
High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients, Tumors, and Data
2.2. Expression and DNA Methylation Analyses
2.3. Data Portrayal Using Self-Organizing Maps
2.4. oposSOM Browser
3. Results
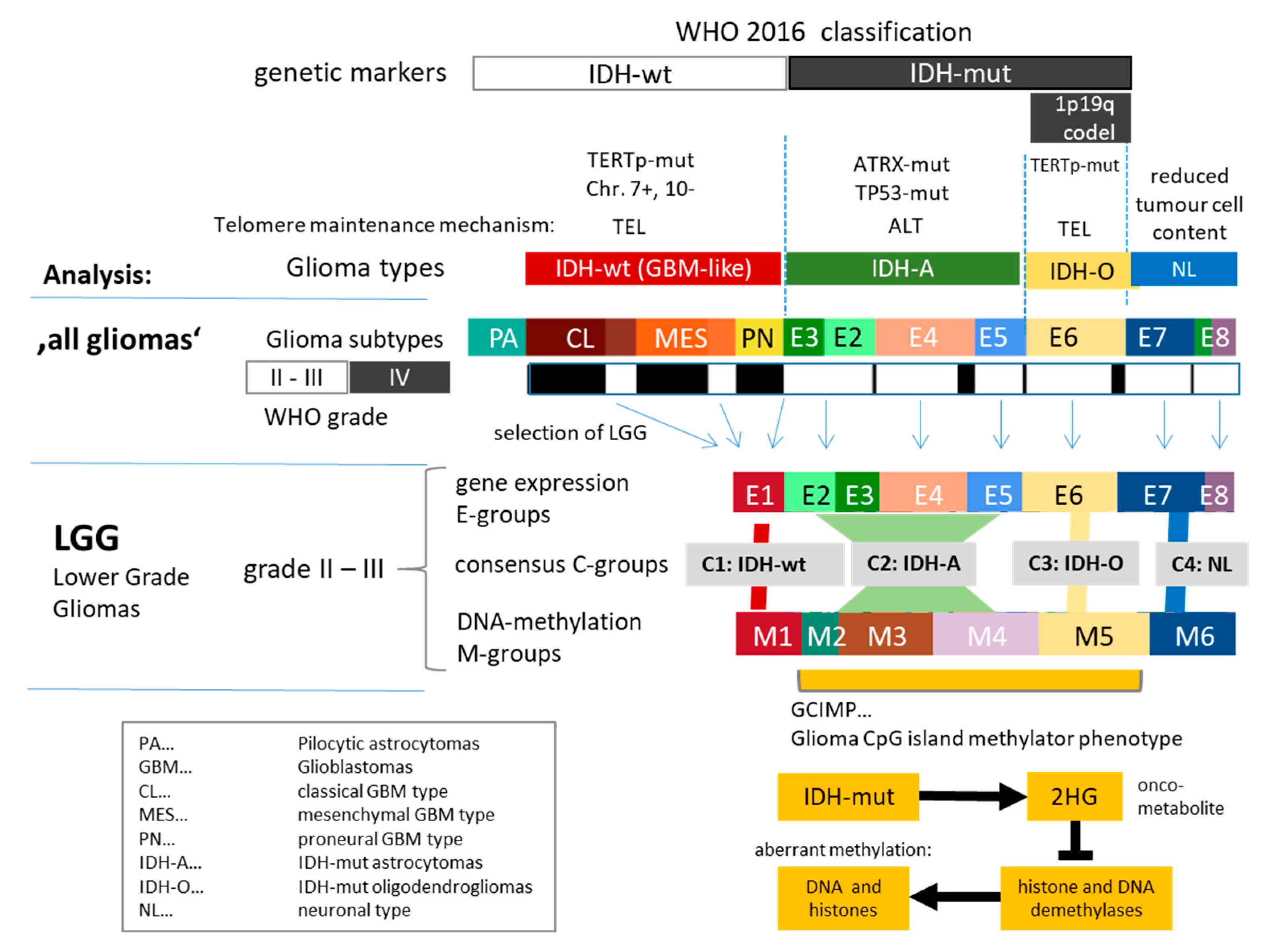
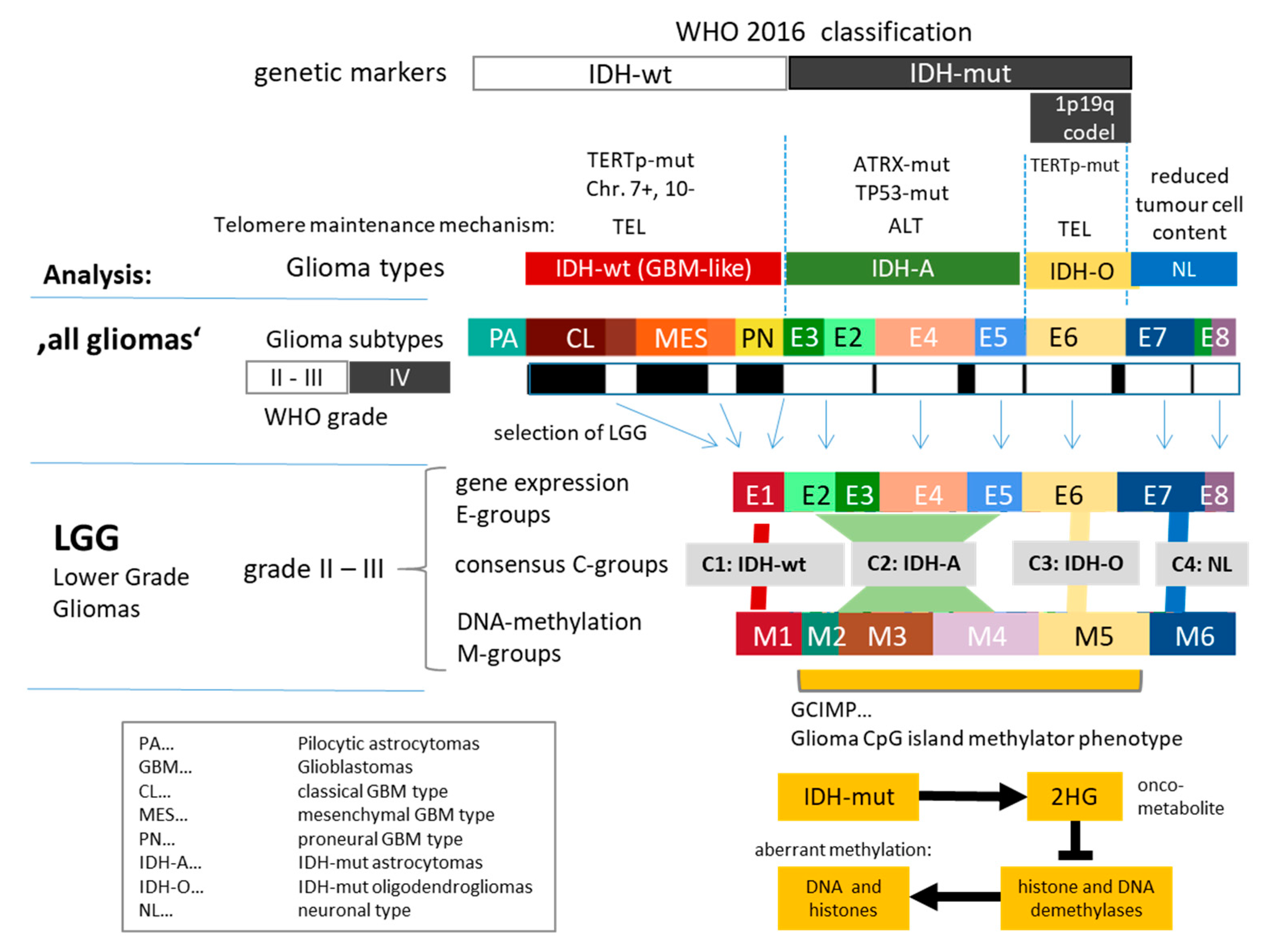
3.1. Cartography of the Transcriptomic States of PA, LGG, and GBM

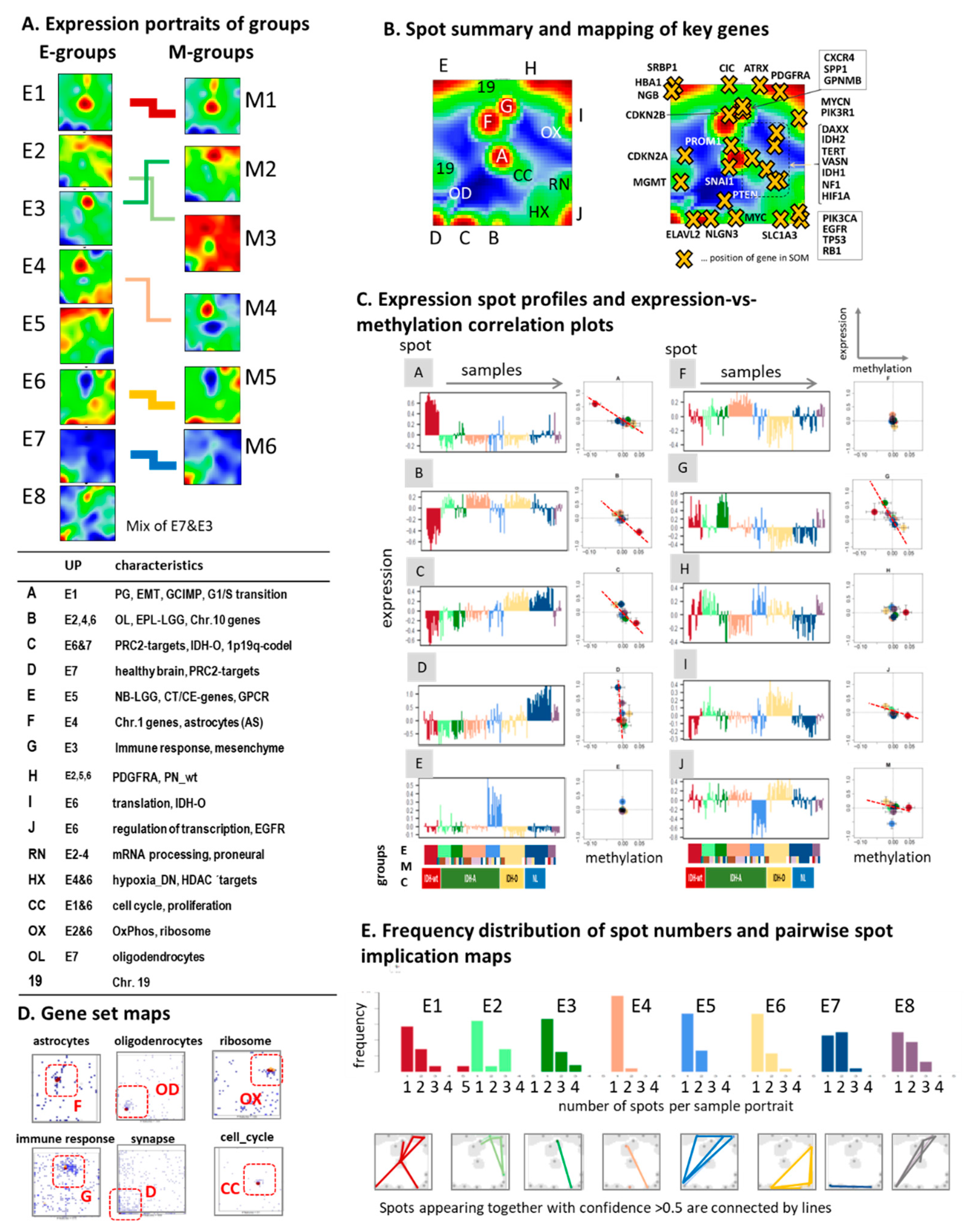
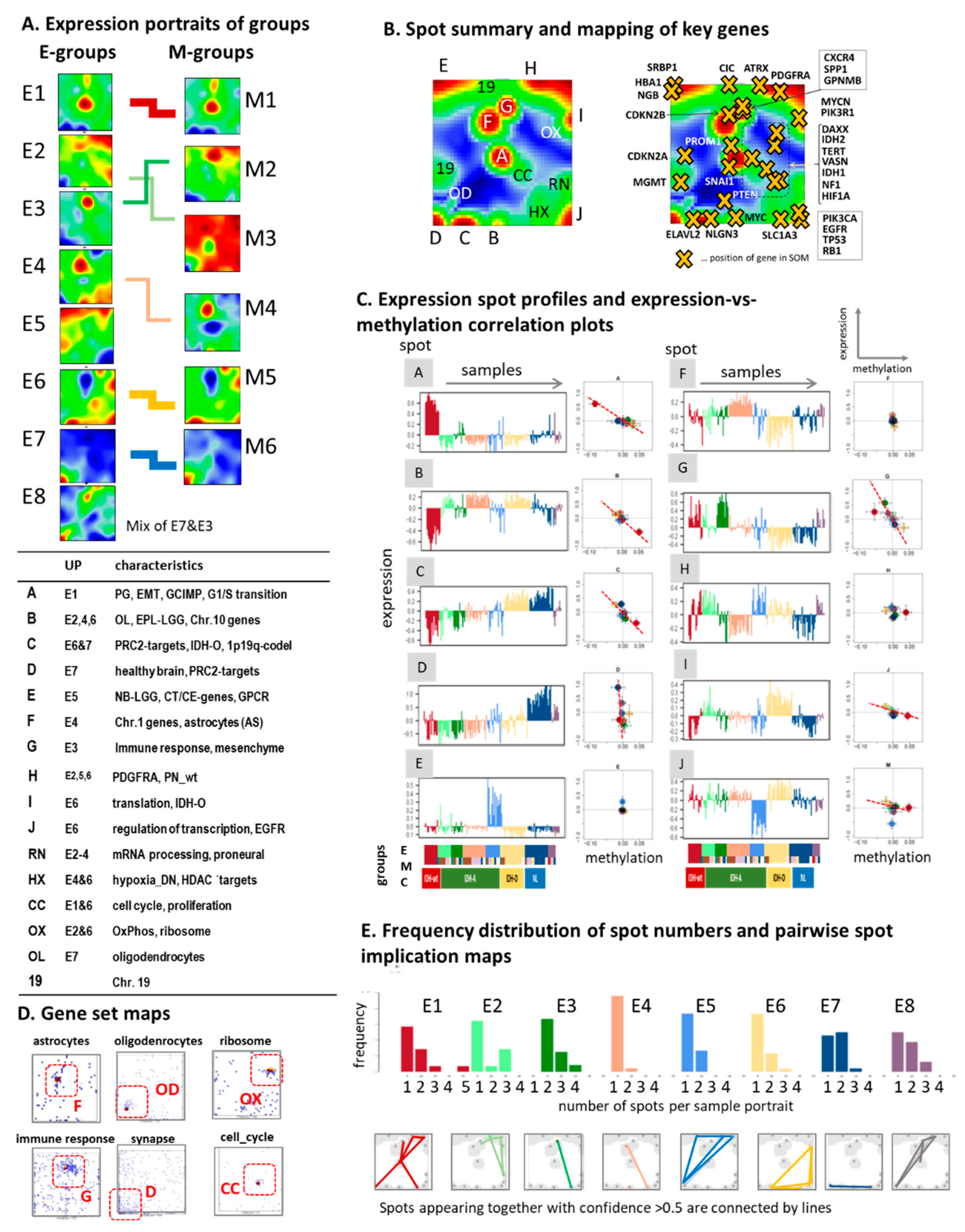
3.2. Cartography of the LGG Transcriptome
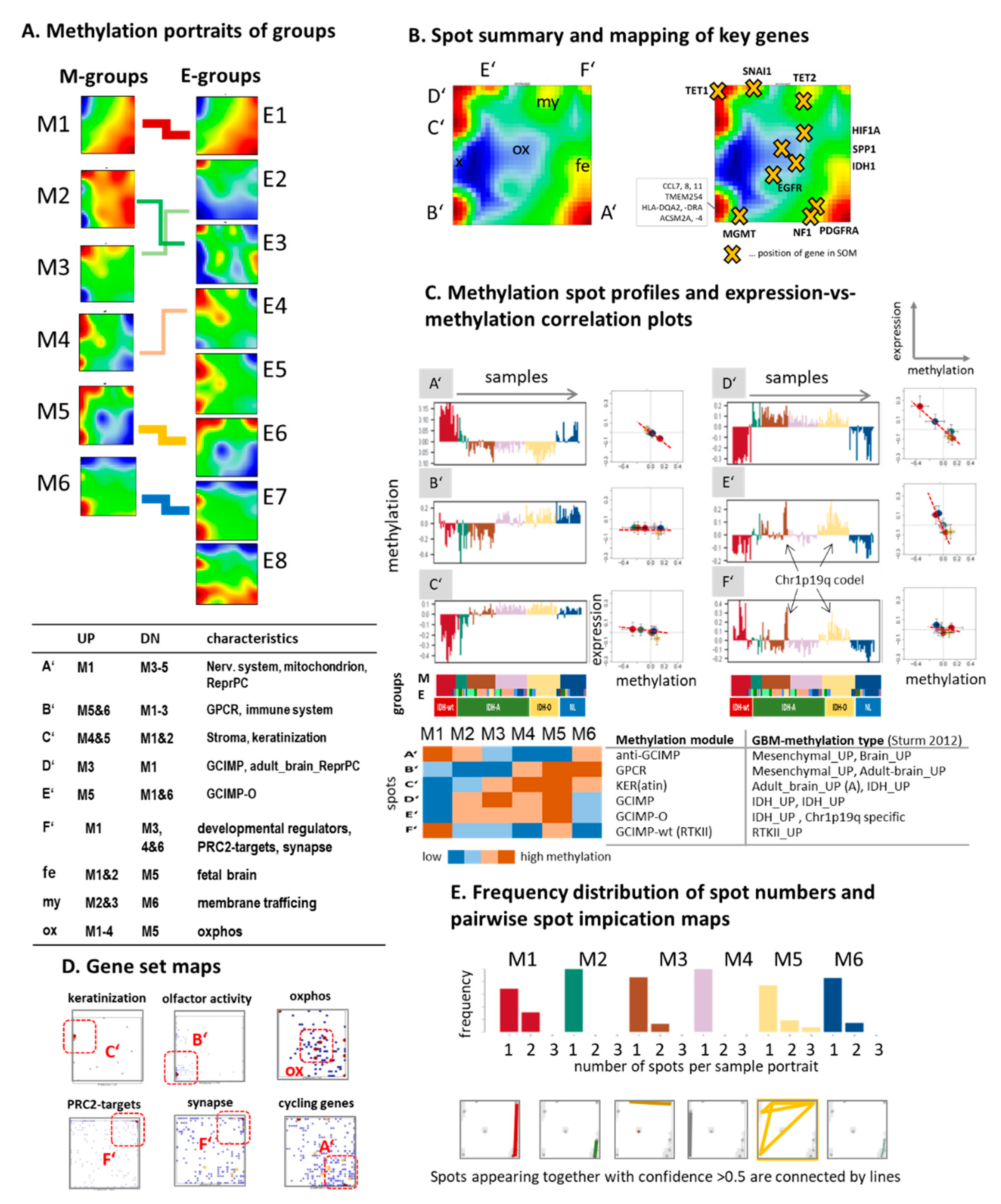
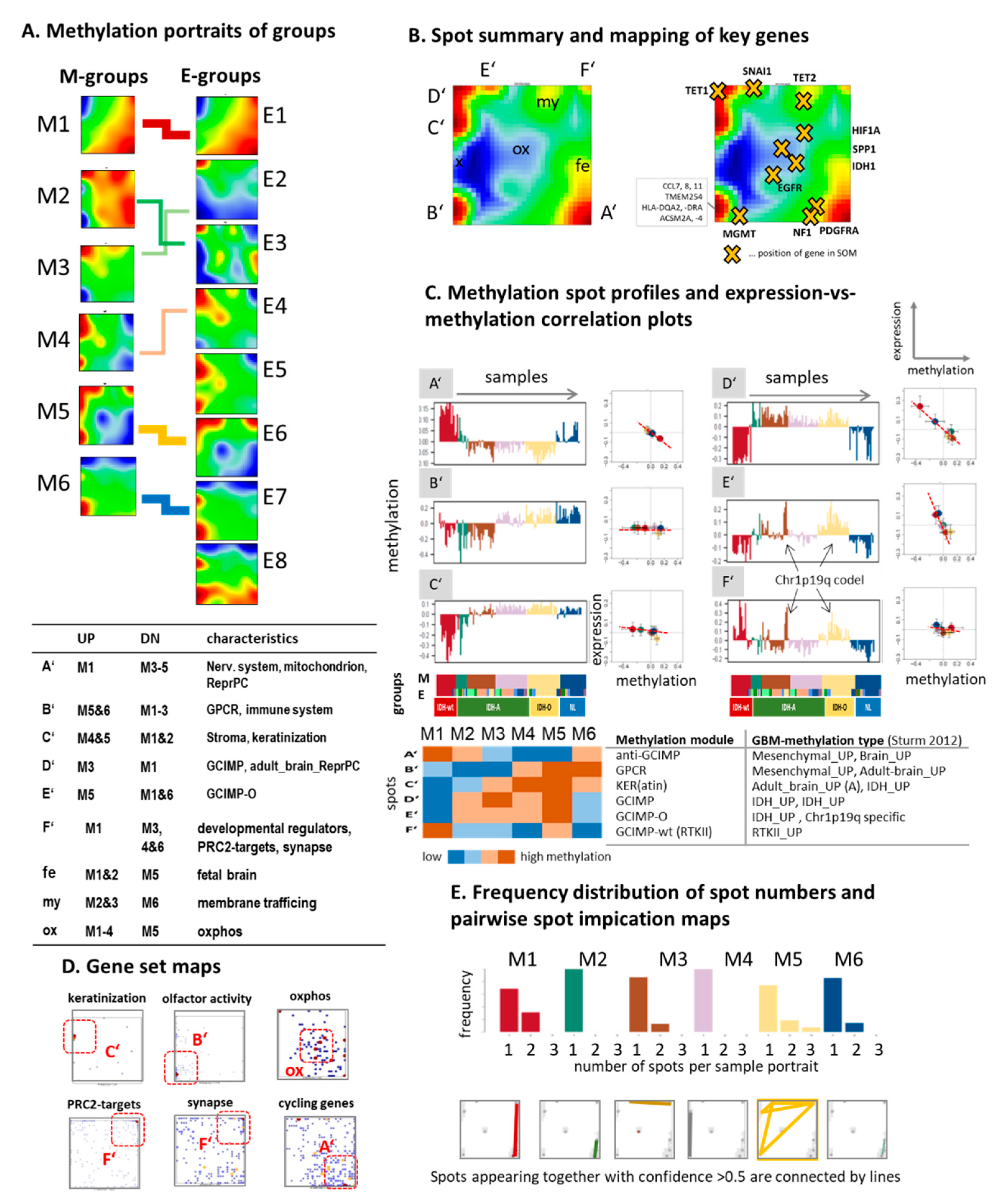
3.3. Cartography of the LGG Methylome
3.4. Glioma Key Genes Support the Functional Impact of the Molecular Maps
3.5. Epi-Genome and -Transcriptome Modifiers, Telomere Maintenance, and Single-Cell Signatures
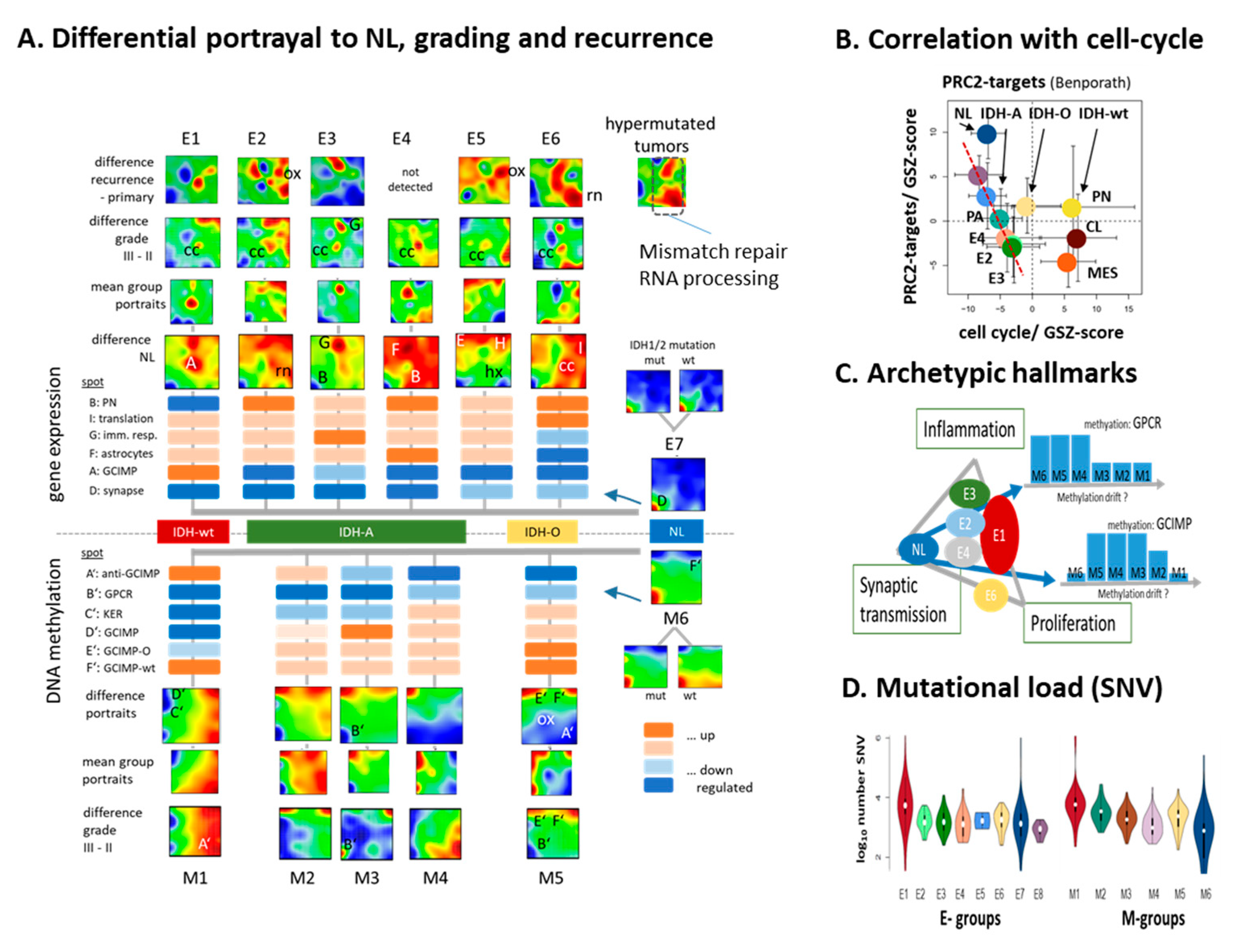
3.6. Glioma Progression and Recurrence
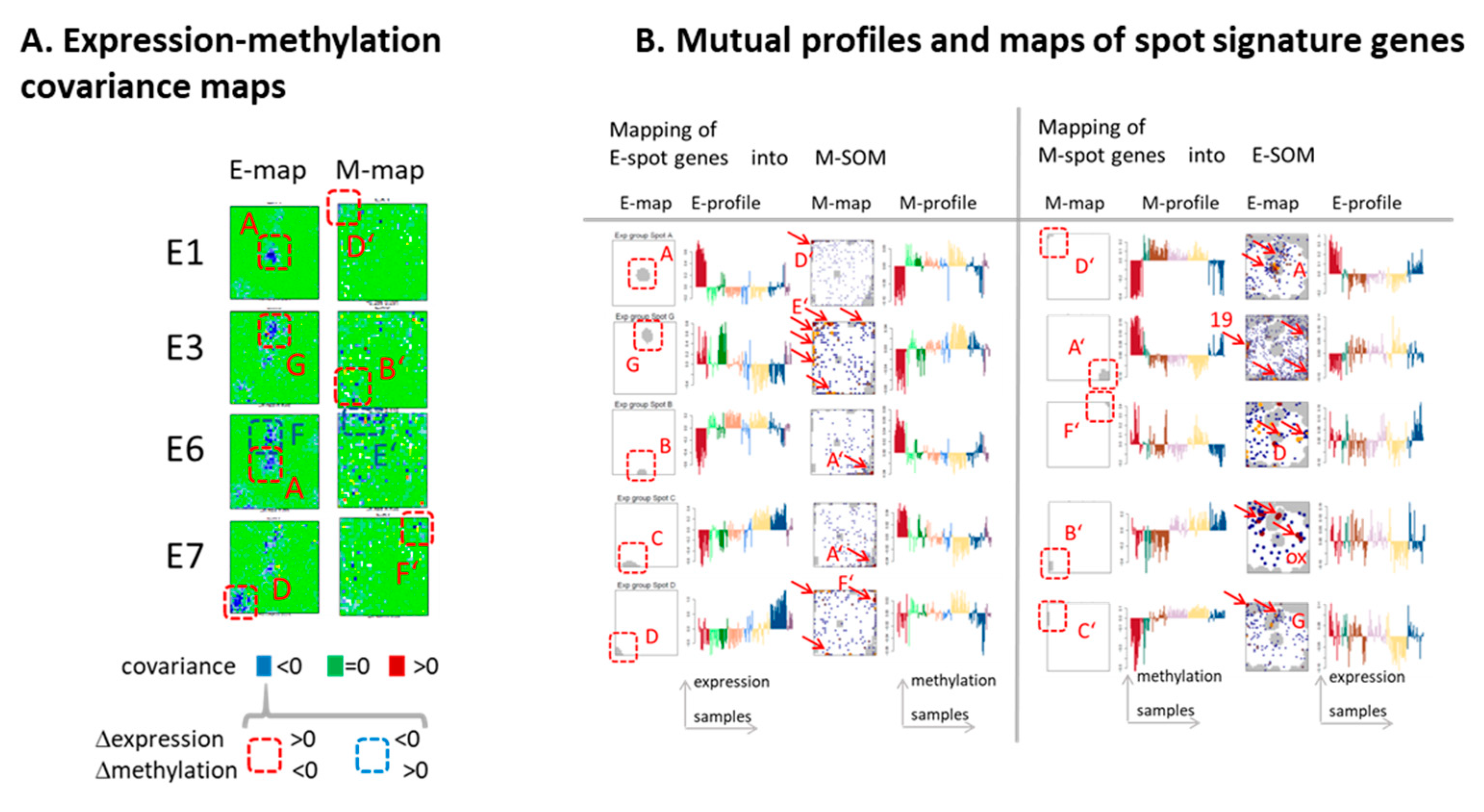
3.7. (De)coupled Alterations of Expression and Methylation Patterns
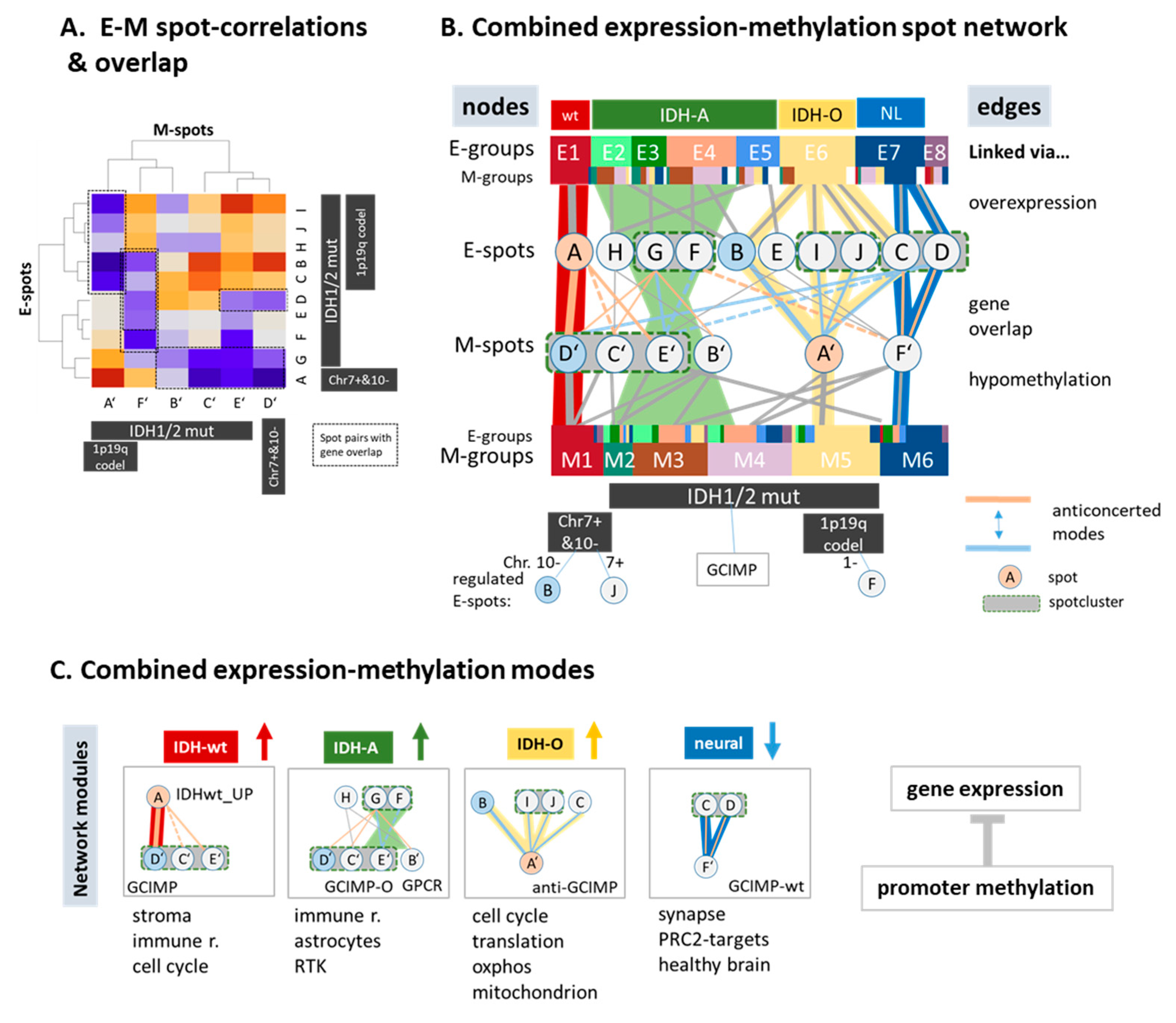
3.8. A Combined Network of Transcriptional and of Methylation Modules
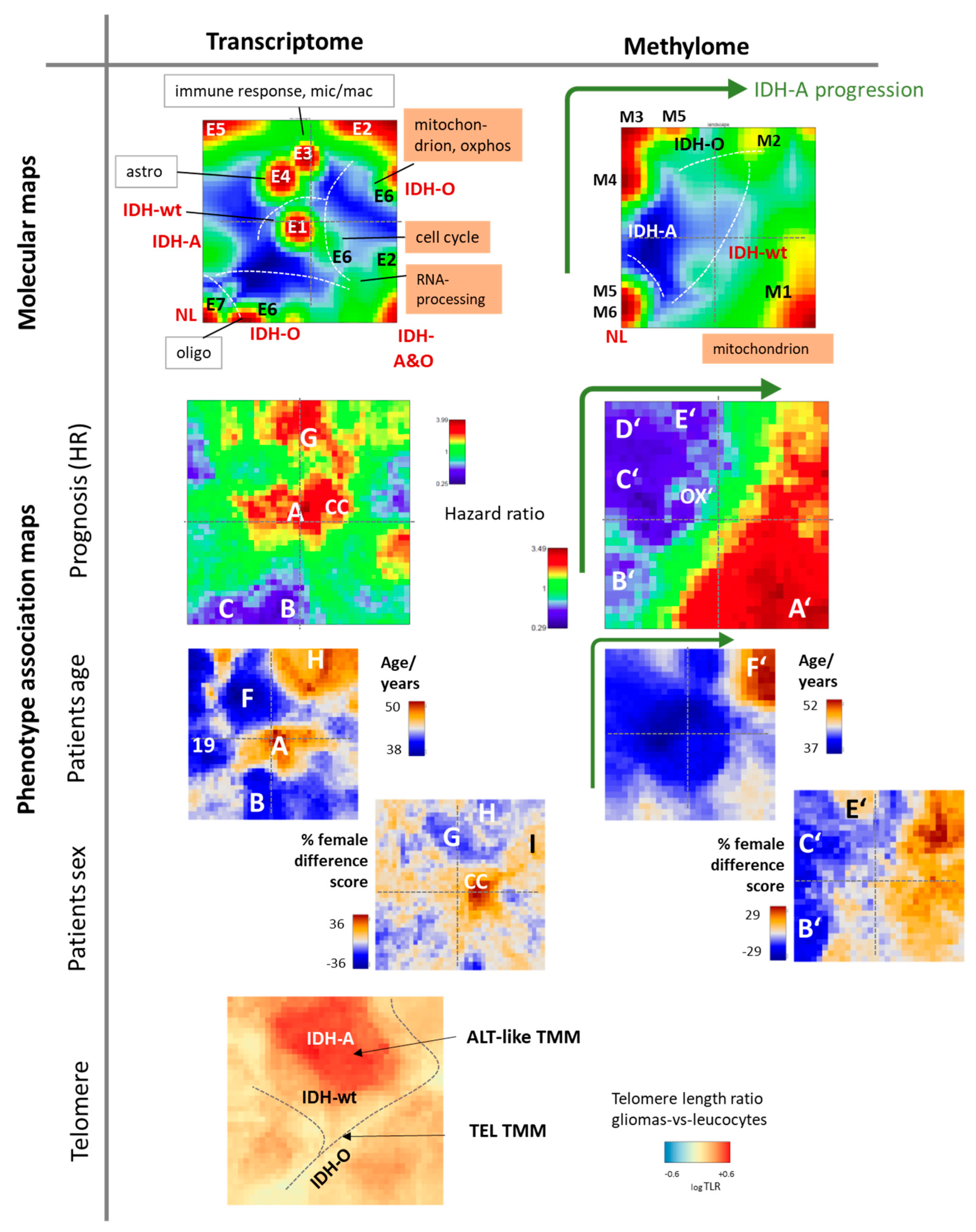
3.9. Phenotype Maps: Prognosis, Age, and Telomere Length
4. Discussion
4.1. Combinations of Proliferative, Inflammatory, and Tissue Remodelling Functions Shape Glioma Phenotypes
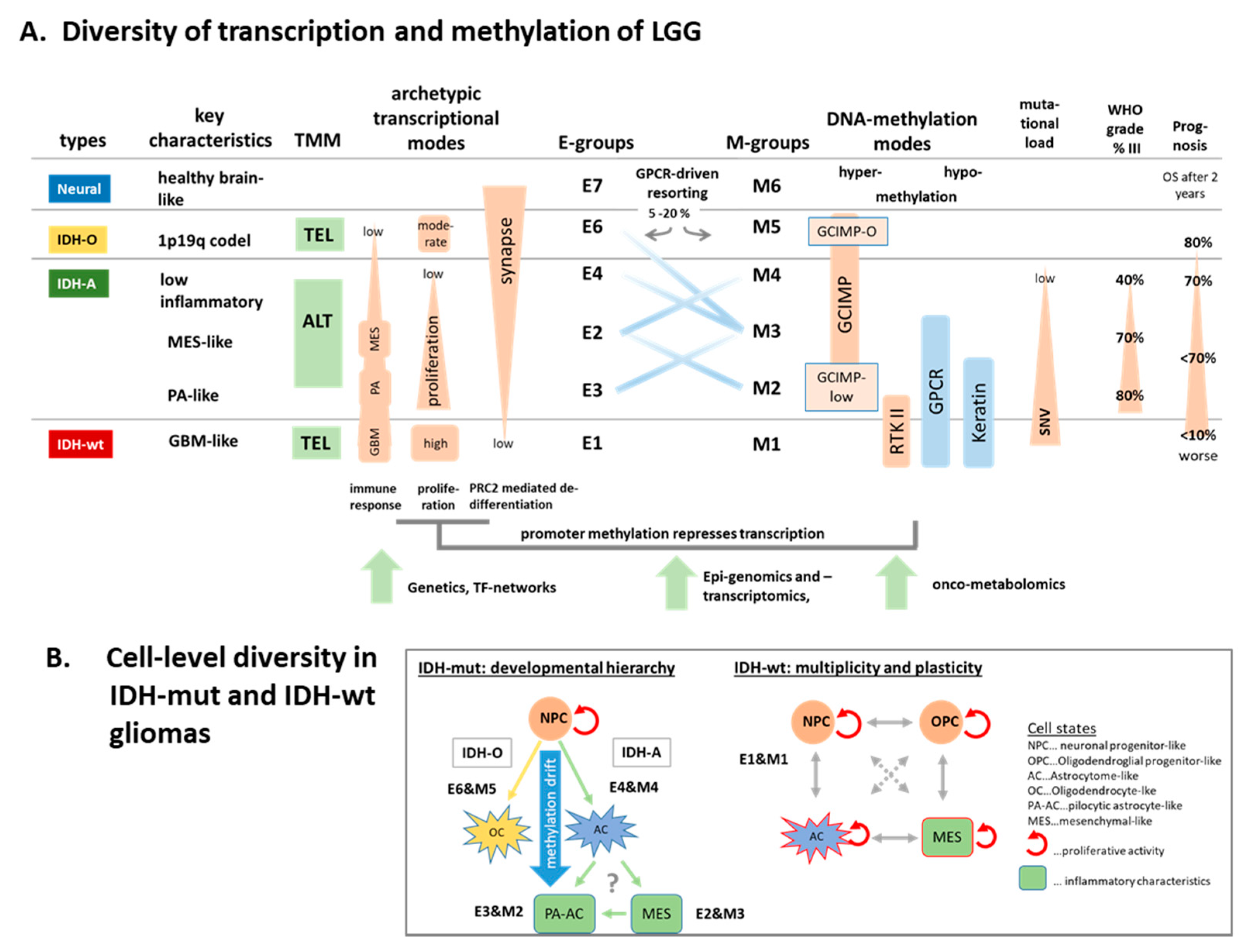
4.2. Glioma Progression, Immuno-Ageing, and Transitory States
4.3. Archetypic Hallmarks, Developmental Hierarchy, and Plasticity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2HG | 2-hydroxyglutarate |
| ALT | alternative lengthening of telomerse |
| BMBF | Federal Ministry of Education and Research |
| BP | biological process |
| C1–C4 | consensus classes of LGG with mutual correspondence between expression and methylation: C1- IDH-wt; C2—IDH-A; C3—IDH-O; C4—NL |
| CC | cell cycle |
| CL | classical subtype of grade IV gliomas according to Verhaak et al. (2010) |
| CHOP | CpG-hypomethylation pattern of IDH-mutated tumors |
| CN | copy number |
| E1–E8 | expression subtypes of LGG |
| EMT | epithelial–mesenchymal transition |
| EPL | early progenitor-like subtype |
| GBM | glioblastoma WHO grade IV |
| GCIMP | glioma CpG-island hypermethylation phenotype |
| GCIMP-O | GCIMP with specific hypermethylation of IDH-O |
| GCIMP-wt | hypermethylation of IDH-wt, particularly of the RTK II type |
| GEO | gene expression omnibus database |
| GGN | German Glioma Network |
| GPCR | G-protein-coupled receptor |
| GSZ | gene set Z-score |
| HLA | human leukocyte antigen |
| HM | hallmarks of cancer |
| HR | hazard ratio |
| hx | hypoxia |
| IDH | isocitrate dehydrogenase |
| IDH-A | malignant astrocyte-like, IDH-mutated with chromosome 1p19q intact |
| IDH-mut-codel | IDH mutant additionally carrying co-deletion of chromosome arm 1p and 19q |
| IDH-mut non-codel | IDH mutant with euploid 1p19q |
| IDH-O | malignant oligodendrocyte-like, IDH-mutated with chromosome 1p19q intact |
| IDH-wt | IDH wild-type |
| LGG | low grade gliomas (WHO grade II and III) |
| M1–M6 | methylation subtypes |
| ME | mesenchymal subtype of grade IV gliomas according to Verhaak et al. (2010) |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| MP | methylation pattern |
| MS | mesenchymal subtype (Sturm et al. (reanalyzed by Hopp et al.)) |
| NGB | neuroglobin |
| NL | neural subtype of grade IV gliomas according to Verhaak et al. (2010) |
| NPC | neural progenitor cells |
| OS HR | overall survival hazard ratio |
| Ox | oxidative phosphorylation |
| PG | pre-glioblastoma subtype of LGG (Gorovets et al., 2012) |
| PN | proneural subtype of grade IV gliomas according to Verhaak et al. (2010) |
| PRC2 | polycomb repressive complex 2 |
| RTK | receptor tyrosine kinase |
| RTKI | GBM methylation class (Sturm et al.) |
| RTKII | GBM methylation class (Sturm et al.) |
| SOM | self-organizing map |
| TEL | telomerase |
| TERT | telomerase reverse transcriptase |
| TF | transcription factors |
| TME | tumor microenvironment |
| TMM | telomere maintenance mechanism |
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Martin-Subero, J.I.; Rohde, V.; Kirsch, M.; Alaminos, M.; Fernandez, A.; Ropero, S.; Schackert, G.; Esteller, M. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics 2009, 4, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Laffaire, J.; Everhard, S.; Idbaih, A.; Crinière, E.; Marie, Y.; De Reyniès, A.; Schiappa, R.; Mokhtari, K.; Hoang-Xuan, K.; Sanson, M.; et al. Methylation profiling identifies 2 groups of gliomas according to their tumorigenesis. Neurooncology 2010, 13, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Kloosterhof, N.K.; Bralten, L.B.; Dubbink, H.J.; French, P.J.; Bent, M.J.V.D. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar] [CrossRef]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiencke, J.K. DNA Methylation, Isocitrate Dehydrogenase Mutation, and Survival in Glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.-H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef]
- Nomura, M.; Saito, K.; Aihara, K.; Nagae, G.; Yamamoto, S.; Tatsuno, K.; Mukasa, A. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci. Rep. 2019, 9, 1903. [Google Scholar] [CrossRef]
- Capper, D.; Stichel, D.; Sahm, F.; Jones, D.T.W.; Schrimpf, D.; Sill, M.; Schmid, S.; Hovestadt, V.; Reuss, D.E.; Koelsche, C.; et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: The Heidelberg experience. Acta Neuropathol. 2018, 136, 181–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.M.; Singal, R. DNA Methylation and Cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.; Bell, R.J.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, B. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, X.; Hu, B.; Muller, F.; Kim, H.; Squatrito, M.; Millelsen, T.; Scarpace, L.; Barthel, F.; Lin, Y.-H.; et al. Tumor evolution of glioma intrinsic gene expression subtype associates with immunological changes in the microenvironment. bioRxiv 2016, 32, 052076. [Google Scholar] [CrossRef]
- Mack, S.C.; Hubert, C.G.; Miller, T.E.; Taylor, M.; Rich, J.N. An epigenetic gateway to brain tumor cell identity. Nat. Neurosci. 2016, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Jones, D.T.W.; Pfister, S.M.; Kreuz, M.; Gramatzki, D.; Fortenbacher, E.; Hentschel, B.; et al. DNA methylation, transcriptome and genetic copy number signatures of diffuse cerebral WHO grade II/III gliomas resolve cancer heterogeneity and development. Acta Neuropathol. Commun. 2019, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Weber, R.; Willscher, E.; Riehmer, V.; Hentschel, B.; Kreuz, M.; Reifenberger, G. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 2015, 129, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, M.; Barthel, F.; Malta, T.M.; Sabedot, T.S.; Salama, S.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reifenberger, G.; Weber, R.G.; Riehmer, V.; Kaulich, K.; Willscher, E.; Wirth, H. for the German Glioma, N. Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling. Int. J. Cancer 2014, 135, 1822–1831. [Google Scholar] [CrossRef]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; The, G.C. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef]
- Richardson, T.; Walker, J.M. The Prognostic Significance of RB and PI3K Pathway Alterations in IDH-Mutant Grade II/III Astrocytomas. J. Neuropathol. Exp. Neurol. 2020, 79, 1019–1023. [Google Scholar] [CrossRef]
- Binder, H.; Wirth, H. Analysis of Large-Scale OMIC Data Using Self Organizing Maps. In Encyclopedia of Information Science and Technology, 3rd ed.; IGI Global: Hershey, PA, USA, 2015; pp. 1642–1653. [Google Scholar]
- Wirth, H.; Löffler, M.; von Bergen, M.; Binder, H. Expression cartography of human tissues using self organizing maps. Nat. Preced. 2011, 12, 306. [Google Scholar] [CrossRef]
- Hopp, L.; Löffler-Wirth, H.; Galle, J.; Binder, H. Combined SOM-portrayal of gene expression and DNA methylation landscapes disentangles modes of epigenetic regulation in glioblastoma. Epigenomics 2018, 10, 745–764. [Google Scholar] [CrossRef]
- Hopp, L.; Willscher, E.; Löffler-Wirth, H.; Binder, H. Function Shapes Content: DNA-Methylation Marker Genes and their Impact for Molecular Mechanisms of Glioma. J. Cancer Res. Updat. 2015, 4, 127–148. [Google Scholar] [CrossRef] [Green Version]
- Hopp, L.; Wirth, H.; Fasold, M.; Binder, H. Portraying the expression landscapes of cancer subtypes: A glioblastoma multiforme and prostate cancer case study. Syst. Biomed. 2013, 1, 99–121. [Google Scholar] [CrossRef]
- Loeffler-Wirth, H.; Kreuz, M.; Hopp, L.; Arakelyan, A.; Haake, A.; Cogliatti, S.B.; Feller, A.C.; Hansmann, M.-L.; Lenze, D.; for the German Cancer Aid consortium Molecular Mechanisms for Malignant Lymphoma; et al. A modular transcriptome map of mature B cell lymphomas. Genome Med. 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopp, L.; Löffler-Wirth, H.; Binder, H. Epigenetic Heterogeneity of B-Cell Lymphoma: DNA Methylation, Gene Expression and Chromatin States. Genes 2015, 6, 812–840. [Google Scholar] [CrossRef] [Green Version]
- Kunz, M.; Löffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef]
- Schmidt, M.; Hopp, L.; Arakelyan, A.; Kirsten, H.; Engel, C.; Wirkner, K.; Krohn, K.; Burkhardt, R.; Thiery, J.; Loeffler, M.; et al. The Human Blood Transcriptome in a Large Population Cohort and Its Relation to Aging and Health. Front. Big Data 2020, 3, 36. [Google Scholar] [CrossRef]
- Hopp, L.; Loeffler-Wirth, H.; Nersisyan, L.; Arakelyan, A.; Binder, H. Footprints of Sepsis Framed Within Community Acquired Pneumonia in the Blood Transcriptome. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, H.; von Bergen, M.; Binder, H. Mining SOM expression portraits: Feature selection and integrating concepts of molecular function. BioData Min. 2012, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on bioconductor: Fig. 1. Bioinformatics 2015, 31, 3225–3227. [Google Scholar] [CrossRef] [PubMed]
- Loeffler-Wirth, H.; Reikowski, J.; Hakobyan, S.; Wagner, J.; Binder, H. oposSOM-Browser: An interactive tool to explore omics data landscapes in health science. BMC Bioinform. 2020, 21, 465. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Aldape, K.; Colman, H.; Figrarella-Branger, D.; Fuller, G.N.; Giannini, C.; Weller, M. cIMPACT-NOW update 5: Recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020, 139, 603–608. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.; Hurt, M.M.; Brown, P.O.; et al. Identification of Genes Periodically Expressed in the Human Cell Cycle and Their Expression in Tumors. Mol. Biol. Cell 2002, 13, 1977–2000. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.W.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Ichimura, K.; Collins, V.P. Tandem Duplication Producing a Novel Oncogenic BRAF Fusion Gene Defines the Majority of Pilocytic Astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.; Kocialkowski, S.; Liu, L.; Pearson, D.; Ichimura, K.; Collins, V.P. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 2009, 28, 2119–2123. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunology 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Nejo, T.; Matsushita, H.; Karasaki, T.; Nomura, M.; Saito, K.; Tanaka, S.; Takayanagi, S.; Hana, T.; Takahashi, S.; Kitagawa, Y.; et al. Reduced Neoantigen Expression Revealed by Longitudinal Multiomics as a Possible Immune Evasion Mechanism in Glioma. Cancer Immunol. Res. 2019, 7, 1148–1161. [Google Scholar] [CrossRef] [Green Version]
- Johanns, T.M.; Bowman-Kirigin, J.A.; Liu, C.; Dunn, G.P. Targeting Neoantigens in Glioblastoma: An Overview of Cancer Immunogenomics and Translational Implications. Neurosurgery 2017, 64, 165–176. [Google Scholar] [CrossRef]
- Johanns, T.M.; Ward, J.P.; Miller, C.A.; Wilson, C.; Kobayashi, D.K.; Bender, D.; Fu, Y.; Alexandrov, A.; Mardis, E.R.; Artyomov, M.N.; et al. Endogenous Neoantigen-Specific CD8 T Cells Identified in Two Glioblastoma Models Using a Cancer Immunogenomics Approach. Cancer Immunol. Res. 2016, 4, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Jühling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Bergthold, G.; Bandopadhayay, P.; Hoshida, Y.; Ramkissoon, S.; Ramkissoon, L.; Rich, B.; Maire, C.L.; Paolella, B.R.; Schumacher, S.E.; Tabak, B.; et al. Expression profiles of 151 pediatric low-grade gliomas reveal molecular differences associated with location and histological subtype. NeuroOncology 2015, 17, 1486–1496. [Google Scholar] [CrossRef]
- Sharma, M.K.; Mansur, D.B.; Reifenberger, G.; Perry, A.; Leonard, J.R.; Aldape, K.D.; Albin, M.G.; Emnett, R.J.; Loeser, S.; Watson, M.A.; et al. Distinct Genetic Signatures among Pilocytic Astrocytomas Relate to Their Brain Region Origin. Cancer Res. 2007, 67, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Hara, A.; Homma, T.; Yonekawa, Y.; Ohgaki, H. Altered Expression of Immune Defense Genes in Pilocytic Astrocytomas. J. Neuropathol. Exp. Neurol. 2005, 64, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Isaac, Y.; Seunggu, J.H.; Michael, E.S.; Tarik, T.; Andrew, T.P. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. JNS 2011, 115, 505–511. [Google Scholar]
- Donson, A.M.; Birks, D.K.; Schittone, S.A.; Kleinschmidt-DeMasters, B.K.; Sun, D.Y.; Hemenway, M.F.; Handler, M.H.; Waziri, A.E.; Wang, M.; Foreman, N.K. Increased Immune Gene Expression and Immune Cell Infiltration in High-Grade Astrocytoma Distinguish Long-Term from Short-Term Survivors. J. Immunol. 2012, 189, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Gorovets, D.; Kannan, K.; Shen, R.; Kastenhuber, E.R.; Islamdoust, N.; Campos, C.; Pentsova, E.; Heguy, A.; Jhanwar, S.C.; Mellinghoff, I.K.; et al. IDH Mutation and Neuroglial Developmental Features Define Clinically Distinct Subclasses of Lower Grade Diffuse Astrocytic Glioma. Clin. Cancer Res. 2012, 18, 2490–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, O.; Caballero, O.L.; Stevenson, B.J.; Chen, Y.-T.; Cohen, T.; Chua, R.; Maher, C.A.; Panji, S.; Schaefer, U.; Kruger, A.; et al. Genome-wide analysis of cancer/testis gene expression. Proc. Natl. Acad. Sci. USA 2008, 105, 20422–20427. [Google Scholar] [CrossRef] [Green Version]
- Ghafouri-Fard, S.; Modarressi, M.-H. Expression of cancer–testis genes in brain tumors: Implications for cancer immunotherapy. Immunotherapy 2012, 4, 59–75. [Google Scholar] [CrossRef]
- Freitas, M.R.P.; Malheiros, S.M.F.; Stávale, J.N.; Biassi, T.P.; Zamunér, F.T.; Begnami, M.D.F.D.S.; Soares, F.; Vettore, A.L. Expression of Cancer/Testis Antigens is Correlated with Improved Survival in Glioblastoma. Oncotarget 2013, 4, 636–646. [Google Scholar] [CrossRef] [Green Version]
- Yawata, T.; Nakai, E.; Park, K.C.; Chihara, T.; Kumazawa, A.; Toyonaga, S.; Masahira, T.; Nakabayashi, H.; Kaji, T.; Shimizu, K. Enhanced expression of cancer testis antigen genes in glioma stem cells. Mol. Carcinog. 2010, 49, 532–544. [Google Scholar] [CrossRef]
- Emara, M.; Salloum, N.; Allalunis-Turner, J. Expression and hypoxic up-regulation of neuroglobin in human glioblastoma cells. Mol. Oncol. 2008, 3, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waitkus, M.S.; Diplas, B.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. NeuroOncology 2016, 18, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, 8478. [Google Scholar] [CrossRef] [Green Version]
- Reitman, Z.J.; Paolella, B.R.; Bergthold, G.; Pelton, K.; Becker, S.; Jones, R.; Sinai, C.E.; Malkin, H.; Huang, Y.; Grimmet, L.; et al. Mitogenic and progenitor gene programmes in single pilocytic astrocytoma cells. Nat. Commun. 2019, 10, 3731. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Li, H.; Shen, X.; He, Z.; He, L.; Guo, Z. Reproducibility and Concordance of Differential DNA Methylation and Gene Expression in Cancer. PLoS ONE 2012, 7, e29686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Shen, Y.; Xue, Z.; Bibikova, M.; April, C.; Liu, Z.; Cheng, L.; Nagy, A.; Pellegrini, M.; Fan, J.-B.; et al. A Panel of CpG Methylation Sites Distinguishes Human Embryonic Stem Cells and Induced Pluripotent Stem Cells. Stem Cell Rep. 2014, 2, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2010, 30, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Polioudaki, H.; Agelaki, S.; Chiotaki, R.; Politaki, E.; Mavroudis, D.; Matikas, A.; Georgoulias, V.; Theodoropoulos, P. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalheim, T.; Hopp, L.; Binder, H.; Aust, G.; Galle, J. On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells. Epigenomes 2018, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Glusman, G.; Yanai, I.; Rubin, I.; Lancet, D. The Complete Human Olfactory Subgenome. Genome Res. 2001, 11, 685–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhang, W.; Chen, D.; Lv, Y.; Zheng, J.; Lilljebjörn, H.; Ran, L.; Bao, Z.; Soneson, C.; Sjögren, H.O.; et al. A glioma classification scheme based on coexpression modules of EGFR and PDGFRA. Proc. Natl. Acad. Sci. USA 2014, 111, 3538–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef]
- Killela, P.J.; Pirozzi, C.J.; Reitman, Z.; Jones, S.; Rasheed, B.A.; Lipp, E.; Friedman, H.; Friedman, A.H.; He, Y.; McLendon, R.E.; et al. The genetic landscape of anaplastic astrocytoma. Oncotarget 2014, 5, 1452–1457. [Google Scholar] [CrossRef] [Green Version]
- Chittaranjan, S.; Chan, S.; Yang, C.; Yang, K.C.; Chen, V.; Moradian, A.; Firme, M.; Song, J.; Go, N.E.; Blough, M.D.; et al. Mutations in CIC and IDH1 cooperatively regulate 2-hydroxyglutarate levels and cell clonogenicity. Oncotarget 2014, 5, 7960–7979. [Google Scholar] [CrossRef] [Green Version]
- Gleize, V.; Alentorn, A.; Connen de Kérillis, L.; Labussière, M.; Nadaradjane, A.A.; Mundwiller, E.; Sanson, M. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann. Neurol. 2015, 78, 355–374. [Google Scholar] [CrossRef]
- Bai, H.; Harmancı, A.S.; Erson-Omay, E.Z.; Li, J.; Coşkun, S.; Simon, M.; Krischek, B.; Özduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nat. Cell Biol. 2017, 549, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Reis, G.F.; Pekmezci, M.; Hansen, H.M.; Rice, T.; Marshall, R.E.; Molinaro, A.M.; Perry, A. CDKN2A Loss Is Associated with Shortened Overall Survival in Lower-Grade (World Health Organization Grades II–III) Astrocytomas. J. Neuropathol. Exp. Neurol. 2015, 74, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Shirahata, M.; Ono, T.; Stichel, D.; Schrimpf, D.; Reuss, D.E.; Sahm, F.; Koelsche, C.; Wefers, A.; Reinhardt, A.; Huang, K.; et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018, 136, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.L.; Boddeke, H.W.G.M.; et al. Glioma-Associated Microglia/Macrophages Display an Expression Profile Different from M1 and M2 Polarization and Highly Express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [Green Version]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Choi, S.A.; Kim, S.-K.; Wang, K.-C.; Park, S.-H. Snail plays an oncogenic role in glioblastoma by promoting epithelial mesenchymal transition. Int. J. Clin. Exp. Pathol. 2014, 7, 1977–1987. [Google Scholar] [PubMed]
- Ward, P.; Cross, J.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R (−)-2-hydroxyglutarate production. Oncogene 2011, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, F.P.; Wesseling, P.; Verhaak, R.G.W. Reconstructing the molecular life history of gliomas. Acta Neuropathol. 2018, 135, 649–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, F.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Szekely, P.; Bar, N.; Zimmer, A.; Sheftel, H.; Caldas, C.; Alon, U. Tumor diversity and the trade-off between universal cancer tasks. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, F.; Liu, L. Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells. Genes 2019, 10, 1030. [Google Scholar] [CrossRef] [Green Version]
- Recagni, M.; Bidzinska, J.; Zaffaroni, N.; Folini, M. The Role of Alternative Lengthening of Telomeres Mechanism in Cancer: Translational and Therapeutic Implications. Cancers 2020, 12, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nersisyan, L.; Hopp, L.; Loeffler-Wirth, H.; Galle, J.; Loeffler, M.; Arakelyan, A.; Binder, H. Telomere Length Maintenance and Its Transcriptional Regulation in Lynch Syndrome and Sporadic Colorectal Carcinoma. Front. Oncol. 2019, 9, 1172. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Wang, K.; Liu, X.; Zhao, Z. Comprehensive profiling identifies a novel signature with robust predictive value and reveals the potential drug resistance mechanism in glioma. Cell Commun. Signal. 2020, 18, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yin, J.; Luo, L.; Huang, D.; Zhai, D.; Wang, G.; Xu, N.; Yang, M.; Song, Y.; Zheng, G.; et al. Integrative Analysis of DNA Methylation and Transcriptome Identifies a Predictive Epigenetic Signature Associated with Immune Infiltration in Gliomas. Front. Cell Dev. Biol. 2021, 9, 1368. [Google Scholar] [CrossRef] [PubMed]
- Duregon, E.; Bertero, L.; Pittaro, A.; Soffietti, R.; Rudà, R.; Trevisan, M.; Papotti, M.; Ventura, L.; Senetta, R.; Cassoni, P. Ki-67 proliferation index but not mitotic thresholds integrates the molecular prognostic stratification of lower grade gliomas. Oncotarget 2016, 7, 21190–21198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvà, M.L.; Tirosh, I. The Glioma Stem Cell Model in the Era of Single-Cell Genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef]
- Tirosh, I.; Venteicher, A.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nat. Cell Biol. 2016, 539, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.21. [Google Scholar] [CrossRef]
- Pérez, R.F.; Tejedor, J.R.; Bayón, G.F.; Fernández, A.F.; Fraga, M.F. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell 2018, 17, e12744. [Google Scholar] [CrossRef]
- Karpinski, P.; Pesz, K.; Sasiadek, M. Pan-cancer analysis reveals presence of pronounced DNA methylation drift in CpG island methylator phenotype clusters. Epigenomics 2017, 9, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 2015, 16, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarone, F.; Tagliatesta, S.; Caiafa, P.; Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms? Mech. Ageing Dev. 2018, 174, 3–17. [Google Scholar] [CrossRef]
- Lowe, R.; Overhoff, M.G.; Ramagopalan, S.V.; Garbe, J.C.; Koh, J.; Stampfer, M.R.; Beach, D.H.; Rakyan, V.K.; Bishop, C.L. The senescent methylome and its relationship with cancer, ageing and germline genetic variation in humans. Genome Biol. 2015, 16, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Otte, P.; Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems and Their Role in Cellular Senescence. Comput. Struct. Biotechnol. J. 2019, 17, 1265–1277. [Google Scholar] [CrossRef]
- Doze, V.A.; Perez, D.M. Chapter Five—GPCRs in Stem Cell Function. In Progress in Molecular Biology and Translational Science; Kenakin, T., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 115, pp. 175–216. [Google Scholar]
- Shlyakhtina, Y.; Moran, K.; Portal, M. Genetic and Non-Genetic Mechanisms Underlying Cancer Evolution. Cancers 2021, 13, 1380. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Turcan, Ş. Epigenetic Reprogramming for Targeting IDH-Mutant Malignant Gliomas. Cancers 2019, 11, 1616. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willscher, E.; Hopp, L.; Kreuz, M.; Schmidt, M.; Hakobyan, S.; Arakelyan, A.; Hentschel, B.; Jones, D.T.W.; Pfister, S.M.; Loeffler, M.; et al. High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas. Cancers 2021, 13, 3198. https://doi.org/10.3390/cancers13133198
Willscher E, Hopp L, Kreuz M, Schmidt M, Hakobyan S, Arakelyan A, Hentschel B, Jones DTW, Pfister SM, Loeffler M, et al. High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas. Cancers. 2021; 13(13):3198. https://doi.org/10.3390/cancers13133198
Chicago/Turabian StyleWillscher, Edith, Lydia Hopp, Markus Kreuz, Maria Schmidt, Siras Hakobyan, Arsen Arakelyan, Bettina Hentschel, David T. W. Jones, Stefan M. Pfister, Markus Loeffler, and et al. 2021. "High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas" Cancers 13, no. 13: 3198. https://doi.org/10.3390/cancers13133198
APA StyleWillscher, E., Hopp, L., Kreuz, M., Schmidt, M., Hakobyan, S., Arakelyan, A., Hentschel, B., Jones, D. T. W., Pfister, S. M., Loeffler, M., Loeffler-Wirth, H., & Binder, H. (2021). High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas. Cancers, 13(13), 3198. https://doi.org/10.3390/cancers13133198