
Blockage of Cholinergic Signaling via Muscarinic Acetylcholine Receptor 3 Inhibits Tumor Growth in Human Colorectal Adenocarcinoma

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
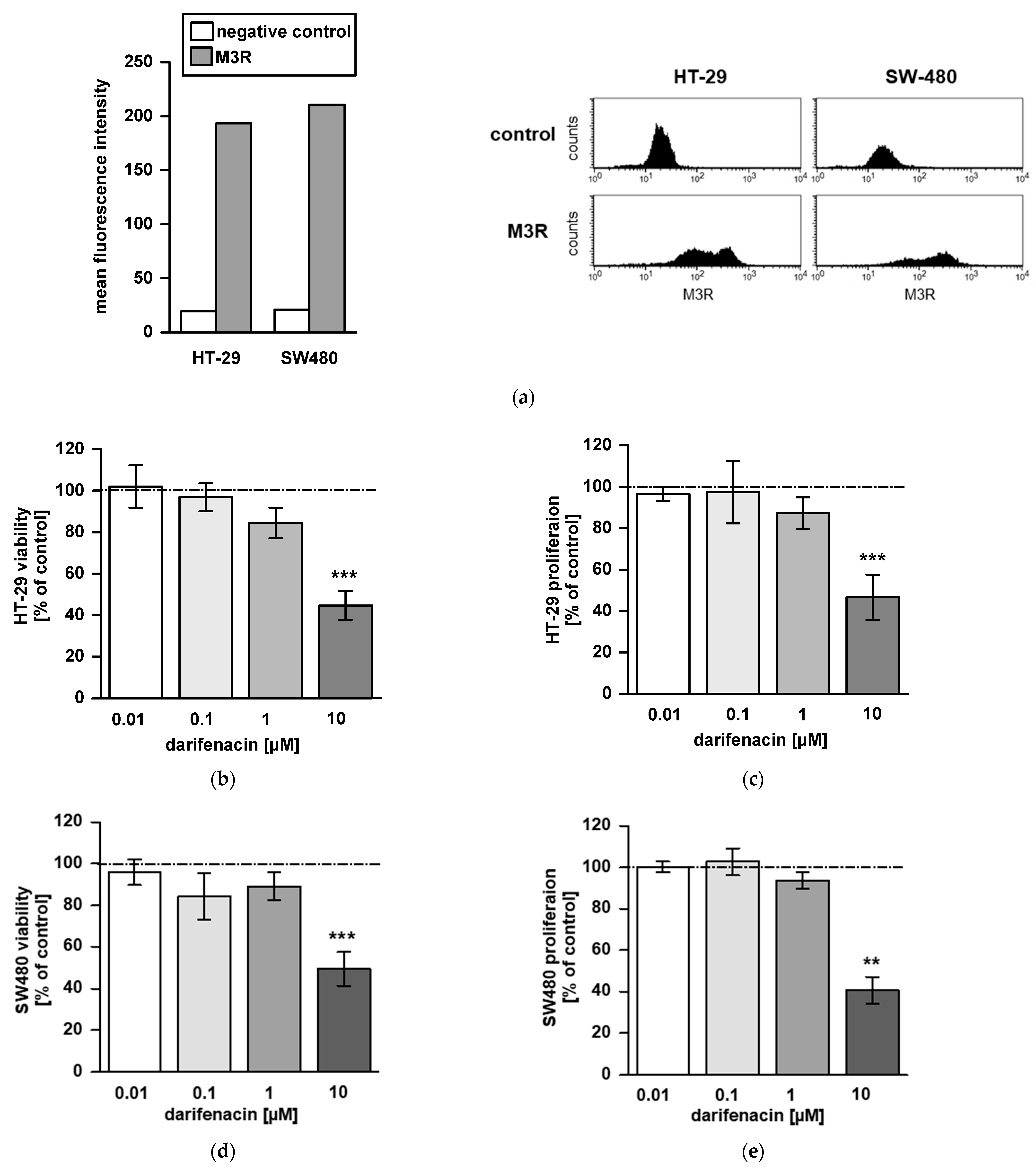
2.1. M3R Is Expressed on CRC Cell Lines
2.2. Darifenacin Inhibits CRC Cell Viability and Proliferation
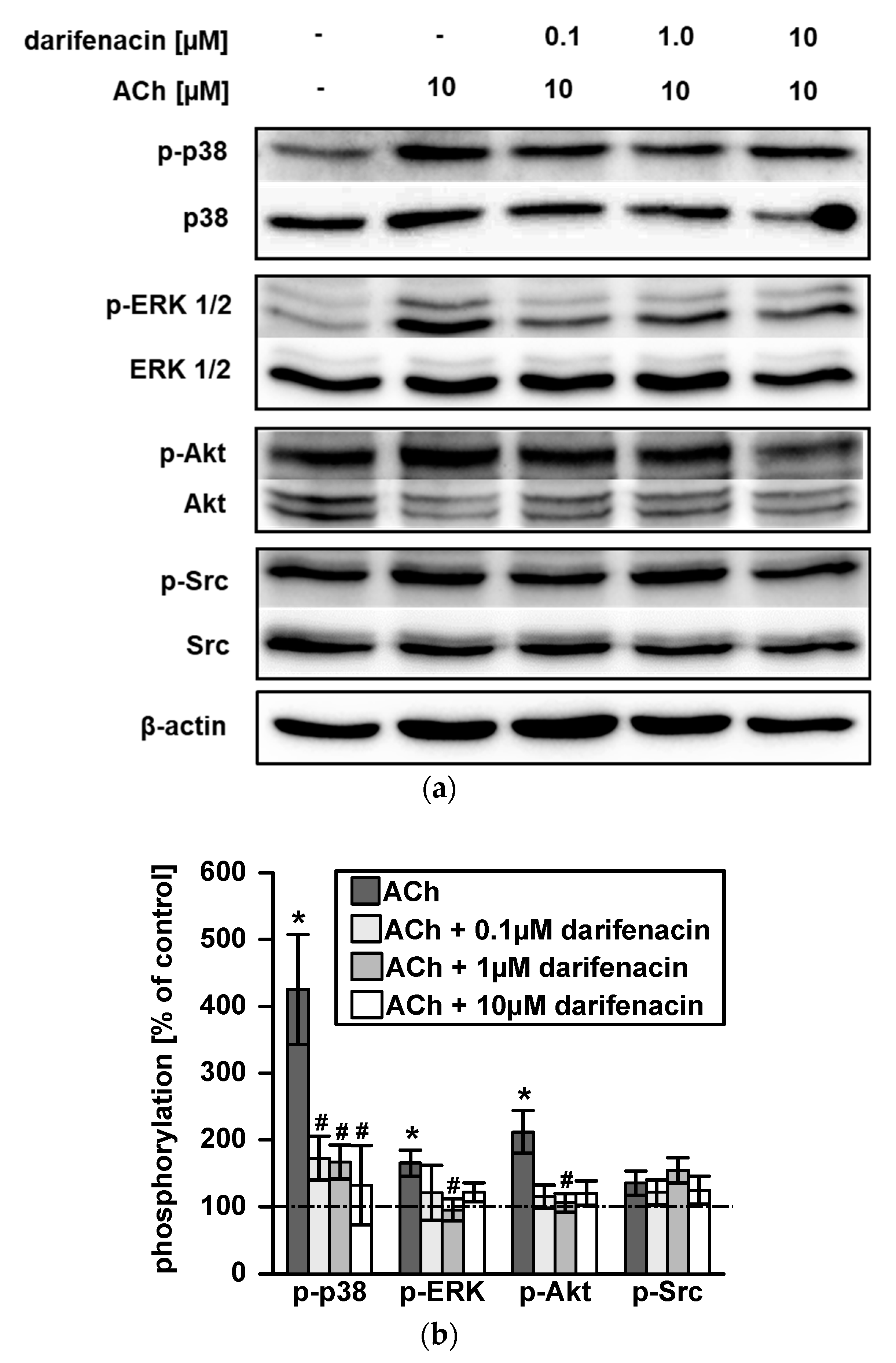
2.3. Darifenacin Inhibits ACh Induced p38, ERK1/2 and Akt Phosphorylation
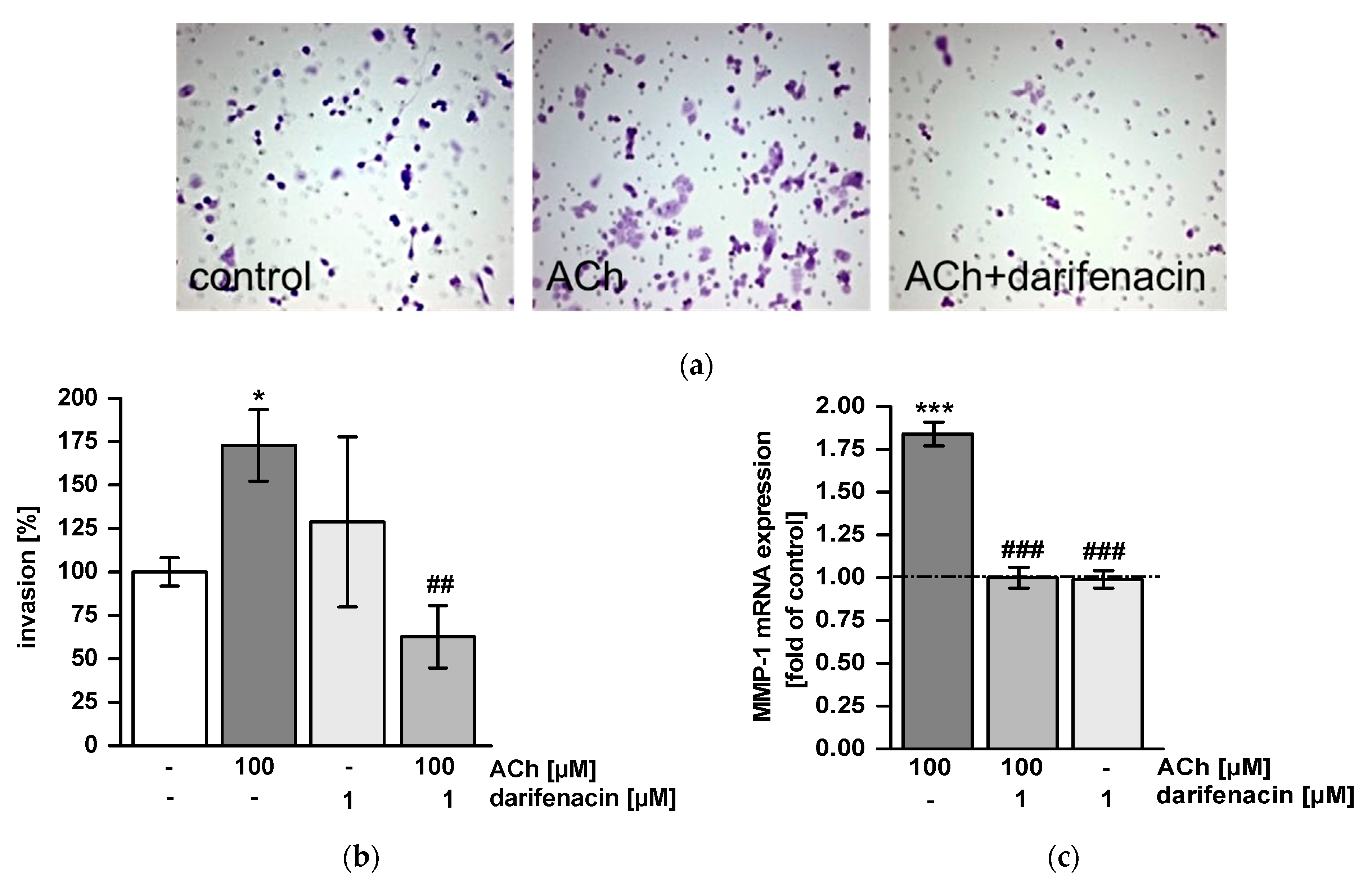
2.4. Darifenacin Inhibits ACh-Induced Invasion and MMP-1 Expression
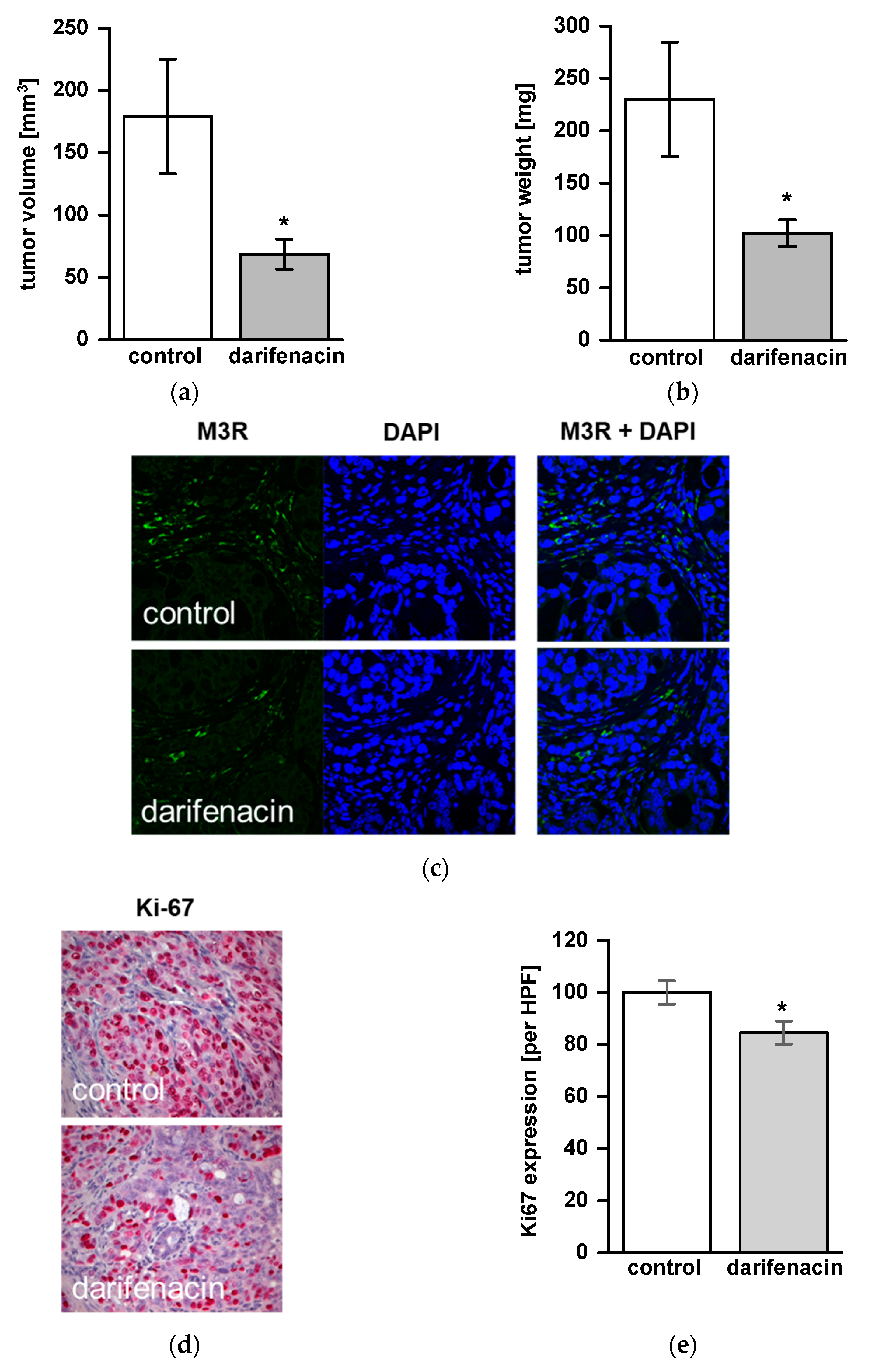
2.5. Darifenacin Inhibits Orthotopic and Metastatic Tumor Growth In Vivo
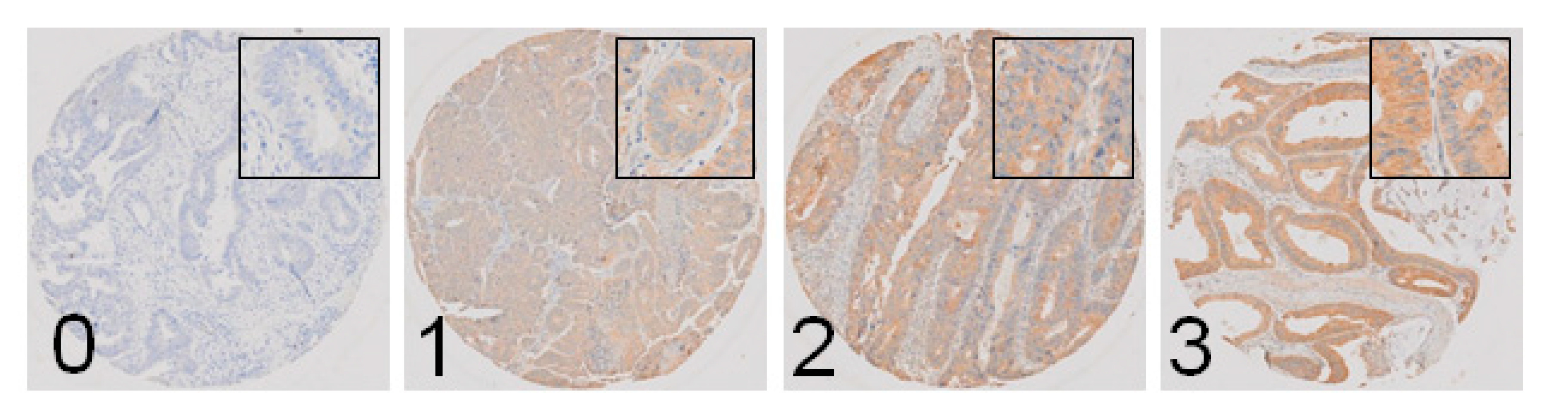
2.6. M3R Is Expressed on Human CRC Samples and Correlates with Infiltrative Tumor Border Configuration and a Non-Mucinous Histological Subtype
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture and Reagents
4.2. Measurement of Cell Viability and Proliferation
4.3. Flow Cytometry
4.4. Western Blotting
4.5. Cell Invasion Assay
4.6. Quantitative Real Time RT-PCR
4.7. Orthotopic Tumor Xenograft Mouse Model
4.8. Immunofluorescence
4.9. Immunohistochemistry
4.10. Tissue Microarray Construction and Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 116. [Google Scholar] [CrossRef] [PubMed]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S. Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 2006, 68, 345–374. [Google Scholar] [CrossRef]
- Cheng, K.; Shang, A.C.; Drachenberg, C.B.; Zhan, M.; Raufman, J.-P. Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget 2017, 8, 21106–21114. [Google Scholar] [CrossRef]
- Frucht, H.; Jensen, R.T.; Dexter, D.; Yang, W.L.; Xiao, Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin. Cancer Res. 1999, 5, 2532–2539. [Google Scholar]
- Cheng, K.; Samimi, R.; Xie, G.; Shant, J.; Drachenberg, C.; Wade, M.; Davis, R.J.; Nomikos, G.; Raufman, J.-P. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G591–G597. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Gutkind, J.S.; Novotny, E.A.; Brann, M.R.; Robbins, K.C. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc. Natl. Acad. Sci. USA 1991, 88, 4703–4707. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; Renz, B.W.; Tailor, Y.; Macchini, M.; Middelhoff, M.; et al. Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell 2017, 31, 21–34. [Google Scholar] [CrossRef]
- Raufman, J.-P.; Samimi, R.; Shah, N.; Khurana, S.; Shant, J.; Drachenberg, C.; Xie, G.; Wess, J.; Cheng, K. Genetic Ablation of M3 Muscarinic Receptors Attenuates Murine Colon Epithelial Cell Proliferation and Neoplasia. Cancer Res. 2008, 68, 3573–3578. [Google Scholar] [CrossRef]
- Raufman, J.-P.; Shant, J.; Xie, G.; Cheng, K.; Gao, X.-M.; Shiu, B.; Shah, N.; Drachenberg, C.B.; Heath, J.; Wess, J.; et al. Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis 2011, 32, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.; Dietrich, A.; Kasparek, M.S.; Benhaqi, P.; Schneider, M.R.; Schemann, M.; Seeliger, H.; Kreis, M.E. Extrinsic intestinal denervation modulates tumor development in the small intestine of Apc(Min/+) mice. J. Exp. Clin. Cancer Res. 2015, 34, 39. [Google Scholar] [CrossRef] [PubMed]
- Maman, K.; Aballea, S.; Nazir, J.; Desroziers, K.; Neine, M.E.; Siddiqui, E.; Odeyemi, I.; Hakimi, Z. Comparative efficacy and safety of medical treatments for the management of overactive bladder: A systematic literature review and mixed treatment comparison. Eur. Urol. 2014, 65, 755–765. [Google Scholar] [CrossRef]
- Yamada, S.; Maruyama, S.; Takagi, Y.; Uchida, S.; Oki, T. In vivo demonstration of M3 muscarinic receptor subtype selectivity of darifenacin in mice. Life Sci. 2006, 80, 127–132. [Google Scholar] [CrossRef]
- Cervantes-Villagrana, R.D.; Albores-Garcia, D.; Cervantes-Villagrana, A.R.; Garcia-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Valès, S.; Bacola, G.; Biraud, M.; Touvron, M.; Bessard, A.; Geraldo, F.; Dougherty, K.A.; Lashani, S.; Bossard, C.; Flamant, M.; et al. Tumor cells hijack enteric glia to activate colon cancer stem cells and stimulate tumorigenesis. EBioMedicine 2019, 49, 172–188. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Knijn, N.; Mogk, S.C.; Teerenstra, S.; Simmer, F.; Nagtegaal, I.D. Perineural Invasion is a Strong Prognostic Factor in Colo-rectal Cancer: A Systematic Review. Am. J. Surg. Pathol. 2016, 40, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Schorn, S.; Demir, I.E.; Haller, B.; Scheufele, F.; Reyes, C.M.; Tieftrunk, E.; Sargut, M.; Goess, R.; Friess, H.; Ceyhan, G.O. The influence of neural invasion on survival and tumor recurrence in pancreatic ductal adenocarcinoma A systematic review and meta-analysis. Surg. Oncol. 2017, 26, 105–115. [Google Scholar] [CrossRef]
- Xie, G.; Raufman, J.-P. Muscarinic receptor signaling and colon cancer progression. J. Cancer Metastasis Treat. 2016, 2, 195. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Jia, Y.; A Keller, J.; Blusztajn, J.K.; Mark, G.P.; Spindel, E.R. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res. 2003, 63, 214–221. [Google Scholar]
- Schütz, B.; Ruppert, A.-L.; Strobel, O.; Lazarus, M.; Urade, Y.; Büchler, M.W.; Weihe, E. Distribution pattern and molecular signature of cholinergic tuft cells in human gastro-intestinal and pancreatic-biliary tract. Sci. Rep. 2019, 9, 17466. [Google Scholar] [CrossRef]
- Yu, H.; Xia, H.; Tang, Q.; Xu, H.; Wei, G.; Chen, Y.; Dai, X.; Gong, Q.; Bi, F. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci. Rep. 2017, 7, 40802. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Lu, A.; Arredondo, J.; Sauer, D.; Gravett, C.; Mark, G.P.; Grando, S.A.; Spindel, E.R. M3 muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer Res. 2007, 67, 3936–3944. [Google Scholar] [CrossRef]
- Tolaymat, M.; Larabee, S.M.; Hu, S.; Xie, G.; Raufman, J.-P. The Role of M3 Muscarinic Receptor Ligand-Induced Kinase Signaling in Colon Cancer Progression. Cancers 2019, 11, 308. [Google Scholar] [CrossRef]
- Raufman, J.P.; Cheng, K.; Saxena, N.; Chahdi, A.; Belo, A.; Khurana, S.; Xie, G. Muscarinic receptor agonists stimulate matrix metalloproteinase 1-dependent invasion of human colon cancer cells. Biochem. Biophys. Res. Commun. 2011, 415, 319–324. [Google Scholar] [CrossRef]
- Hu, S.; Raufman, J.-P. Targeting M3 Muscarinic Receptors for Colon Cancer Therapy. Curr. Mol. Pharmacol. 2018, 11, 184–190. [Google Scholar] [CrossRef]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfán-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. 2019, 137, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Xie, G.; Khurana, S.; Heath, J.; Drachenberg, C.B.; Timmons, J.; Shah, N.; Raufman, J.-P. Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol. Cancer 2014, 13, 77. [Google Scholar] [CrossRef]
- Sunami, E.; Tsuno, N.; Osada, T.; Saito, S.; Kitayama, J.; Tomozawa, S.; Tsuruo, T.; Shibata, Y.; Muto, T.; Nagawa, H. MMP-1 is a Prognostic Marker for Hematogenous Metastasis of Colorectal Cancer. Oncologist 2000, 5, 108–114. [Google Scholar] [CrossRef]
- Cremolini, C.; Di Bartolomeo, M.; Amatu, A.; Antoniotti, C.; Moretto, R.; Berenato, R.; Perrone, F.; Tamborini, E.; Aprile, G.; Lonardi, S.; et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at fa-vorable prognosis. Ann. Oncol. 2015, 26, 2092–2097. [Google Scholar] [CrossRef]
- Cheng, K.; Zimniak, P.; Raufman, J.P. Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of H508 human colon cancer cells. Cancer Res. 2003, 63, 6744–6750. [Google Scholar] [CrossRef]
- Said, A.H.; Hu, S.; Abutaleb, A.; Watkins, T.; Cheng, K.; Chahdi, A.; Kuppusamy, P.; Saxena, N.; Xie, G.; Raufman, J.-P. Interacting post-muscarinic receptor signaling pathways potentiate matrix metalloproteinase-1 expression and invasion of human colon cancer cells. Biochem. J. 2017, 474, 647–665. [Google Scholar] [CrossRef]
- Xu, R.; Shang, C.; Zhao, J.; Han, Y.; Liu, J.; Chen, K.; Shi, W. Activation of M3 muscarinic receptor by acetylcholine pro-motes non-small cell lung cancer cell proliferation and invasion via EGFR/PI3K/AKT pathway. Tumour Biol. 2015, 36, 4091–4100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014, 6, 250ra115. [Google Scholar] [CrossRef]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic Signaling via Mus-carinic Receptors Directly and Indirectly Suppresses Pancreatic Tumorigenesis and Cancer Stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef]
- Kühn, T.; Stepien, M.; López-Nogueroles, M.; Damms-Machado, A.; Sookthai, D.; Johnson, T.; Roca, M.; Hüsing, A.; Maldonado, S.G.; Cross, A.J.; et al. Prediagnostic Plasma Bile Acid Levels and Colon Cancer Risk: A Prospective Study. J. Natl. Cancer Inst. 2020, 112, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Chen, Y.; Cheng, K.; Compadre, C.; Compadre, L.; Zimniak, P. Selective interaction of bile acids with muscarinic receptors: A case of molecular mimicry. Eur. J. Pharmacol. 2002, 457, 77–84. [Google Scholar] [CrossRef]
- Shiozawa, J.; Ito, M.; Nakayama, T.; Nakashima, M.; Kohno, S.; Sekine, I. Expression of matrix metalloproteinase-1 in hu-man colorectal carcinoma. Mod. Pathol. 2000, 13, 925–933. [Google Scholar] [CrossRef]
- Luo, C.; Cen, S.; Ding, G.; Wu, W. Mucinous colorectal adenocarcinoma: Clinical pathology and treatment options. Cancer Commun. 2019, 39, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hirano, Y.; Heng, G.; Ishii, T.; Kondo, H.; Hara, K.; Obara, N.; Asari, M.; Kato, T.; Yamaguchi, S. Mucinous Ade-nocarcinoma as a High-risk Factor in Stage II Colorectal Cancer: A Propensity Score-matched Study from Japan. Anticancer Res. 2020, 40, 1651–1659. [Google Scholar] [CrossRef]
- Kang, H.; Min, B.S.; Lee, K.Y.; Kim, N.K.; Kim, S.N.; Choi, J.; Kim, H. Loss of E-cadherin and MUC2 expressions correlated with poor survival in patients with stages II and III colorectal carcinoma. Ann. Surg. Oncol. 2011, 18, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-W.; Tao, L.-Y.; Jiang, Y.-S.; Yang, J.-Y.; Huo, Y.-M.; Liu, D.-J.; Li, J.; Fu, X.-L.; He, R.; Lin, C.; et al. Perineural invasion reprograms the immune microenvironment through cholinergic signaling in pancreatic ductal adenocarcinoma. Cancer Res. 2020, 80, 1991–2003. [Google Scholar] [CrossRef]
- Cienfuegos, J.A.; Martínez, P.; Baixauli, J.; Beorlegui, C.; Rosenstone, S.; Sola, J.J.; Rodríguez, J.; Hernández-Lizoáin, J.L. Perineural Invasion is a Major Prognostic and Predictive Factor of Response to Adjuvant Chemotherapy in Stage I–II Colon Cancer. Ann. Surg. Oncol. 2016, 24, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Kim, S.Y.; Lee, J.S.; Nam, B.-H.; Kim, K.-P.; Kim, J.E.; Park, Y.S.; Park, J.O.; Baek, J.Y.; Kim, T.-Y.; et al. Oxaliplatin-Based Adjuvant Chemotherapy for Rectal Cancer After Preoperative Chemoradiotherapy (ADORE): Long-Term Results of a Randomized Controlled Trial. J. Clin. Oncol. 2019, 37, 3111–3123. [Google Scholar] [CrossRef]
- Löfling, L.; Sundström, A.; Kieler, H.; Bahmanyar, S.; Linder, M. Exposure to antimuscarinic medications for treatment of overactive bladder and risk of lung cancer and colon cancer. Clin. Epidemiol. 2019, 11, 133–143. [Google Scholar] [CrossRef]
- Stojanovska, V.; McQuade, R.M.; Miller, S.; Nurgali, K. Effects of Oxaliplatin Treatment on the Myenteric Plexus Innervation and Glia in the Murine Distal Colon. J. Histochem. Cytochem. 2018, 66, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, M.; Iwasaki, Y.; Inoue, S.; Saito, S.; Kawashima, K.; Fujii, T. Acetylcholine released from T cells regulates intracellular Ca2+, IL-2 secretion and T cell proliferation through nicotinic acetylcholine receptor. Life Sci. 2017, 172, 13–18. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Seeliger, H.; Guba, M.; Koehl, G.E.; Doenecke, A.; Steinbauer, M.; Bruns, C.J.; Wagner, C.; Frank, E.; Jauch, K.W.; Geissler, E.K. Blockage of 2-deoxy-D-ribose-induced angiogenesis with rapamycin counteracts a thymidine phosphorylase-based es-cape mechanism available for colon cancer under 5-fluorouracil therapy. Clin. Cancer Res. 2004, 10, 1843–1852. [Google Scholar] [CrossRef][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | n | M3R Low | M3R High |
|---|---|---|---|
| Total | 585 | 63 (10.8%) | 522 (89.2%) |
| Age [years] | 68.6 (30–96) a | 67.2 (30–87) a | 68.8 (36–96) a |
| Sex | |||
| Female | 322 (55.0%) | 32 (9.9%) | 290 (90.1%) |
| Male | 263 (45.0%) | 31 (11.8%) | 232 (88.2%) |
| Tumor location b | |||
| Left sided | 405 (69.2%) | 41 (10.1%) | 364 (89.9%) |
| Right sided | 178 (30.4%) | 22 (12.4%) | 156 (87.6%) |
| Histologic subtype | |||
| Mucinous | 28 (4.8%) | 6 (21.4%) | 22 (78.6%) |
| Non-mucinous | 557 (95.2%) | 57 (10.2%) | 500 (89.8%) |
| pT status b | |||
| pT1/2 | 111 (19.0%) | 14 (12.6%) | 97 (87.4%) |
| pT3/4 | 464 (79.3%) | 44 (9.5%) | 420 (90.5%) |
| pN status | |||
| pN0 | 311 (53.2%) | 32 (10.3%) | 279 (89.7%) |
| pN1-2 | 274 (46.8%) | 31 (11.3%) | 243 (88.7%) |
| Grading b | |||
| G1/2 | 559 (95.6%) | 55 (9.8%) | 504 (90.2%) |
| G3 | 16 (2.7%) | 3 (18.8%) | 13 (81.3%) |
| Vascular invasion b | |||
| Present | 161 (27.5%) | 19 (11.8%) | 142 (88.2%) |
| Absent | 414 (70.8%) | 39 (9.4%) | 375 (90.6%) |
| Tumor border configuration b | |||
| Infiltrative | 396 (67.7%) | 46 (11.6%) | 350 (88.4%) |
| Pushing | 58 (9.9%) | 12 (20.7%) | 46 (79.3%) |
| Microsatellite instability | |||
| Deficient | 68 (11.6%) | 8 (11.8%) | 60 (88.2%) |
| Proficient | 517 (88.4%) | 55 (10.6%) | 462 (89.4%) |
| Parameter | Univariate Regression | Multivariate Regression | ||
|---|---|---|---|---|
| OR (95% CI) | p Value | OR (95% CI) | p Value | |
| Age | ||||
| <60 | Reference | Reference | ||
| >=60 | 1.29 (0.72–2.32) | 0.381 | 1.01 (0.98–1.04) | 0.438 |
| Sex | ||||
| Female | Reference | Reference | ||
| Male | 0.83 (0.49–1.39) | 0.473 | 0.75 (0.42–1.32) | 0.315 |
| Tumor location | ||||
| Right sided | Reference | Reference | ||
| Left sided | 0.80 (0.46–1.39) | 0.424 | 0.77 (0.41–1.44) | 0.415 |
| Histologic subtype | ||||
| Non-mucinous | Reference | Reference | ||
| Mucinous | 0.418 (0.16–1.07) | 0.070 | 0.26 (0.10–0.74) | 0.011 |
| pT status | ||||
| pT1/2 | Reference | Reference | ||
| pT3/4 | 1.38 (0.73–2.61) | 0.327 | 1.89 (0.88–4.03) | 0.102 |
| pN status | ||||
| pN0 | Reference | Reference | ||
| pN1–2 | 0.90 (0.53–1.52) | 0.690 | 1.03 (0.55–1.92) | 0.926 |
| Grading | ||||
| G1 | Reference | Reference | ||
| G2 | 2.43 (0.67–8.87) | 0.177 | 1.98 (0.48–8.12) | 0.346 |
| G3 | 0.67 (0.16–2.84) | 0.558 | 0.49 (0.10–2.55) | 0.399 |
| Vascular invasion | ||||
| Present | Reference | Reference | ||
| Absent | 0.77 (0.43–1.39) | 0.396 | 1.02 (0.52–2.02) | 0.956 |
| Tumor border configuration | ||||
| Infiltrative | Reference | Reference | ||
| Pushing | 0.55 (0.29–1.07) | 0.080 | 0.43 (0.20–0.89) | 0.024 |
| Microsatellite instability | ||||
| Deficient | Reference | Reference | ||
| Proficient | 1.12 (0.51–2.47) | 0.778 | 0.96 (0.40–2.28) | 0.928 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hering, N.A.; Liu, V.; Kim, R.; Weixler, B.; Droeser, R.A.; Arndt, M.; Pozios, I.; Beyer, K.; Kreis, M.E.; Seeliger, H. Blockage of Cholinergic Signaling via Muscarinic Acetylcholine Receptor 3 Inhibits Tumor Growth in Human Colorectal Adenocarcinoma. Cancers 2021, 13, 3220. https://doi.org/10.3390/cancers13133220
Hering NA, Liu V, Kim R, Weixler B, Droeser RA, Arndt M, Pozios I, Beyer K, Kreis ME, Seeliger H. Blockage of Cholinergic Signaling via Muscarinic Acetylcholine Receptor 3 Inhibits Tumor Growth in Human Colorectal Adenocarcinoma. Cancers. 2021; 13(13):3220. https://doi.org/10.3390/cancers13133220
Chicago/Turabian StyleHering, Nina A., Verena Liu, Rayoung Kim, Benjamin Weixler, Raoul A. Droeser, Marco Arndt, Ioannis Pozios, Katharina Beyer, Martin E. Kreis, and Hendrik Seeliger. 2021. "Blockage of Cholinergic Signaling via Muscarinic Acetylcholine Receptor 3 Inhibits Tumor Growth in Human Colorectal Adenocarcinoma" Cancers 13, no. 13: 3220. https://doi.org/10.3390/cancers13133220
APA StyleHering, N. A., Liu, V., Kim, R., Weixler, B., Droeser, R. A., Arndt, M., Pozios, I., Beyer, K., Kreis, M. E., & Seeliger, H. (2021). Blockage of Cholinergic Signaling via Muscarinic Acetylcholine Receptor 3 Inhibits Tumor Growth in Human Colorectal Adenocarcinoma. Cancers, 13(13), 3220. https://doi.org/10.3390/cancers13133220