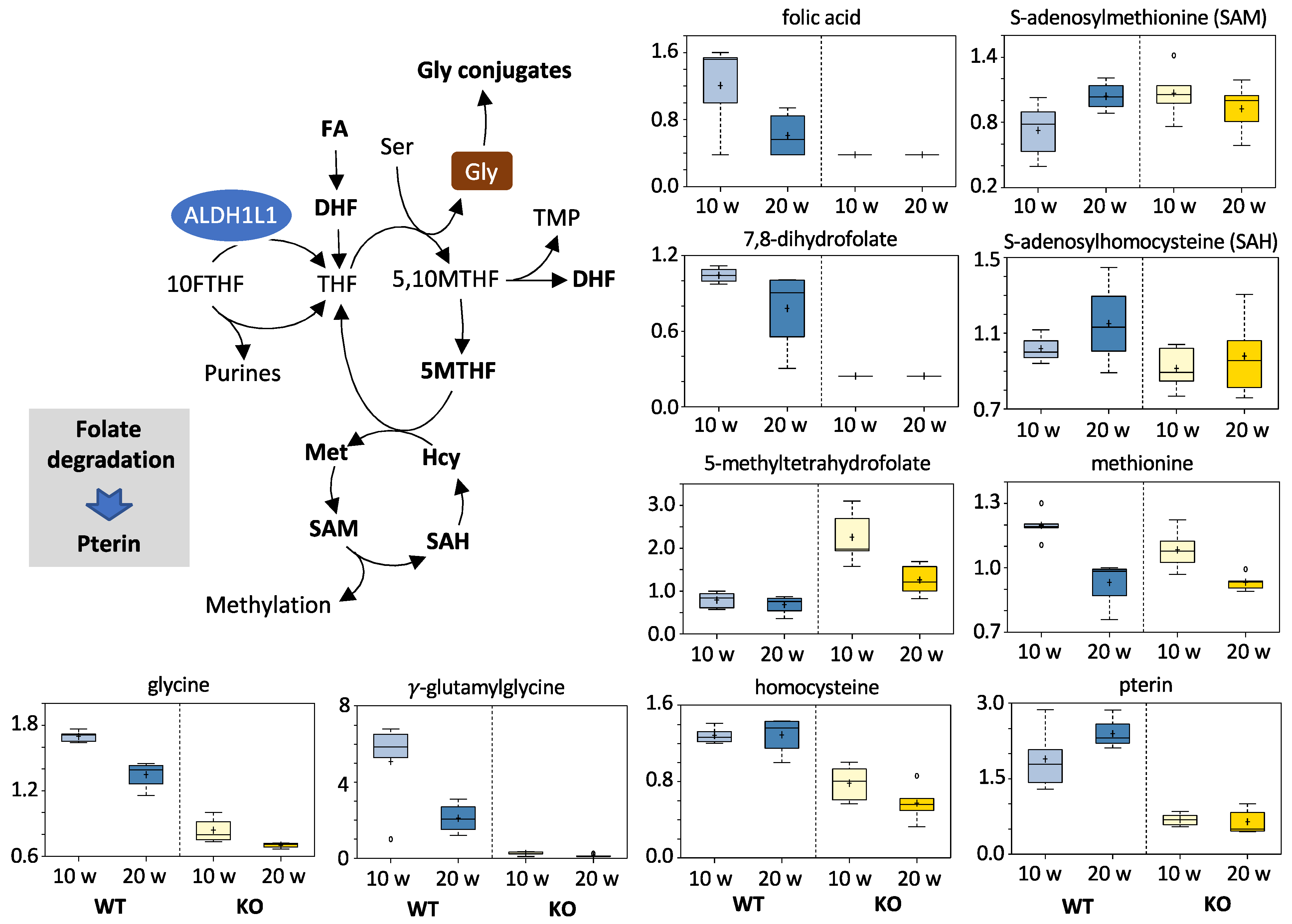
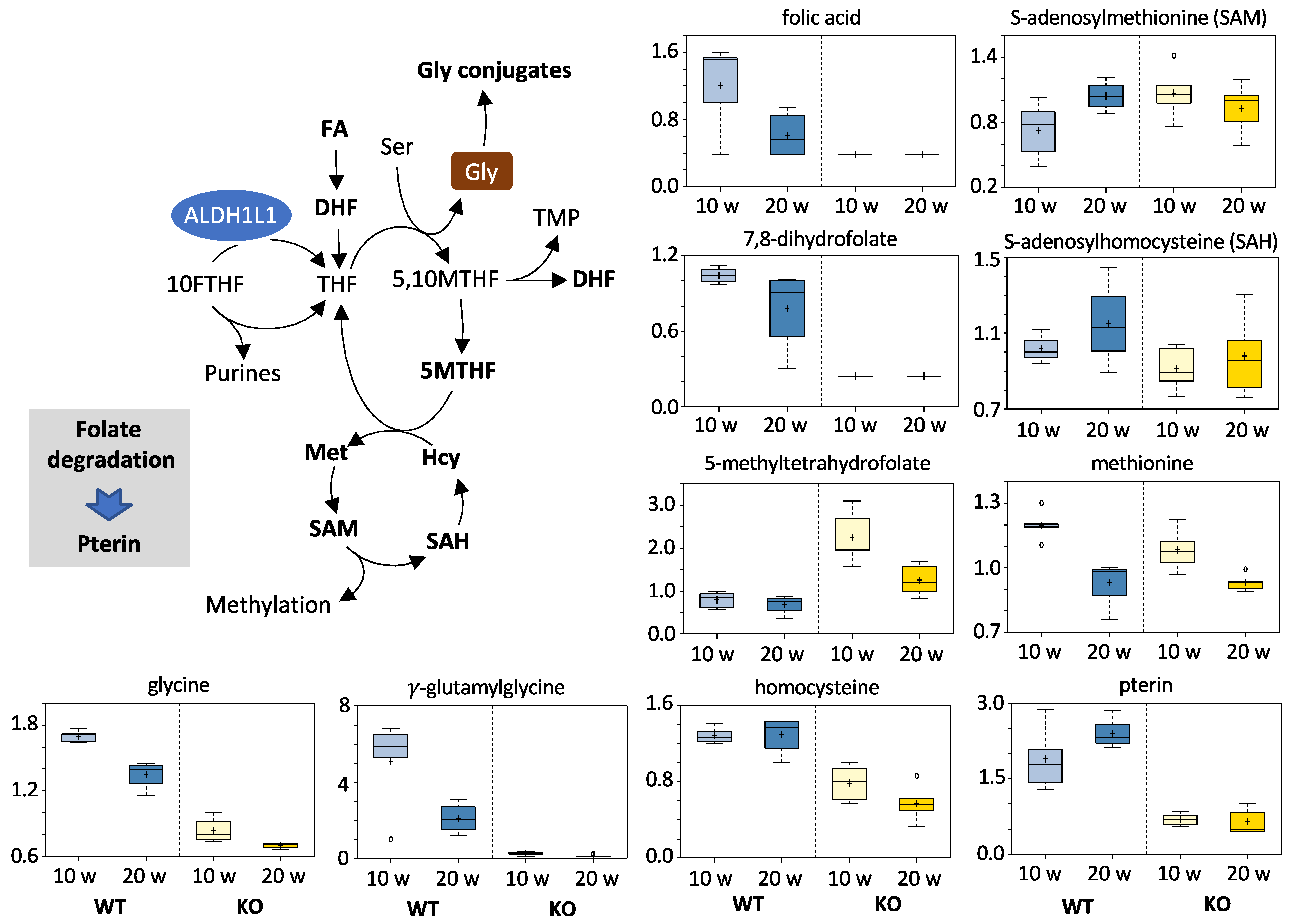
3.5.2. Folates and Related Metabolites
3.5.3. Acylglycine Conjugates
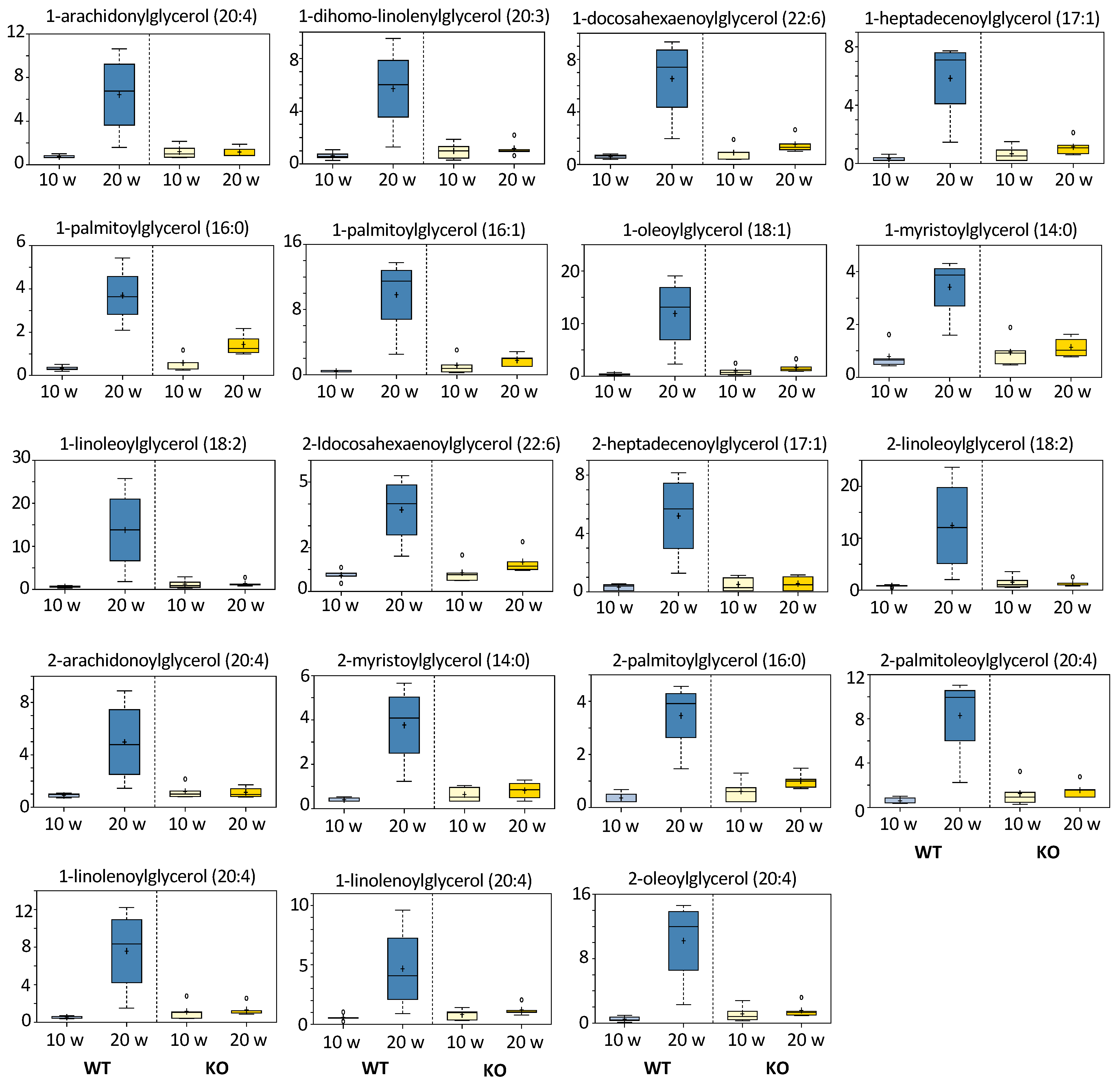
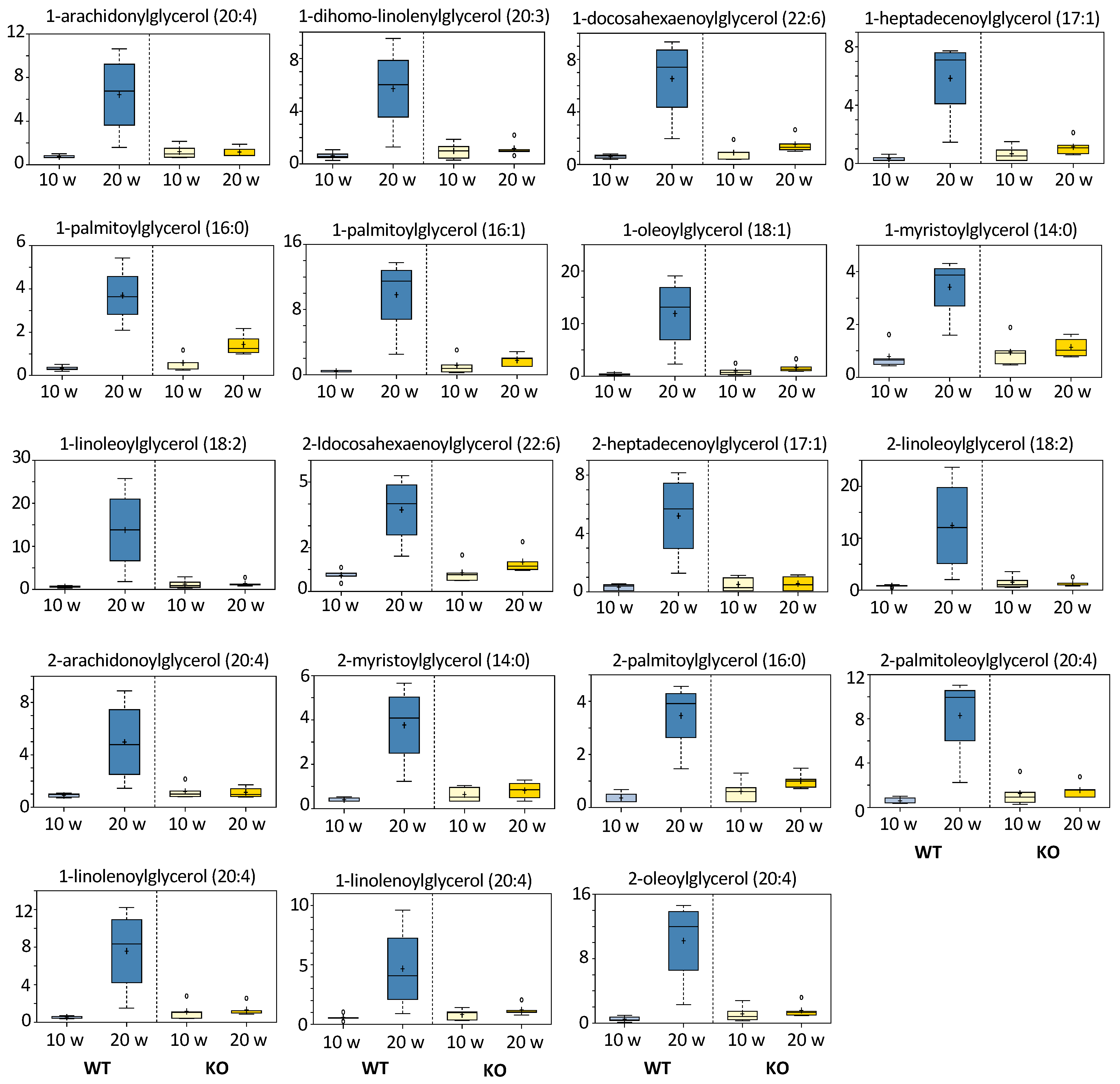
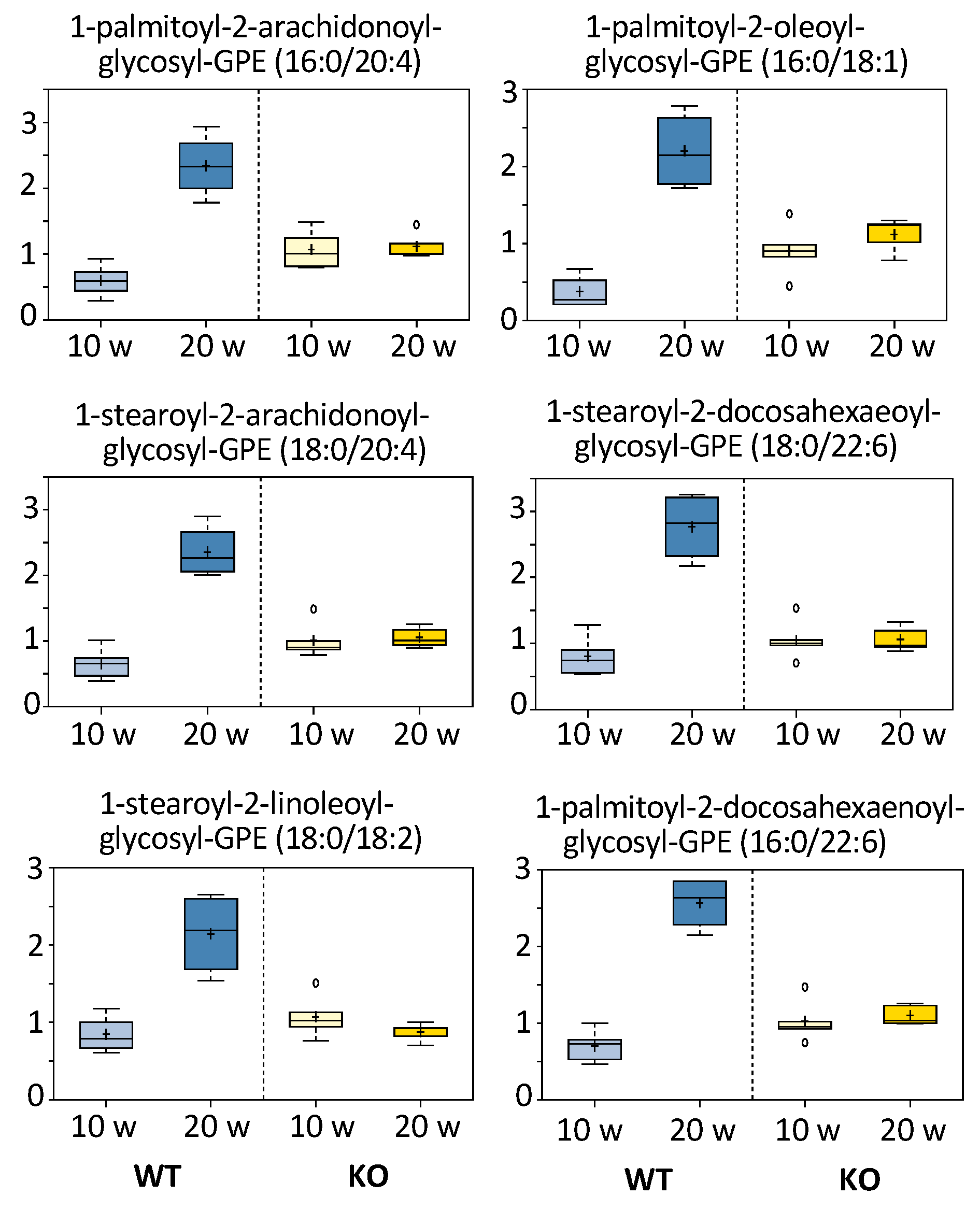
3.5.4. Monoacylglycerols and Fatty Acids
3.5.5. Carbohydrates
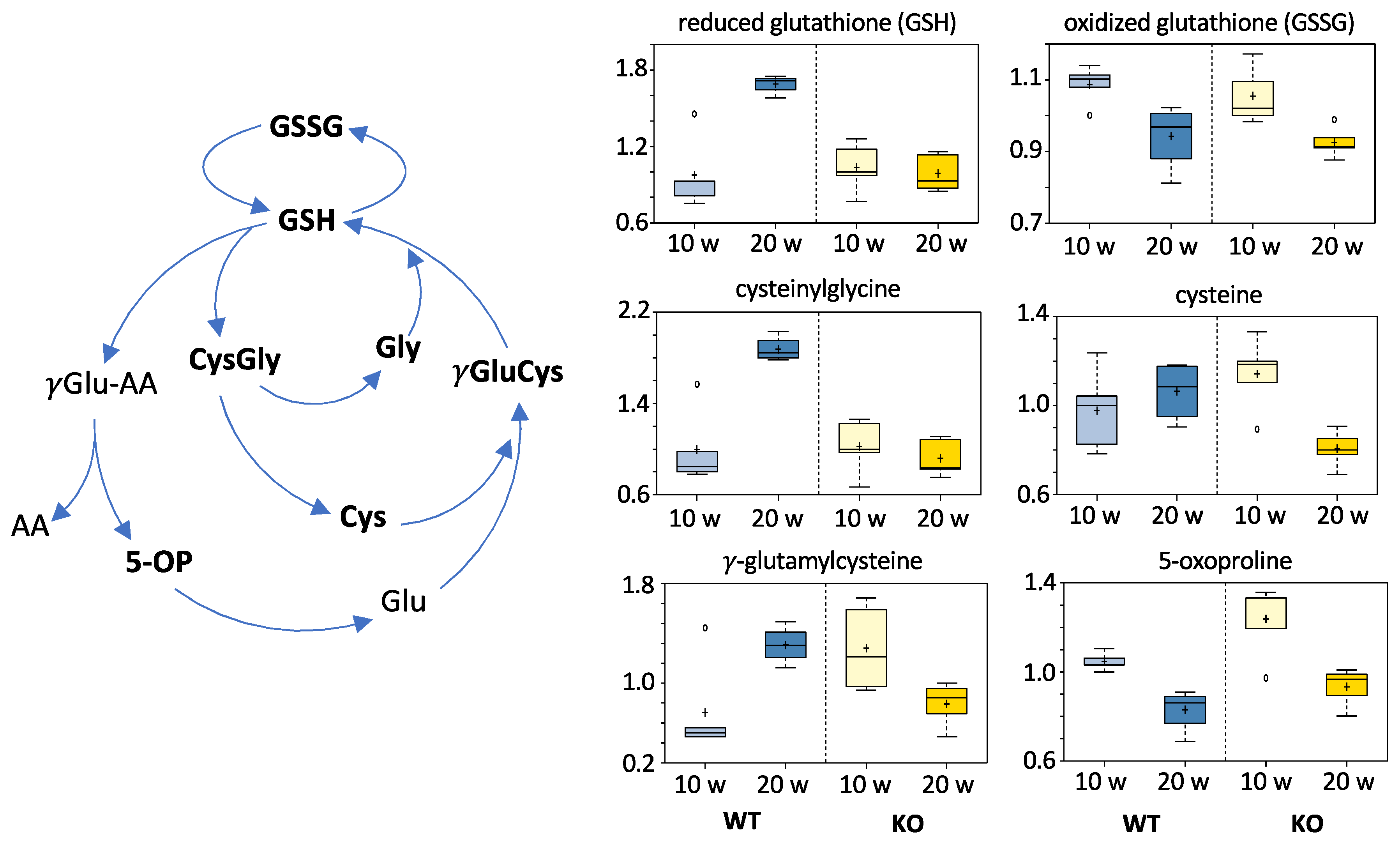
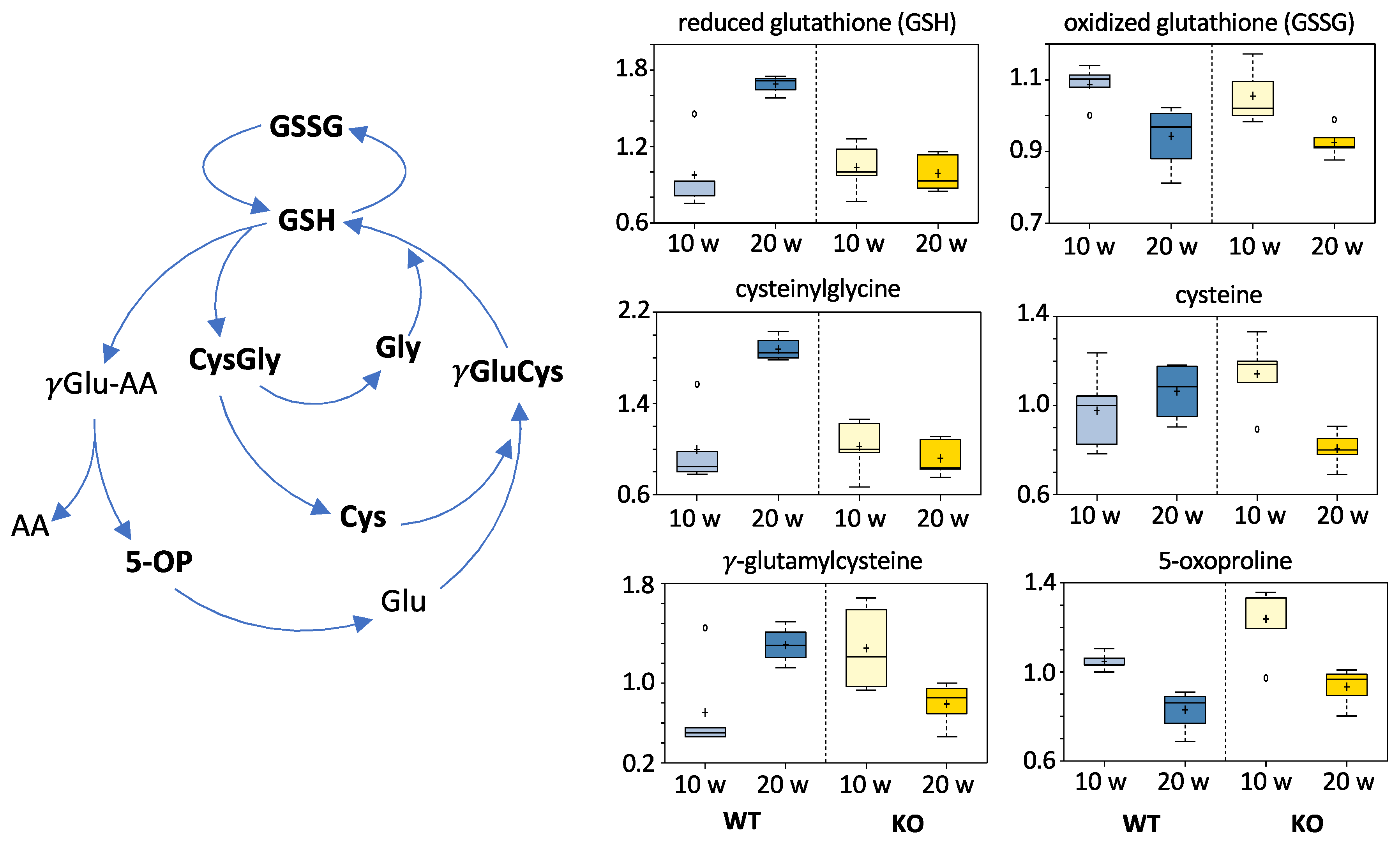
3.5.6. Redox Homeostasis Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor, D., III; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauffer, J.K.; Scarzello, A.J.; Jiang, Q.; Wiltrout, R.H. Chronic inflammation, immune escape, and oncogenesis in the liver: A unique neighborhood for novel intersections. Hepatology 2012, 56, 1567–1574. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Garmire, L.; Calvisi, D.F.; Chua, M.S.; Kelley, R.K.; Chen, X. Harnessing big ‘omics’ data and AI for drug discovery in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Desert, R.; Nieto, N.; Musso, O. Dimensions of hepatocellular carcinoma phenotypic diversity. World J. Gastroenterol. 2018, 24, 4536–4547. [Google Scholar] [CrossRef]
- Erstad, D.J.; Fuchs, B.C.; Tanabe, K.K. Molecular signatures in hepatocellular carcinoma: A step toward rationally designed cancer therapy. Cancer 2018, 124, 3084–3104. [Google Scholar] [CrossRef] [Green Version]
- Tackels-Horne, D.; Goodman, M.D.; Williams, A.J.; Wilson, D.J.; Eskandari, T.; Vogt, L.M.; Boland, J.F.; Scherf, U.; Vockley, J.G. Identification of differentially expressed genes in hepatocellular carcinoma and metastatic liver tumors by oligonucleotide expression profiling. Cancer 2001, 92, 395–405. [Google Scholar] [CrossRef]
- Li, M.; Sun, Q.; Wang, X. Transcriptional landscape of human cancers. Oncotarget 2017, 8, 34534–34551. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Q.; He, J.R.; Wang, H.Y. Decreased expression of ALDH1L1 is associated with a poor prognosis in hepatocellular carcinoma. Med. Oncol. 2012, 29, 1843–1849. [Google Scholar] [CrossRef]
- Krupenko, S.A. FDH: An aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem. Biol. Interact. 2009, 178, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupenko, S.A.; Oleinik, N.V. 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ. 2002, 13, 227–236. [Google Scholar] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.T.; Stover, P.J. Folate-mediated one-carbon metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.J.; Hershman, S.G.; Kohrer, C.; Belcher-Timme, C.A.; Patel, J.; Goldberger, O.A.; Christodoulou, J.; Silberstein, J.M.; McKenzie, M.; Ryan, M.T.; et al. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011, 14, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Strickland, K.C.; Krupenko, N.I.; Krupenko, S.A. Molecular mechanisms underlying the potentially adverse effects of folate. Clin. Chem. Lab. Med. 2013, 51, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [Green Version]
- Odin, E.; Wettergren, Y.; Nilsson, S.; Willen, R.; Carlsson, G.; Spears, C.P.; Larsson, L.; Gustavsson, B. Altered gene expression of folate enzymes in adjacent mucosa is associated with outcome of colorectal cancer patients. Clin. Cancer Res. 2003, 9, 6012–6019. [Google Scholar] [PubMed]
- Lee, D.; Xu, I.M.; Chiu, D.K.; Lai, R.K.; Tse, A.P.; Lan Li, L.; Law, C.T.; Tsang, F.H.; Wei, L.L.; Chan, C.Y.; et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. J. Clin. Investig. 2017, 127, 1856–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. Epigenetic silencing of ALDH1L1, a metabolic regulator of cellular proliferation, in cancers. Genes Cancer 2011, 2, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Beniaminov, A.D.; Puzanov, G.A.; Krasnov, G.S.; Kaluzhny, D.N.; Kazubskaya, T.P.; Braga, E.A.; Kudryavtseva, A.V.; Melnikova, N.V.; Dmitriev, A.A. Deep Sequencing Revealed a CpG Methylation Pattern Associated with ALDH1L1 Suppression in Breast Cancer. Front. Genet. 2018, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Dmitriev, A.A.; Kashuba, V.I.; Haraldson, K.; Senchenko, V.N.; Pavlova, T.V.; Kudryavtseva, A.V.; Anedchenko, E.A.; Krasnov, G.S.; Pronina, I.V.; Loginov, V.I.; et al. Genetic and epigenetic analysis of non-small cell lung cancer with NotI-microarrays. Epigenetics 2012, 7, 502–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmitriev, A.A.; Rudenko, E.E.; Kudryavtseva, A.V.; Krasnov, G.S.; Gordiyuk, V.V.; Melnikova, N.V.; Stakhovsky, E.O.; Kononenko, O.A.; Pavlova, L.S.; Kondratieva, T.T.; et al. Epigenetic alterations of chromosome 3 revealed by NotI-microarrays in clear cell renal cell carcinoma. Biomed. Res. Int. 2014, 2014, 735292. [Google Scholar] [CrossRef]
- Senchenko, V.N.; Kisseljova, N.P.; Ivanova, T.A.; Dmitriev, A.A.; Krasnov, G.S.; Kudryavtseva, A.V.; Panasenko, G.V.; Tsitrin, E.B.; Lerman, M.I.; Kisseljov, F.L.; et al. Novel tumor suppressor candidates on chromosome 3 revealed by NotI-microarrays in cervical cancer. Epigenetics 2013, 8, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleinik, N.V.; Krupenko, N.I.; Priest, D.G.; Krupenko, S.A. Cancer cells activate p53 in response to 10-formyltetrahydrofolate dehydrogenase expression. Biochem. J. 2005, 391, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Krupenko, S.A.; Krupenko, N.I. Loss of ALDH1L1 folate enzyme confers a selective metabolic advantage for tumor progression. Chem. Biol. Interact. 2019, 302, 149–155. [Google Scholar] [CrossRef]
- Anguera, M.C.; Field, M.S.; Perry, C.; Ghandour, H.; Chiang, E.P.; Selhub, J.; Shane, B.; Stover, P.J. Regulation of Folate-mediated One-carbon Metabolism by 10-Formyltetrahydrofolate Dehydrogenase. J. Biol. Chem. 2006, 281, 18335–18342. [Google Scholar] [CrossRef] [Green Version]
- Khan, Q.A.; Pediaditakis, P.; Malakhau, Y.; Esmaeilniakooshkghazi, A.; Ashkavand, Z.; Sereda, V.; Krupenko, N.I.; Krupenko, S.A. CHIP E3 ligase mediates proteasomal degradation of the proliferation regulatory protein ALDH1L1 during the transition of NIH3T3 fibroblasts from G0/G1 to S-phase. PLoS ONE 2018, 13, e0199699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesselinovitch, S.D.; Koka, M.; Mihailovich, N.; Rao, K.V. Carcinogenicity of diethylnitrosamine in newborn, infant, and adult mice. J. Cancer Res. Clin. Oncol. 1984, 108, 60–65. [Google Scholar] [CrossRef]
- Vesselinovitch, S.D.; Mihailovich, N. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1983, 43, 4253–4259. [Google Scholar] [PubMed]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Duan, T.; Sun, W.; Zhang, M.; Ge, J.; He, Y.; Zhang, J.; Zheng, Y.; Yang, W.; Shen, H.M.; Yang, J.; et al. Dietary restriction protects against diethylnitrosamine-induced hepatocellular tumorigenesis by restoring the disturbed gene expression profile. Sci. Rep. 2017, 7, 43745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupenko, N.I.; Sharma, J.; Pediaditakis, P.; Fekry, B.; Helke, K.L.; Du, X.; Sumner, S.; Krupenko, S.A. Cytosolic 10-formyltetrahydrofolate dehydrogenase regulates glycine metabolism in mouse liver. Sci. Rep. 2019, 9, 14937. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Nakajima, T.; Kagawa, K.; Deguchi, T.; Kakusui, M.; Katagishi, T.; Okanoue, T.; Kashima, K.; Ashihara, T. Angiogenesis in hepatocellular carcinoma as evaluated by CD34 immunohistochemistry. Liver 1998, 18, 14–19. [Google Scholar] [CrossRef]
- Yan, Z.X.; Gao, X.J.; Li, T.; Wei, B.; Wang, P.P.; Yang, Y.; Yan, R. Fecal Microbiota Transplantation in Experimental Ulcerative Colitis Reveals Associated Gut Microbial and Host Metabolic Reprogramming. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Nakano, D.; Koga, H.; Torimura, T. Effects of a DPP4 Inhibitor on Progression of NASH-related HCC and the p62/Keap1/Nrf2-Pentose Phosphate Pathway in a Mouse Model. Liver Cancer 2019, 8, 359–372. [Google Scholar] [CrossRef]
- Oleinik, N.V.; Krupenko, S.A. Ectopic expression of 10-formyltetrahydrofolate dehydrogenase in a549 cells induces g(1) cell cycle arrest and apoptosis. Mol. Cancer Res. 2003, 1, 577–588. [Google Scholar] [PubMed]
- Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene 2007, 26, 7222–7230. [Google Scholar] [CrossRef] [Green Version]
- Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. ALDH1L1 inhibits cell motility via dephosphorylation of cofilin by PP1 and PP2A. Oncogene 2010, 29, 6233–6244. [Google Scholar] [CrossRef] [Green Version]
- Ghose, S.; Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. 10-formyltetrahydrofolate dehydrogenase-induced c-Jun-NH2-kinase pathways diverge at the c-Jun-NH2-kinase substrate level in cells with different p53 status. Mol. Cancer Res. 2009, 7, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakasam, A.; Ghose, S.; Oleinik, N.V.; Bethard, J.R.; Peterson, Y.K.; Krupenko, N.I.; Krupenko, S.A. JNK1/2 regulate Bid by direct phosphorylation at Thr59 in response to ALDH1L1. Cell Death Dis. 2014, 5, e1358. [Google Scholar] [CrossRef]
- Hoeferlin, L.A.; Fekry, B.; Ogretmen, B.; Krupenko, S.A.; Krupenko, N.I. Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6. J. Biol. Chem. 2013, 288, 12880–12890. [Google Scholar] [CrossRef] [Green Version]
- Hoeferlin, L.A.; Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. Activation of p21-Dependent G1/G2 Arrest in the Absence of DNA Damage as an Antiapoptotic Response to Metabolic Stress. Genes Cancer 2011, 2, 889–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luka, Z.; Pakhomova, S.; Loukachevitch, L.V.; Egli, M.; Newcomer, M.E.; Wagner, C. 5-methyltetrahydrofolate is bound in intersubunit areas of rat liver folate-binding protein glycine N-methyltransferase. J. Biol. Chem. 2007, 282, 4069–4075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.J.; Wagner, C. Tissue distribution of glycine N-methyltransferase, a major folate-binding protein of liver. Proc. Natl. Acad. Sci. USA 1994, 91, 210–214. [Google Scholar] [CrossRef] [Green Version]
- DebRoy, S.; Kramarenko, I.I.; Ghose, S.; Oleinik, N.V.; Krupenko, S.A.; Krupenko, N.I. A novel tumor suppressor function of glycine N-methyltransferase is independent of its catalytic activity but requires nuclear localization. PLoS ONE 2013, 8, e70062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Chantar, M.L.; Vazquez-Chantada, M.; Ariz, U.; Martinez, N.; Varela, M.; Luka, Z.; Capdevila, A.; Rodriguez, J.; Aransay, A.M.; Matthiesen, R.; et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 2008, 47, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timperi, E.; Barnaba, V. Viral Hepatitides, Inflammation and Tumour Microenvironment. Adv. Exp. Med. Biol. 2020, 1263, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers 2019, 11, 1704. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Karin, M. NF-kappaB and STAT3–key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [Green Version]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Tang, B.; Lang, J.; Du, Y.; Cao, B.; Jin, L.; Fang, M.; Hu, Z.; Cheng, C.; Liu, X.; et al. High-Fat Diet Promotes Macrophage-Mediated Hepatic Inflammation and Aggravates Diethylnitrosamine-Induced Hepatocarcinogenesis in Mice. Front. Nutr. 2020, 7, 585306. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef]
- Hall, Z.; Chiarugi, D.; Charidemou, E.; Leslie, J.; Scott, E.; Pellegrinet, L.; Allison, M.; Mocciaro, G.; Anstee, Q.M.; Evan, G.I.; et al. Lipid Remodeling in Hepatocyte Proliferation and Hepatocellular Carcinoma. Hepatology 2020. [Google Scholar] [CrossRef]
- Evangelista, E.B.; Kwee, S.A.; Sato, M.M.; Wang, L.; Rettenmeier, C.; Xie, G.; Jia, W.; Wong, L.L. Phospholipids are A Potentially Important Source of Tissue Biomarkers for Hepatocellular Carcinoma: Results of a Pilot Study Involving Targeted Metabolomics. Diagnostics 2019, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hong, Z.; Tan, G.; Dong, X.; Yang, G.; Zhao, L.; Chen, X.; Zhu, Z.; Lou, Z.; Qian, B.; et al. NMR and LC/MS-based global metabolomics to identify serum biomarkers differentiating hepatocellular carcinoma from liver cirrhosis. Int. J. Cancer 2014, 135, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Casadei-Gardini, A.; Del Coco, L.; Marisi, G.; Conti, F.; Rovesti, G.; Ulivi, P.; Canale, M.; Frassineti, G.L.; Foschi, F.G.; Longo, S.; et al. (1)H-NMR Based Serum Metabolomics Highlights Different Specific Biomarkers between Early and Advanced Hepatocellular Carcinoma Stages. Cancers 2020, 12, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepien, M.; Keski-Rahkonen, P.; Kiss, A.; Robinot, N.; Duarte-Salles, T.; Murphy, N.; Perlemuter, G.; Viallon, V.; Tjonneland, A.; Rostgaard-Hansen, A.L.; et al. Metabolic perturbations prior to hepatocellular carcinoma diagnosis: Findings from a prospective observational cohort study. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Chen, J.; Li, H.; Fan, T.; Zheng, X.; Wang, H.; Zhang, N.; Liu, Y.; Luo, X.; Wang, J.; et al. Role of the Mitochondrial Citrate-malate Shuttle in Hras12V-Induced Hepatocarcinogenesis: A Metabolomics-Based Analysis. Metabolites 2020, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Florio, A.A.; Koshiol, J.; Pfeiffer, R.M.; Yang, B.; Yu, K.; Chen, C.J.; Yang, H.I.; Lee, M.H.; McGlynn, K.A. Prediagnostic concentrations of circulating bile acids and hepatocellular carcinoma risk: REVEAL-HBV and HCV studies. Int. J. Cancer 2020, 147, 2743–2753. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Raymond, V.A.; Lacoste, B.; Lapierre, P.; Bilodeau, M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget 2018, 9, 26868–26883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Liu, H.; Wang, C.; Li, B. Phenotypic Characterization Analysis of Human Hepatocarcinoma by Urine Metabolomics Approach. Sci. Rep. 2016, 6, 19763. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.U.; Shariff, M.I.; Crossey, M.M.; Gomez-Romero, M.; Holmes, E.; Cox, I.J.; Fye, H.K.; Njie, R.; Taylor-Robinson, S.D. Hepatocellular carcinoma: Review of disease and tumor biomarkers. World J. Hepatol. 2016, 8, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, A.; Sun, H. Power of metabolomics in diagnosis and biomarker discovery of hepatocellular carcinoma. Hepatology 2013, 57, 2072–2077. [Google Scholar] [CrossRef]
- Han, J.; Qin, W.X.; Li, Z.L.; Xu, A.J.; Xing, H.; Wu, H.; Zhang, H.; Wang, M.D.; Li, C.; Liang, L.; et al. Tissue and serum metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Clin. Chim. Acta 2019, 488, 68–75. [Google Scholar] [CrossRef]
- Chen, T.; Xie, G.; Wang, X.; Fan, J.; Qiu, Y.; Zheng, X.; Qi, X.; Cao, Y.; Su, M.; Wang, X.; et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Mol. Cell. Proteom. 2011, 10, M110004945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyoglu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman-Rossi, J.; Dufour, J.F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, F.F.; Song, Y.N.; Lu, Y.Y.; Zhang, Y.; Hu, Y.Y.; Su, S.B. Analysis of plasma metabolic profile, characteristics and enzymes in the progression from chronic hepatitis B to hepatocellular carcinoma. Aging 2020, 12, 14949–14965. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Agarwal, A.; Singh, A.K.; Gautam, A.K.; Chakraborti, S.; Kumar, U.; Kumar, D.; Bhattacharya, B.; Panda, P.; Saha, B.; et al. Antineoplastic properties of zafirlukast against hepatocellular carcinoma via activation of mitochondrial mediated apoptosis. Regul. Toxicol. Pharmacol. 2019, 109, 104489. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lu, S.; Zheng, H.; Xu, M.; Song, J.; Yang, W.; Weng, Q.; Zheng, L.; Fan, X.; Cheng, X.; et al. Identification of the Potential Metabolic Pathways Involved in the Hepatic Tumorigenesis of Rat Diethylnitrosamine-Induced Hepatocellular Carcinoma via (1)H NMR-Based Metabolomic Analysis. Biomed. Res. Int. 2019, 2019, 9367082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, D.; Qiao, S.; Wu, X.; Cao, S.; Wang, L.; Su, X.; Li, L. Metabolic and microbial signatures in rat hepatocellular carcinoma treated with caffeic acid and chlorogenic acid. Sci. Rep. 2017, 7, 4508. [Google Scholar] [CrossRef]
- Nelson, M.E.; Lahiri, S.; Chow, J.D.; Byrne, F.L.; Hargett, S.R.; Breen, D.S.; Olzomer, E.M.; Wu, L.E.; Cooney, G.J.; Turner, N.; et al. Inhibition of hepatic lipogenesis enhances liver tumorigenesis by increasing antioxidant defence and promoting cell survival. Nat. Commun. 2017, 8, 14689. [Google Scholar] [CrossRef]
- Mason, J.B. Unraveling the complex relationship between folate and cancer risk. Biofactors 2011, 37, 253–260. [Google Scholar] [CrossRef]
- Miller, J.W.; Ulrich, C.M. Folic acid and cancer—Where are we today? Lancet 2013, 381, 974–976. [Google Scholar] [CrossRef]
- Oleinik, N.V.; Helke, K.L.; Kistner-Griffin, E.; Krupenko, N.I.; Krupenko, S.A. Rho GTPases RhoA and Rac1 mediate effects of dietary folate on metastatic potential of A549 cancer cells through the control of cofilin phosphorylation. J. Biol. Chem. 2014, 289, 26383–26394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bopp, A.; Wartlick, F.; Henninger, C.; Schwarz, M.; Kaina, B.; Fritz, G. Rac1 promotes diethylnitrosamine (DEN)-induced formation of liver tumors. Carcinogenesis 2015, 36, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Yan, S.; Khambu, B.; Chen, X.; Dong, Z.; Luo, J.; Michalopoulos, G.K.; Wu, S.; Yin, X.M. Gene Expression Analysis Indicates Divergent Mechanisms in DEN-Induced Carcinogenesis in Wild Type and Bid-Deficient Livers. PLoS ONE 2016, 11, e0155211. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Balague, E.; Matilla-Duenas, A. Ataxin-1 regulates the cerebellar bioenergetics proteome through the GSK3beta-mTOR pathway which is altered in Spinocerebellar ataxia type 1 (SCA1). Hum. Mol. Genet. 2016, 25, 4021–4040. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Paula Santos, N.; Colaco, A.; Gil da Costa, R.M.; Manuel Oliveira, M.; Peixoto, F.; Alexandra Oliveira, P. N-diethylnitrosamine mouse hepatotoxicity: Time-related effects on histology and oxidative stress. Exp. Toxicol. Pathol. 2014, 66, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Ashkavand, Z.; O’Flanagan, C.; Hennig, M.; Du, X.; Hursting, S.D.; Krupenko, S.A. Metabolic Reprogramming by Folate Restriction Leads to a Less Aggressive Cancer Phenotype. Mol. Cancer Res. 2017, 15, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.F.; O’Flanagan, C.H.; Pfeil, A.J.; Chen, X.; Pearce, J.B.; Sumner, S.; Krupenko, S.A.; Hursting, S.D. Metabolic Response of Triple-Negative Breast Cancer to Folate Restriction. Nutrients 2021, 13, 1637. [Google Scholar] [CrossRef] [PubMed]
- Dow, M.; Pyke, R.M.; Tsui, B.Y.; Alexandrov, L.B.; Nakagawa, H.; Taniguchi, K.; Seki, E.; Harismendy, O.; Shalapour, S.; Karin, M.; et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E9879–E9888. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krupenko, N.I.; Sharma, J.; Fogle, H.M.; Pediaditakis, P.; Strickland, K.C.; Du, X.; Helke, K.L.; Sumner, S.; Krupenko, S.A. Knockout of Putative Tumor Suppressor Aldh1l1 in Mice Reprograms Metabolism to Accelerate Growth of Tumors in a Diethylnitrosamine (DEN) Model of Liver Carcinogenesis. Cancers 2021, 13, 3219. https://doi.org/10.3390/cancers13133219
Krupenko NI, Sharma J, Fogle HM, Pediaditakis P, Strickland KC, Du X, Helke KL, Sumner S, Krupenko SA. Knockout of Putative Tumor Suppressor Aldh1l1 in Mice Reprograms Metabolism to Accelerate Growth of Tumors in a Diethylnitrosamine (DEN) Model of Liver Carcinogenesis. Cancers. 2021; 13(13):3219. https://doi.org/10.3390/cancers13133219
Chicago/Turabian StyleKrupenko, Natalia I., Jaspreet Sharma, Halle M. Fogle, Peter Pediaditakis, Kyle C. Strickland, Xiuxia Du, Kristi L. Helke, Susan Sumner, and Sergey A. Krupenko. 2021. "Knockout of Putative Tumor Suppressor Aldh1l1 in Mice Reprograms Metabolism to Accelerate Growth of Tumors in a Diethylnitrosamine (DEN) Model of Liver Carcinogenesis" Cancers 13, no. 13: 3219. https://doi.org/10.3390/cancers13133219
APA StyleKrupenko, N. I., Sharma, J., Fogle, H. M., Pediaditakis, P., Strickland, K. C., Du, X., Helke, K. L., Sumner, S., & Krupenko, S. A. (2021). Knockout of Putative Tumor Suppressor Aldh1l1 in Mice Reprograms Metabolism to Accelerate Growth of Tumors in a Diethylnitrosamine (DEN) Model of Liver Carcinogenesis. Cancers, 13(13), 3219. https://doi.org/10.3390/cancers13133219