1. Introduction
Despite advancements in pediatric cancer care, neuroblastoma, a childhood malignancy resulting from abnormal neural crest cell development, remains the cause of over 15% of pediatric cancer related deaths [
1]. Current treatment regimens for high-risk disease include chemotherapy, surgical resection, autologous stem cell transplant, radiation, immunotherapy, and retinoic acid [
2]. Despite these extensive therapies, outcomes remain dismal for patients with high-risk disease as less than 50% will attain a 5 year survival [
3]. It is crucial to continue to investigate this disease in order to develop new therapies, especially for the cohort of patients with high-risk disease.
Serine-threonine kinase receptor-associated protein (STRAP) is a scaffolding protein that facilitates protein to protein interactions [
4]. Investigators began to study the role of STRAP in cancer when they found that STRAP inhibited the tumor suppressor, TGF-β [
5]. STRAP is overexpressed in several malignancies, including colorectal [
6] and lung cancer [
7] and the pediatric bone cancer, osteosarcoma [
8]. In these malignancies, STRAP enhanced cancer cell proliferation and tumor growth.
STRAP expression supports tumorigenicity by promoting the Wnt/β-catenin pathway in colorectal cancer. In cells overexpressing STRAP, silencing the STRAP gene with short hairpin RNAs (shRNA) resulted in decreased invasion and metastasis [
6]. In lung cancer, STRAP led to tumor progression by downregulating tumor suppressors, E-cadherin, and the CDK inhibitor, p21Cip1, through the modulation of the transcription factor Sp1 [
9]. Pruksakorn et al. found STRAP was upregulated in osteosarcoma and inhibiting STRAP with small interfering RNA (siRNA) decreased migration and invasion; this suggests that STRAP contributed to osteosarcoma metastasis [
8]. These discoveries lend support for investigating STRAP in neuroblastoma. We hypothesized that inhibition of STRAP protein expression in neuroblastoma would result in a decrease in the malignant phenotype in vitro and in vivo. In the current study, we demonstrate STRAP inhibition with siRNA, shRNA, and CRISPR-Cas9 knockout (KO) decreased tumor cell viability, proliferation, growth, stemness, and motility in vitro and tumor growth in vivo. To our knowledge, researchers have not performed extensive study of STRAP in neuroblastoma and our findings provide evidence for STRAP as a potential driver for neuroblastoma tumorigenesis.
2. Materials and Methods
2.1. Cells and Cell Culture
The human neuroblastoma cell lines SK-N-AS (AS, CRL-2137, and MYCN non-amplified), SK-N-BE (2) (BE, CRL-2271, and MYCN amplified), and SH-SY5Y (CRL-2266 and MYCN non-amplified) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The isogenic SH-EP (SHEP) and WAC (2) (WAC), MYCN non-amplified and amplified cell lines, respectively, were a gift from M. Schwab (Deutsches Krebsforschungszentrum, Heidelberg, Germany) [
10]. Cells were maintained under standard culture conditions at 37 °C and 5% CO
2. AS cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, 30-2601, ATCC) containing 10% fetal bovine serum (FBS, Hyclone, Suwanee, GA, USA), 4 mM L-glutamine (Thermo Fisher Scientific Inc., Waltham, MA, USA), 1 µM non-essential amino acids, and 1 µg/mL penicillin/streptomycin (Gibco, Carlsbad, CA, USA), which is referred to as media. BE cells were maintained in a 1:1 mixture of minimum Eagle medium and Ham F-12 medium (30–2004, ATCC) with 10% FBS (Hyclone), 1 μM non-essential amino acids, 2 mM l-glutamine (Thermo Fisher Scientific Inc.), and 1/μg/mL penicillin/streptomycin (Gibco). SH-EP and WAC (2) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (30–2001, ATCC) with 10% FBS (Hyclone), 2/mM l-glutamine (Thermo Fisher Scientific Inc.), and 1 μg/mL penicillin/streptomycin (Gibco). All cell lines were verified within the last 12 months using short tandem repeat analysis (University of Alabama at Birmingham (UAB) Genomics Core, Birmingham, AL, USA) and tested for and deemed free of mycoplasma infection.
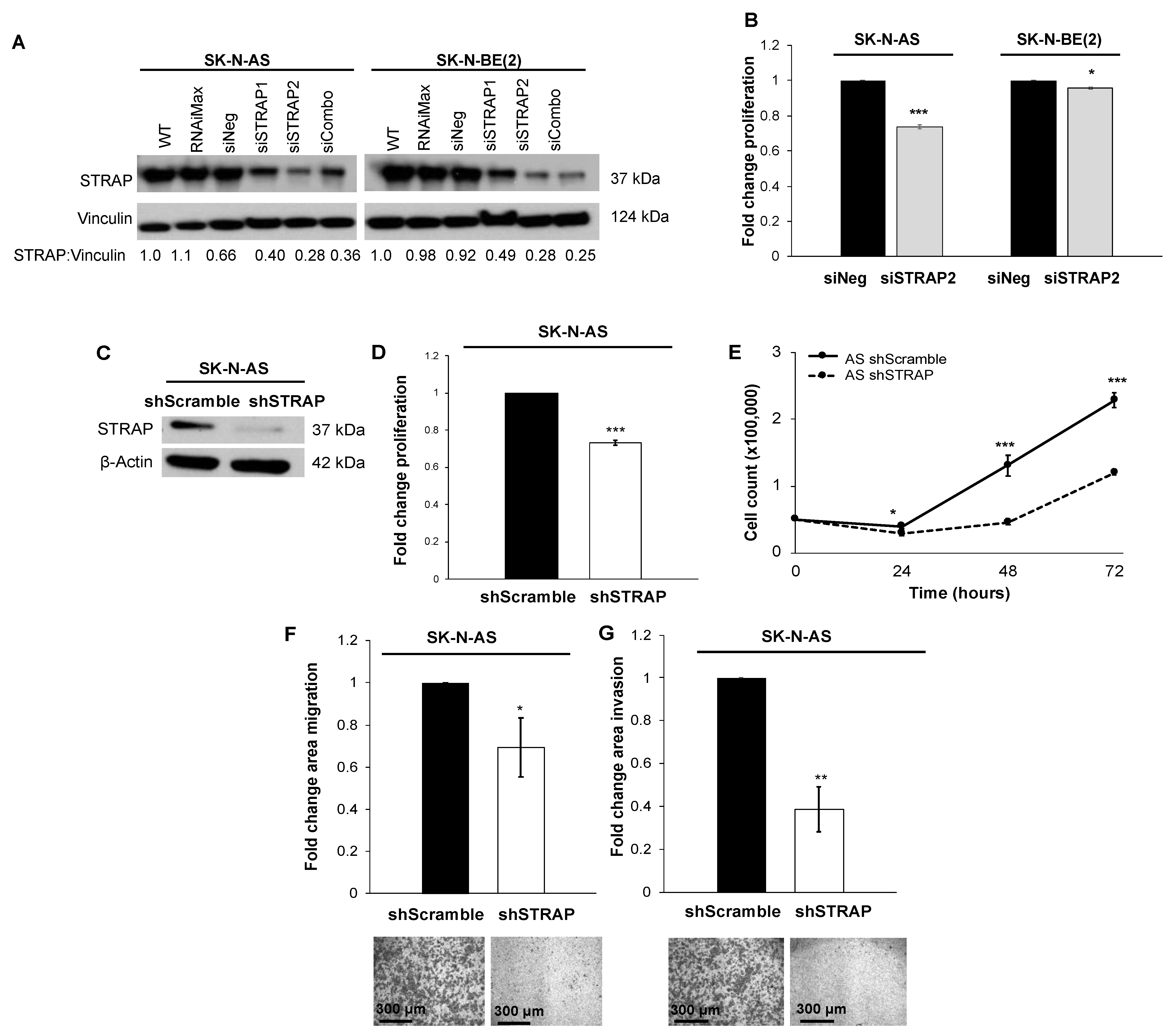
2.2. Small Interfering RNA (siRNA) Inhibition of STRAP
AS or BE cells (1 × 106) were plated in 6-well plates and transfected for 72 h with Lipofectamine RNAiMax (Thermo Fisher Scientific) or siRNA directed to either control (siNeg), siSTRAP1, siSTRAP2, or a combination of the two STRAP siRNAs at 20 µM concentration with Lipofectamine RNAiMax (Thermo Fisher Scientific). SiNeg (ON-TARGETplus Non-targeting siRNA #1) was obtained from Dharmacon (GE Dharmacon, Thermo Fisher Scientific, Lafayette, CO, USA). The siSTRAP1 (SASI_Hs01_00016957) and siSTRAP2 (SASI_Hs02_00343131) were obtained from Sigma Aldrich (St. Louis, MO, USA).
2.3. Short Hairpin RNA (shRNA) Inhibition of STRAP
The shScramble and shSTRAP cell lines were generously provided by Dr. Pran Datta’s laboratory and were established as previously described [
11]. The shScramble served as the control for shSTRAP cells. AS cells that underwent lentiviral transfection with shScramble or shSTRAP were cultured in AS media with the addition of puromycin (5 µM, P8833, Sigma-Aldrich, St. Louis, MO, USA) for selection.
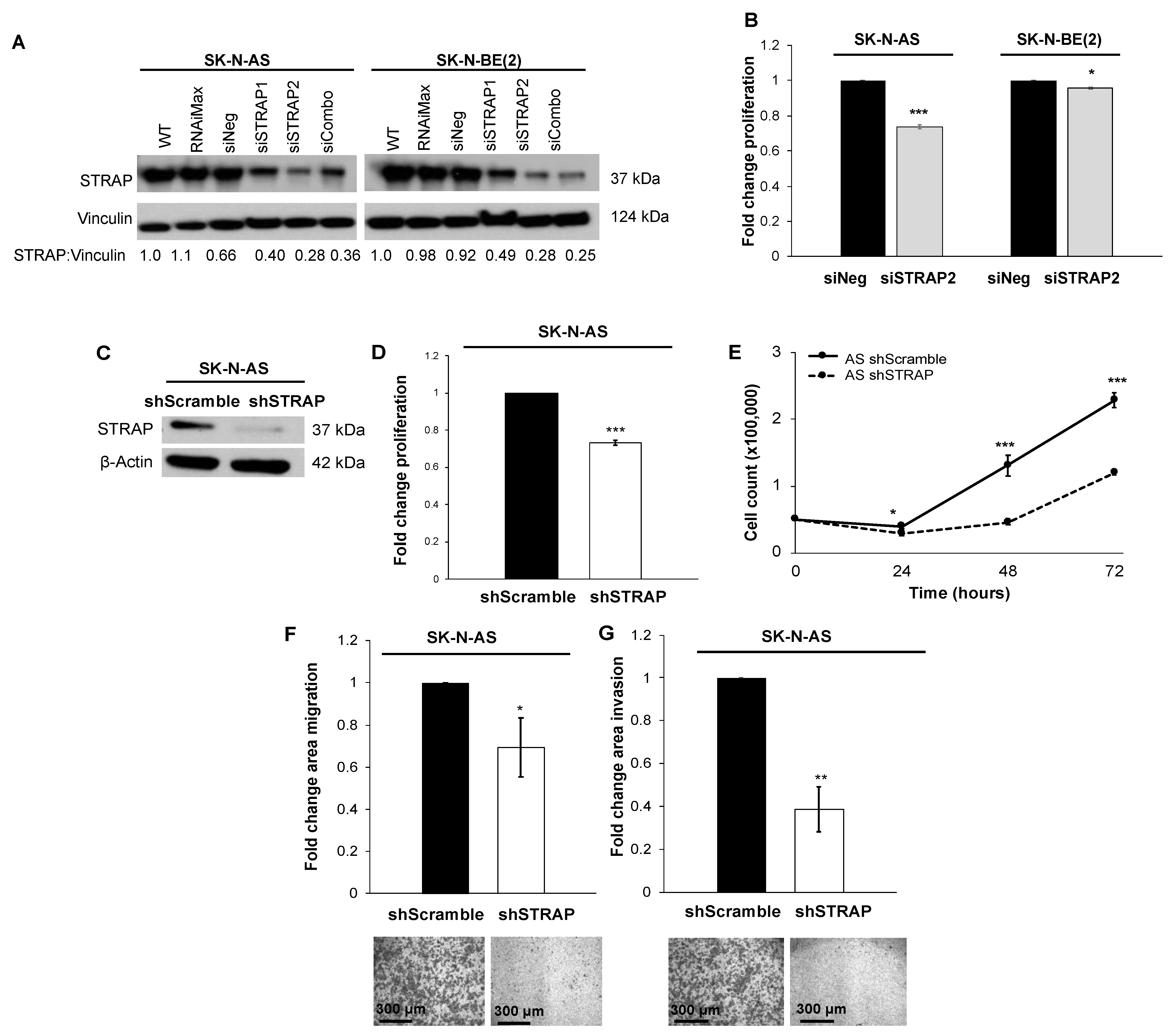
2.4. CRISPR-Cas9 Knockout (KO) of STRAP
The CRISPR vector, pSpCas9(BB)-2A-GFP (pX458), was a gift from Dr. A. Joseph Tector and developed by Dr. Feng Zhang (Addgene plasmid #48138) [
12]. We used Geneious software (Biomatters, Auckland, New Zealand) to design guide RNAs (gRNAs) to regions from the 5′ untranslated region through exon 3 of the STRAP gene. The selected gRNAs were evaluated with the MIT CRISPR Design Tool (
http://crispr.mit.edu/) to assess for potential off-target sequences. The oligonucleotides (5′-CACCGTTGGGGTGCAACACTGAATA-3′ and 5′-CAACCCCACGTTGTGACTTATCAAAA-3′) were annealed at 37 °C for 30 min, 95 °C for 5 min, and ramped down to 25 °C at 5 °C per minute. Annealed oligonucleotides were cloned into the CRISPR plasmid by digesting 1 µg of plasmid pX458 with BbsI (New England Biolabs, Ipswich, MA, USA) in the presence of annealed oligonucleotides, T7 ligase, and ATP in a MyCycler™ thermal cycler (Bio-Rad, Hercules, CA, USA) for 37 °C for 5 min and 23 °C for 5 min for 6 cycles. Ligation reaction was transformed into Invitrogen MAX Efficiency™ DH5α™ competent
E. coli cells (Invitrogen) following the manufacturer’s protocol. Plasmids were isolated from one colony per treatment using the QIAprep
® Miniprep (Qiagen, Germantown, MD, USA). DNA was isolated using QIAprep Spin Miniprep Kit (Qiagen) and sequenced by the UAB Genomics Core. AS or BE cells (2 × 10
6) were plated in 6-well plates and 24 h later, transfection was carried out using FuGENE
® HD Transfection Reagent (Promega, Madison, WI, USA) per the manufacturer’s protocol. The STRAP gRNA plasmid was incubated for 15 min at room temperature in OptiMEM™ media (Thermo Fisher Scientific) with FuGENE
® HD Transfection Reagent (Promega) in 3:2 ratio of the transfection reagent to DNA. Plasmid DNA (10 µg) was added to the cells while swirling the flask. Forty-eight hours after transfection, cells from the plasmid transfection were sorted based on green fluorescent protein (GFP) expression using a FACSAria II cell sorter (BD Biosciences, San Jose, CA, USA) into 96-well plates with a single cell per well (Comprehensive Flow Cytometry Core, UAB, Birmingham, AL, USA). In order to screen for CRISPR-Cas9 mediated deletions of the STRAP gene, genomic DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen) from AS or BE wild-type (WT) cells and those clones that had grown to confluence and survived passage into larger flasks. Pwo SuperYield DNA Polymerase, dNTPack (Sigma Aldrich) with GC-rich solution were utilized per manufacturer’s protocol to amplify the region of interest within the STRAP gene using the following primers, (forward: 5′-TTAGTGCCTTCAGTGGGTGG-3′, reverse: 5′-GGTGGGATCAAACATGCGTTC-3′), which were designed using Primer-Blast (
https://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 3 September 2018)). PCR products were assessed using gel electrophoresis on a 1% agarose gel. Individual bands were cut and DNA was purified using the QIAquick Gel Extraction Kit (Qiagen). Nucleotide sequences of these DNA fragments were analyzed by Sanger sequencing (UAB Genomics Core) and aligned to the human reference sequence using the basic local alignment search tool (BLAST, NCBI,
https://pubmed.ncbi.nlm.nih.gov/22708584/ (accessed on 27 February 2019)). STRAP protein expression in the AS or BE WT cells and selected STRAP KO clones was assessed by Western blotting to confirm the absence of the STRAP protein.
2.5. Rescue of STRAP Expression
In order to validate that the phenotypic changes noted with STRAP KO cells were not secondary to off-target effects of the CRISPR-Cas9 system, we performed rescue experiments by transfecting AS STRAP KO cells with c-Flag pc-DNA empty vector (EV) or pc-DNA STRAP plasmid [
13]. STRAP KO cells (3 × 10
3) were plated in 6-well plates and transfected with either FuGENE
® HD, FuGENE
® HD and EV plasmid, or FuGENE
® HD and STRAP plasmid for 72 h. Stable rescue cells were selected and maintained in AS media with the addition of G418 (600 µg/mL, A1720, Sigma-Aldrich). Cells were used in proliferation studies with CellTiter 96
® assay as described below to examine the phenotype of the KO cells following the re-introduction of STRAP.
2.6. Reagents and Antibodies
Trypan blue stain was obtained from Life Technologies Corporation (Grand Island, NY, USA). Primary antibodies used for Western blotting included the following: polyclonal rabbit anti-STRAP (18277-1-AP) from Proteintech (Rosemont, IL, USA), monoclonal rabbit anti-PGDFRβ (#3169) and monoclonal rabbit anti-vinculin (E1E9V, #13901) from Cell Signaling (Danvers, MA, USA), and monoclonal mouse anti-FLAG (F3165) and monoclonal mouse anti-β-actin (A1978) from Sigma Aldrich. Antibodies were used according to manufacturers’ suggestions.
2.7. Immunoblotting
Cells or homogenized tumor specimens were lysed on ice in a buffer consisting of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton x-100, 1% sodium deoxcycholate, 0.1% SDS, phosphatase inhibitor (P5726, Sigma Aldrich), protease inhibitor (P8340, Sigma Aldrich), and phenylmethylsulfonyl fluoride (PMSF, P7626, Sigma Aldrich) for 30 min. Lysates were centrifuged at 17,000×
g for 30 min at 4 °C. Protein concentrations were determined using a Micro BCA™ Protein Assay Kit (Thermo Fisher Scientific). Proteins were separated on SDS-PAGE gels by electrophoresis and transferred to Immobilon
®-P polyvinylidene fluoride (PVDF) transfer membrane (EMD Millipore, Burlington, MA, USA). In order to confirm the expected size of target proteins, Precision Plus Protein Kaleidoscope Standards (161-0375, Bio-Rad) molecular weight markers were used. Antibodies were used per the manufacturers’ recommended protocol. Luminata Classico or Luminata Crescendo (EMD Millipore) substrates were used to visualize immunoblots by enhanced chemiluminescence (ECL) of horseradish peroxidase (HPR)-conjugated secondary antibodies. β-actin or vinculin served as a control to ensure equal protein loading. We performed densitometry of Western blots using ImageJ software (Ver 1.49,
http://imagej.nih.gov/ij (accessed on 7 July 2018).
2.8. Proliferation
Proliferation was examined using the CellTiter 96® Aqueous One Solution Cell Proliferation assay (Promega). Cells (5 × 103 cells) were plated onto 96-well plates. After 24 h, CellTiter 96® dye (10 µL) was added to each well and the absorbance was measured at 490 nm using a microplate reader (Epoch Microplate Spectrophotometer, BioTek Instruments, Winooski, VT, USA). For the siRNA experiments, we transfected the cells with siRNA for 72 h as described above, then we lifted and plated transfected cells onto 96-well plates for 24 h, added CellTiter96® dye (Promega), and read the plates. Proliferation experiments were completed with at least three biologic replicates and data reported as fold change ± standard error of the mean (SEM).
2.9. Growth Curve
Cells (5 × 104) were plated in a 12-well plate in adherent conditions in 12-well plates. In order to measure cell growth over time, cells were lifted and live cells were counted after being stained with trypan blue at 1, 2, 3, and 4 days for AS cells and at 5, 7, and 9 days for BE cells.
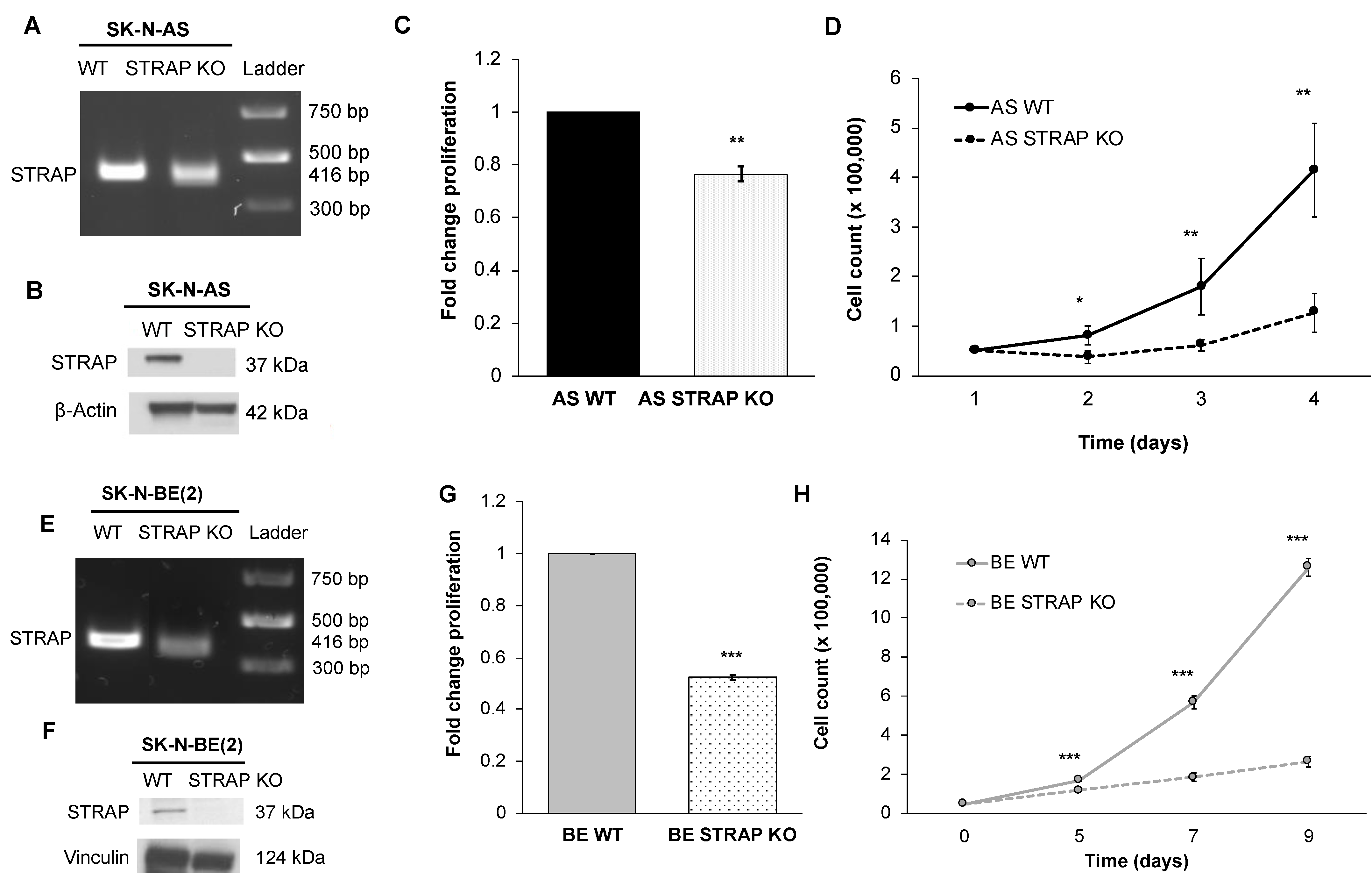
2.10. Cell Cycle
AS WT and STRAP KO cells were plated in 6-well plates and maintained in AS media with decreased FBS (4%). After 24 h, cells (5 × 105) were washed with PBS and fixed with 100% ethanol at 4 °C for at least 30 min. After a second wash with PBS, cells were stained with propidium iodide (Invitrogen), 0.1% TritonX (Active Motif, Carlsbad, CA, USA), and RNAse A (0.1 mg/mL, Qiagen) and cell cycle data were obtained using the FACSCalibur™ Flow Cytometer (BD Biosciences) and analyzed using the FlowJo software (FlowJo, LLC, Ashland, OR, USA).
2.11. Migration and Invasion
The effect of STRAP KO or shRNA inhibition on migration was assessed using modified Boyden chamber assays. Cells (4 × 10
4) were seeded onto 8 µM pore inserts (TransWell
®, Corning, Corning, NY, USA) and were allowed to migrate for 24 h with laminin (10 μg/mL, 100 μL, Trevigen, Gaithersburg, MD, USA) used in the outer well as a chemoattractant. Inserts were fixed with 4% paraformaldehyde for 10 min, stained with 1% crystal violet for 15 min, and photographed. Photographs were analyzed using ImageJ software (Ver 1.49,
http://imagej.nih.gov/ij (accessed on 17 March 2019) to quantitate migration.
Invasion was evaluated similarly, except for a layer of Matrigel™ (1 mg/mL, 50 µL, BD Biosciences) which was used to coat the top of the insert membrane. Cells (4 × 104) were seeded in the upper chamber and allowed to invade through the Matrigel™ layer for 24 h toward the laminin chemoattractant in the outer well. Inserts were then fixed, stained, and photographed as described for migration. Photographs were analyzed using ImageJ to quantitate invasion.
2.12. Anchorage-Independent Growth
A soft agar assay was utilized to assess for anchorage-independent growth. A base layer of 1% noble agar mixed with culture media was established in 60 mm culture dishes. AS WT or STRAP KO cells (1 × 104) were plated in the top layer in the agar and culture media mixture. After 6 weeks, colonies were stained with crystal violet, imaged, and quantified using ImageJ.
2.13. RNA Sequencing and Analysis
Total cellular RNA was extracted from AS WT and AS STRAP KO cells using the RNAeasy kit (Qiagen) according to the manufacturer’s protocol. UAB Genomics Core performed sample quality control, library preparation, and RNA sequencing. The Agilent 2100 Bioanalyzer was used to assess the total RNA quality, which was followed by two rounds of poly A+ selection and conversion to cDNA. The NEBNext
® Ultra™ Directional RNA Library Prep Kit for Illumina
® library generation kit (New England Biolabs) was used per the manufacturer’s instructions. qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit (Kapa Biosystems, Woburn, MA, USA) was used for library quantitation. The Illumina NextSeq500 was used to perform the sequencing using the latest versions of the sequencing reagents and flow cells with single-end 75 bp reads. Raw and processed data were deposited in the Gene Expression Omnibus (GEO, Accession GSE169322) [
14].
STAR (version 2.7.3a) was used to align the raw RNA-Seq fastq reads to the human reference genome (GRCh38 p13 Release 32) from Gencode using parameters the following parameters: outReadsUnmapped Fastx; outSAMtype BAM SortedByCoordinate; outSAMattributes All --outFilterIntronMotifs RemoveNoncanonicalUnannotated [
15]. Following alignment, Cufflinks (version 2.2.1) was used to assemble transcripts, estimate their abundances, and test for differential expression and regulation using parameters—library-type fr-firststrand-G–L [
16]. Cuffmerge, which is part of Cufflinks, merged the Cufflinks transcripts across multiple samples using the default parameters. Cuffdiff found significant changes in transcript expression, splicing, and promoter usage using default parameters. For generating pathway analysis of biological processes, a data set containing gene identifiers and corresponding expression values was uploaded into Reactome Pathway Database [
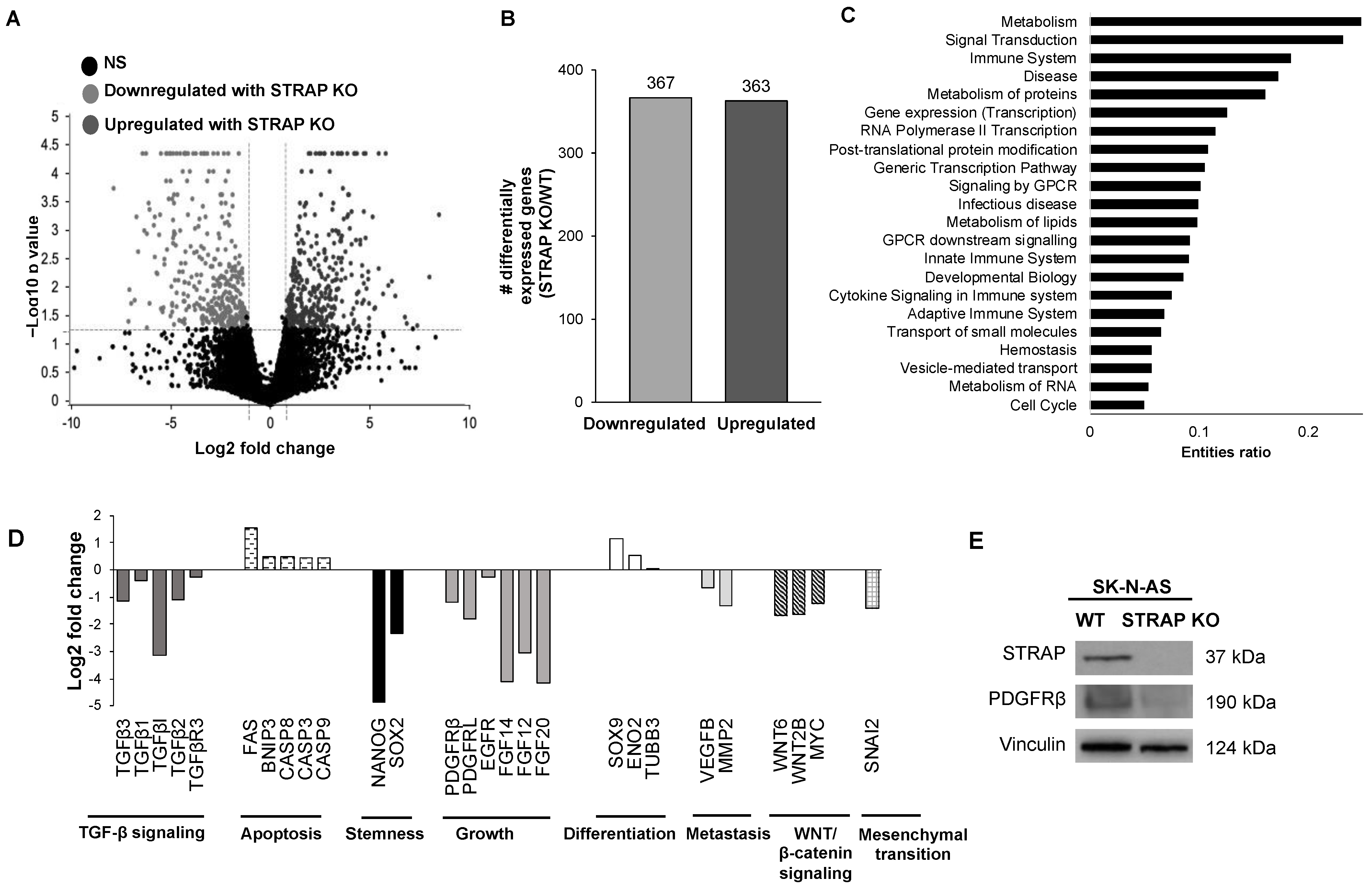
17]. Differentially expressed genes that met the fold change cutoff of ±2 were considered for the analysis. Each identifier is mapped to its corresponding molecule in the Reactome database and pathways identified at a false discovery rate (FDR) of 0.05. Entities ratio is defined as the proportion of Reactome pathway molecules represented in the dataset.
2.14. Real-Time PCR (qPCR)
iScript cDNA Synthesis kit (Bio-Rad) was used to synthesize cDNA with 1 μg of RNA used in a 20 μL reaction. The reverse transcription products were stored at −20 °C until further use. SsoAdvanced™ SYBR
® Green Supermix (Bio-Rad) was utilized according to the manufacturer’s protocol for quantitative real-time PCR (qPCR). Primers specific for Octamer-binding transcription factor 4 (Oct4), homeobox protein Nanog, and β-actin were utilized (Applied Biosystems, Foster City, CA, USA). Nestin primers (forward: 5′-TCCAGGAACGGAAAATCAAG-3′, reverse: 5′-GCCTCCTCATCCCCTACTTC-3′) were designed using Primer3 web version 4.1.0 [
18] and examined for non-specific binding using BLAST (NCBI). qPCR was performed with 10 ng cDNA in 20 μL reaction volume. Amplification was performed using an Applied Biosystems 7900HT cycler (Applied Biosystems). Cycling conditions were 95 °C for 2 min, followed by 39-cycle amplification at 95 °C for 5 s and 60 °C for 30 s. β-actin was utilized as an internal control. Gene expression was calculated using the ΔΔCT method [
19] and reported as mean fold change ± SEM.
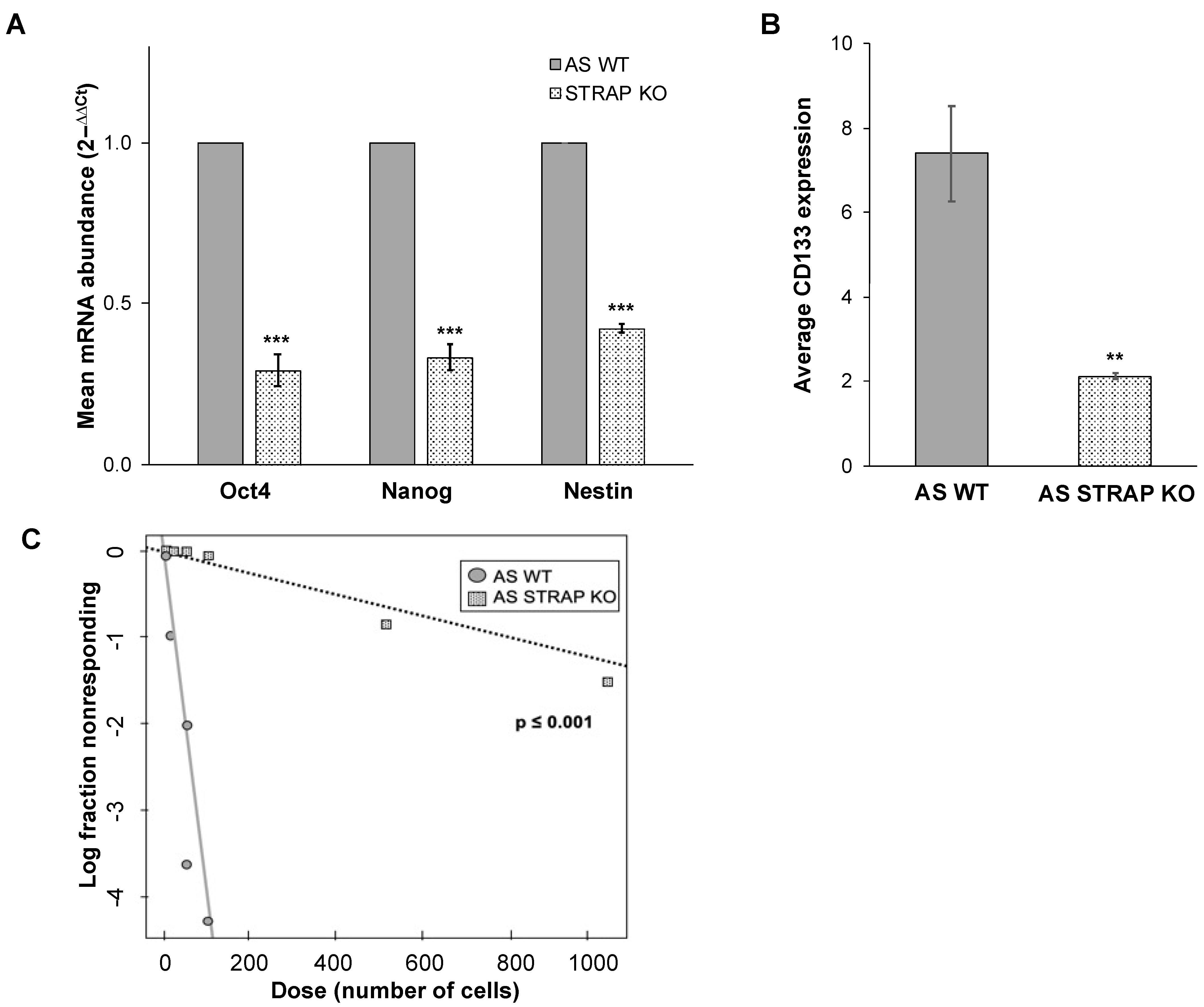
2.15. CD133 Cell Surface Expression
AS WT or STRAP KO cells (1 × 106) were labeled with CD133/1 (AC133)-APC (Miltenyi Biotec, San Diego, CA, USA) according to the manufacturer’s instructions. Unlabeled cells served as negative controls. The percent of cells positive for APC was determined via flow cytometry using the FACSCalibur™ Flow Cytometer (BD Biosciences) and analyzed using the FlowJo software (FlowJo, LLC FlowJo, Ashland, OR, USA).
2.16. Extreme Limiting Dilution Analysis
AS WT or STRAP KO cells were plated in conditioned media in 96-well plates with a decreasing number of cells in each row of 12 wells (1000 to 10 cells). After one week, each well was assessed for formation of tumorspheres. The number of wells containing spheres in each group was counted and analyzed using the extreme limiting dilution analysis (ELDA) software [
20] and a plot of the log proportion of negative wells versus the number of cells plated was generated. The slope of the line is the estimated log-active sphere-forming fraction. Tables showing estimated and 95% confidence intervals for the 1/(stem cell frequency) for each group were also generated.
2.17. Animal Statement
Animal experiments were approved by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee (IACUC-09363) and were conducted within institutional, national, and NIH guidelines and in compliance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
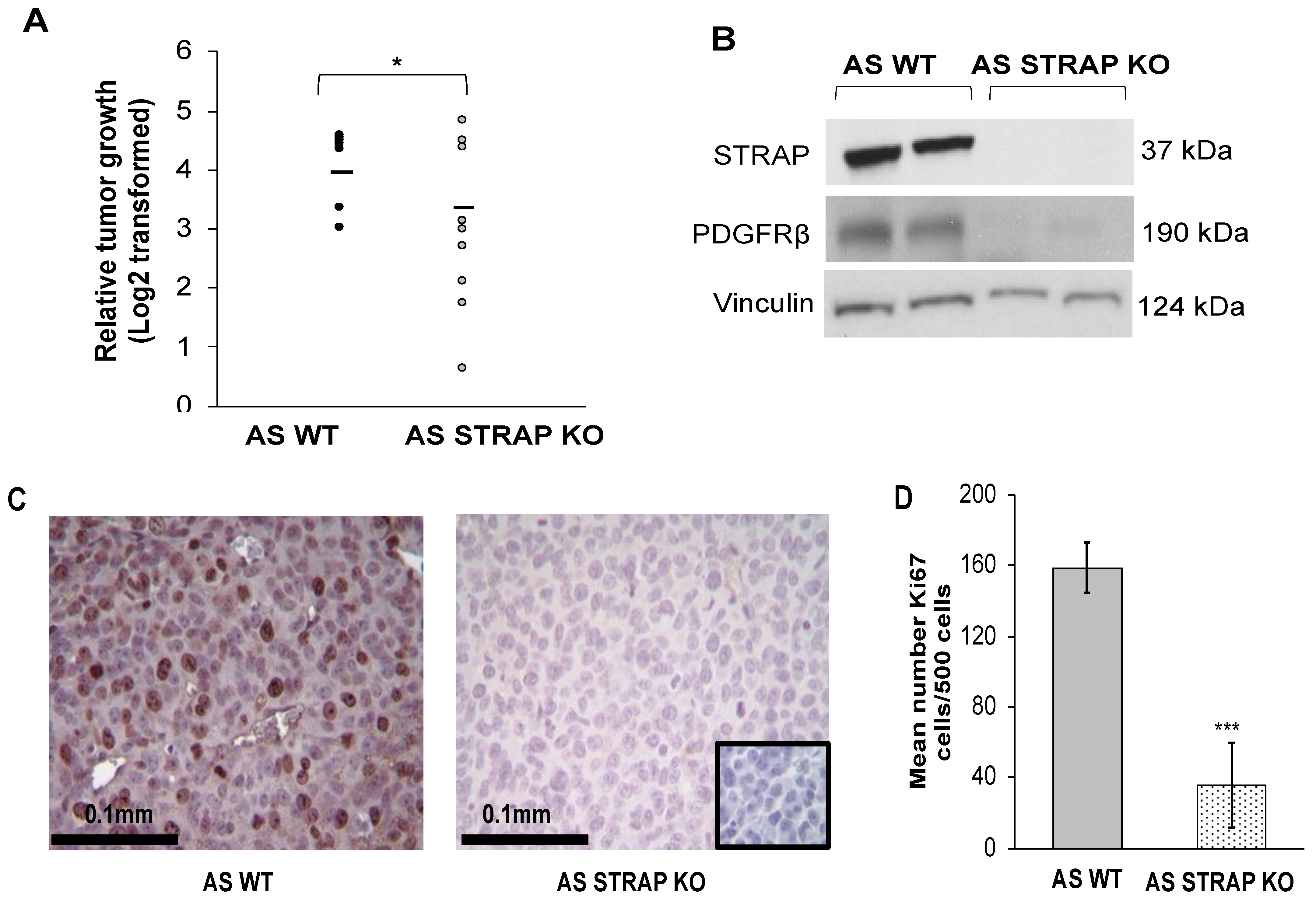
2.18. In Vivo Tumor Growth
AS WT or STRAP KO cells (1.8 × 106 cells in 25% Matrigel™, Corning, Inc.) were injected into the right flank of 6-week-old female athymic nude mice (n = 9 per group, Envigo, Prattville, AL, USA). Tumors were measured three times weekly, and tumor volumes calculated with the formula [(width2 × length)/2] mm3 with width being the smallest measurement. After 21 days post-injection or when animals met IACUC parameters for euthanasia, the animals were humanely euthanized in their home cages with CO2 and cervical dislocation. Tumors were harvested and prepared for further study.
2.19. Immunohistochemistry
In order to evaluate proliferation in the tumors, immunohistochemistry staining for Ki67 [
21] was performed. Formalin-fixed paraffin-embedded samples of AS WT and STRAP KO flank tumors were cut (5 μm sections), baked for 1 h at 70 °C, deparaffinized, rehydrated, and steamed. The sections were quenched with 3% hydrogen peroxide and blocked with PBS-blocking buffer (BSA, powdered milk, Triton X-100, PBS) for 30 min at 4 °C. Ki67 staining was completed by adding the primary antibody anti-Ki67 (rabbit polyclonal, 1:100, AB9260, Millipore Sigma) and incubated overnight at room temperature. After PBS washing, secondary antibody for rabbit (R.T.U. biotinylated universal antibody, Vector Laboratories, Burlingame, CA, USA) was added for 1 h at 22 °C. VECTASTAIN Elite ABC reagent (PK-7100, Vector Laboratories) and Metal Enhanced DAB Substrate (Thermo Fisher Scientific) was used to develop the staining reaction. Slides were counterstained with hematoxylin. For each run, a negative control (rabbit IgG, 1 μg/mL, EMD Millipore) was included. A board-certified pediatric pathologist (EMM), blinded to the treatment groups, evaluated the Ki67 staining which was quantified by counting the number of Ki67 positive cells per 500 cells in a representative section of each tumor [
21]. The mean was calculated and results reported as mean ± SEM.
2.20. Statistical Analysis
ImageJ was utilized to perform densitometry of immunoblots with each protein band being normalized to the background (
http://www.yorku.ca/yisheng/Internal/Protocols/ImageJ.pdf and the protein band in question then normalized to the internal control (vinculin or β-actin). Normalized bands were compared to that of WT cells. All experiments were performed with a minimum of three biologic replicates. Data were reported as mean ± SEM of separate experiments. Student’s
t test or analysis of variance (ANOVA) were used where appropriate. Statistical significance was defined as
p ≤ 0.05.
4. Discussion
High-risk neuroblastoma continues to carry a poor prognosis and continued investigations are crucial to improve our understanding of the disease. STRAP is overexpressed in several malignancies including another pediatric cancer, osteosarcoma [
7,
8,
32,
36]. Recently, Jin et al. found that STRAP functions in neuronal development and STRAP knockout in Xenopus resulted in neural tube defects [
37]. These findings were relevant to the present study, since neuroblastoma is an embryologic tumor derived from neural crest cells [
2]. These previous studies supported the investigation of the potential for oncogenic function of STRAP in neuroblastoma. In the current study, we demonstrated that STRAP knockdown decreased neuroblastoma proliferation, growth, and motility. By establishing two stable neuroblastoma cell lines with the genetic knockout of STRAP and examining their malignant phenotype as well as the effect on the transcriptome, we provide evidence to suggest a role for STRAP in promoting neuroblastoma tumorigenicity in vitro and in vivo as well as maintaining the stem cell-like phenotype.
RNA interference is a method for transiently inhibiting gene expression and/or translation by targeting the corresponding RNA. Small interfering RNA (siRNA) and short hairpin RNA (shRNA) are two applications of RNA interference which have been utilized to achieve specific knockdown of a specific protein [
38]. STRAP siRNA knockdown decreased migration and invasion of osteosarcoma cells [
8]. However, Wu and colleagues found that knockdown of STRAP utilizing siRNA promoted hepatocellular carcinoma tumorgenicity in vitro and in vivo [
36], suggesting that STRAP’s role in cancer may not be tumor-specific. We initially investigated the knockdown of STRAP by utilizing siRNA in two neuroblastoma cell lines, which are AS (MYCN non-amplified) and BE (MYCN amplified), and found that STRAP knockdown resulted in decreased proliferation. These findings led us to further explore the role of STRAP in neuroblastoma.
Although siRNA and shRNA may achieve similar knockdown, the mechanism of action is different. SiRNA is a transient transfection, whereas shRNA carrying cells may be selected with an antibiotic for stable transfection. It is hypothesized that shRNA may have higher potency and fewer off-target effects than siRNA [
38], enabling us to investigate the knockdown of STRAP utilizing shRNA in addition to siRNA. Datta et al. demonstrated decreased tumorigenicity in colorectal cancer cells following transfection with STRAP shRNA [
6]. Similarly, we found that shRNA-mediated stable knockdown of STRAP resulted in decreased neuroblastoma cell proliferation, growth, and motility. The same investigators also demonstrated an increase in the sensitivity to 5-FU and oxaliplatin in colorectal cancer following shRNA STRAP knockdown [
11]. Studying the effects of STRAP knockdown in combination with neuroblastoma chemotherapeutics will be an exciting avenue of future studies.
RNA interference has several limitations including a decrease in expression of the protein of interest, but not a total absence of that protein [
39]. Due to these limitations, we proceeded to investigate the genetic knockout of STRAP. Using CRISPR-Cas9 gene editing technology, we established a stable cell line of STRAP KO cells in the neuroblastoma cell line SK-N-AS. Wang et al. also utilized CRISPR-Cas9 gene editing technology to establish a stable line of STRAP KO cells in hepatocellular carcinoma and demonstrated that STRAP KO led to decreased Wnt/β-catenin signaling, which was associated with decreased colony formation and stemness markers in vitro [
32]. In order to further validate our findings of STRAP’s role in neuroblastoma, we established an additional CRISPR-Cas9 stable knockout of STRAP in SK-N-BE (2) human neuroblastoma cells. We found that STRAP KO resulted in decreased growth, proliferation, motility, and anchorage-independent growth.
Using the findings from genetic sequencing of AS STRAP KO and AS WT cells, we found that numerous pathways were affected by the loss of STRAP. Specifically, pathways with the highest entities ratio that were associated with downregulated genes following the STRAP KO included those involved in metabolism, signal transduction, and cell cycles. We also found genes associated with stemness, growth, metastasis, and the known oncogenic pathways previously shown to be associated with STRAP by other investigators, such as TGF-β [
22] and WNT/β-catenin [
6], to be downregulated while apoptotic and differentiation genes were upregulated in the STRAP KO cells. Many of these pathways are important in neuroblastoma. Prior investigators showed that by stimulating neuroblastoma with TGF-β, there was an increase in cell migration and invasion [
40]. Similarly, Tran et al. demonstrated the restoration of natural killer cells’ cytotoxicity following treatment with a TGF-β inhibitor in neuroblastoma [
41]. SiRNA knockdown of the WNT/β-catenin signaling pathway has been shown to decrease neuroblastoma viability, growth [
42], and motility [
43]. Therefore, the inhibition of these oncogenic signaling pathways observed with STRAP KO provides support for the inhibition of STRAP in neuroblastoma as a therapeutic strategy.
Platelet-derived growth factor receptors (PDGFRs) have been associated with growth, angiogenesis, cell viability, and proliferation in numerous malignancies, including glioma, prostate, breast, and pancreatic cancers [
44]. By inhibiting PDGFRβ signaling, other investigators decreased cell viability, increased apoptosis, and induced cell cycle arrest in mesothelioma [
45]. In colorectal cancer, lower PDGFRβ expression was associated with better prognosis and PDGFRβ knockdown using siRNA resulted in decreased colorectal cancer proliferation, growth, and invasion [
46]. PDGFRβ was one of the downregulated genes observed with AS STRAP KO and we confirmed decreased protein expression in the AS STRAP KO cells. These studies provide evidence that STRAP may exert its phenotypic effects on cell proliferation and growth in neuroblastoma through its effect on expression of PDGFRβ. PDGFRβ has become a receptor of interest in neuroblastoma. Targeting PDGFRs with imantinib, which is a tyrosine kinase inhibitor, decreased neuroblastoma survival and proliferation as well as enhanced apoptosis when combined with doxorubicin [
47]. Similarly, other investigators inhibited neuroblastoma growth in vivo and decreased tumor angiogenesis using SU11657, a tyrosine kinase inhibitor that targets PDGFRs [
30]. In the current study, when STRAP was knocked out, we observed a decrease in PDGFRβ which suggests the presence of a potential mechanism by which STRAP may promote the malignant phenotype.
Stem cell-like cancer cells (SCLCCs) are a subpopulation of cancer cells that have been shown to be important for neuroblastoma progression, therapeutic resistance, and disease recurrence [
34]. Therefore, therapies that target SCLCCs could greatly affect the course of the disease. STRAP has been implicated in maintaining colorectal cancer cell stemness. Jin and colleagues found that STRAP inhibition with shRNA decreased CD133 positive colorectal cancer cells and tumorsphere forming ability [
11]. STRAP promoted the stem cell-like characteristics of colorectal cancer cells by activating the NOTCH pathway and the silencing of STRAP in these cells resulted in decreased stem cell phenotype [
11]. In addition, STRAP knockout in hepatocellular carcinoma decreased mRNA abundance of stemness markers and liver progenitor genes, including AXIN2, LGR5, CD133, and CD44 [
32]. In the current study, genetic evaluation demonstrated a downregulation of genes associated with neuroblastoma stemness following the knockout of STRAP. Further phenotypic evaluation of AS STRAP KO cells demonstrated a significant decrease in mRNA abundance of stemness markers, a decrease in CD133 cell surface expression, and decreased ability to form tumorspheres; all of these indicate that STRAP plays a role in promoting neuroblastoma cancer cell stemness.
Other investigators have examined STRAP knockdown and its effects on in vivo tumor growth. In colorectal cancer, STRAP knockdown with shRNA led to decreased tumor growth [
11] and metastasis [
6] in vivo. In addition, the knockdown of STRAP with shRNA in hepatocellular carcinoma resulted in decreased tumorigenicity and metastasis [
36]. To our knowledge, the current study is the first to investigate the effect of STRAP knockdown and CRISPR KO on neuroblastoma and the first investigation of STRAP CRIPSPR KO on neuroblastoma tumor growth in vivo. Similar to results described in other in vivo studies, we demonstrated that the genetic knock out of STRAP resulted in decreased neuroblastoma tumor growth with significantly decreased proliferation in the KO tumors.
One limitation of CRISPR-Cas9 genetic knockout is the possibility for off-target activity or non-specific gene editing [
12,
48,
49]. Validation of the target gene’s role in the observed phenotype may be achieved by reintroducing the knocked-out gene in a rescue experiment. Other investigators have utilized this method. Chen and colleagues showed that CRISPR-Cas9 knockout of EZH2 in neuroblastoma resulted in decreased cell viability. They subsequently re-introduced EZH2, resulting in the rescue of the phenotype observed following EZH2 deletion. These findings confirmed EZH2′s importance in neuroblastoma survival [
50]. In the present study, STRAP re-expression in the AS STRAP KO cells restored the phenotype to that observed in AS WT cells.
In the current study, we demonstrate that the knockdown and knockout of STRAP resulted in a decrease in the malignant phenotype in neuroblastoma. Our pre-clinical findings support the potential use of STRAP as a therapeutic target; however, there are currently no STRAP inhibitors available. Therefore, collaborations and development of inhibitors targeting STRAP are areas of interest and future studies. Interestingly, knocking down STRAP using siRNA had a more drastic effect on proliferation in AS cells compared to BE. One explanation for these findings is the difference in MYCN amplification in the two cell lines, which is related with a more aggressive phenotype in neuroblastoma [
51]. An association between STRAP and MYCN has not yet been explored, providing us an additional avenue for future studies. STRAP is overexpressed in other cancers and has been associated with a more aggressive phenotype [
6,
8]; this provides an exciting avenue for future work to investigate STRAP expression levels in patient neuroblastoma specimens as a potential predictor for high-risk disease.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}