
Maximum Tolerated Dose and Anti-Tumor Activity of Intraperitoneal Cantrixil (TRX-E-002-1) in Patients with Persistent or Recurrent Ovarian Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer: Phase I Study Results

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
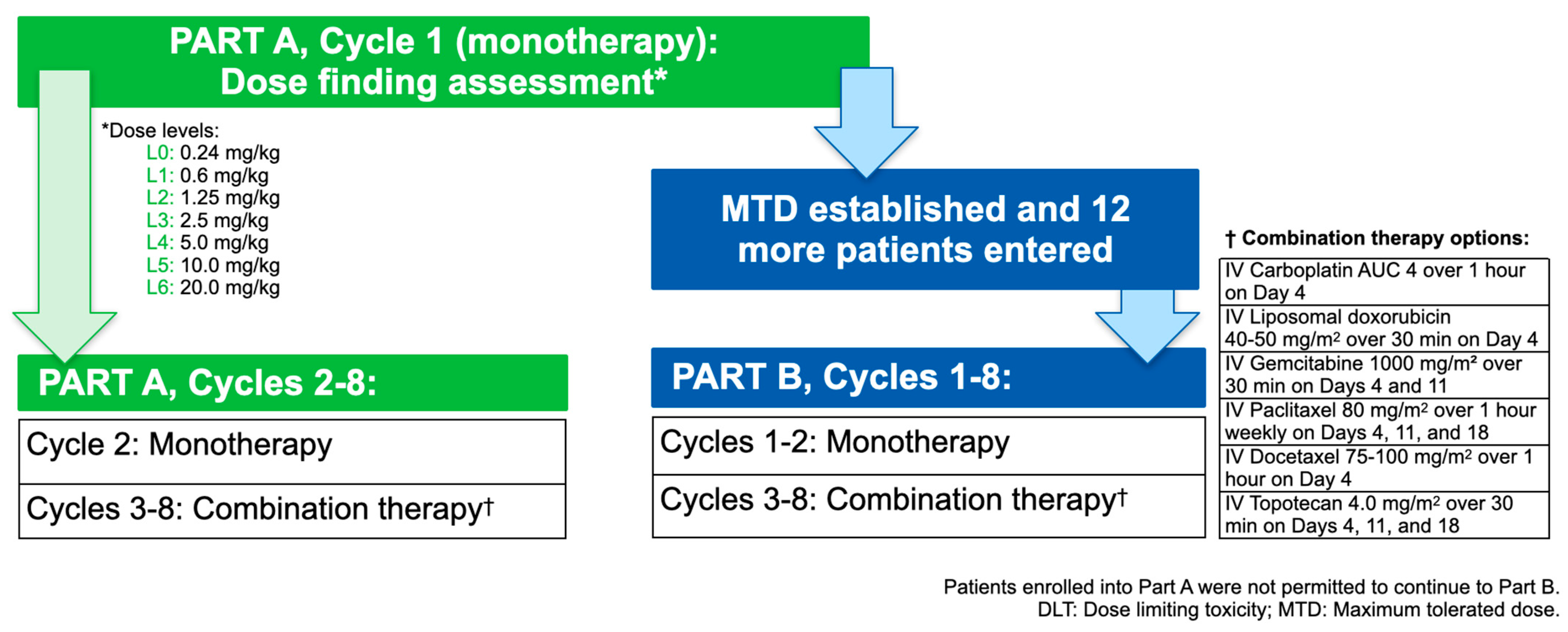
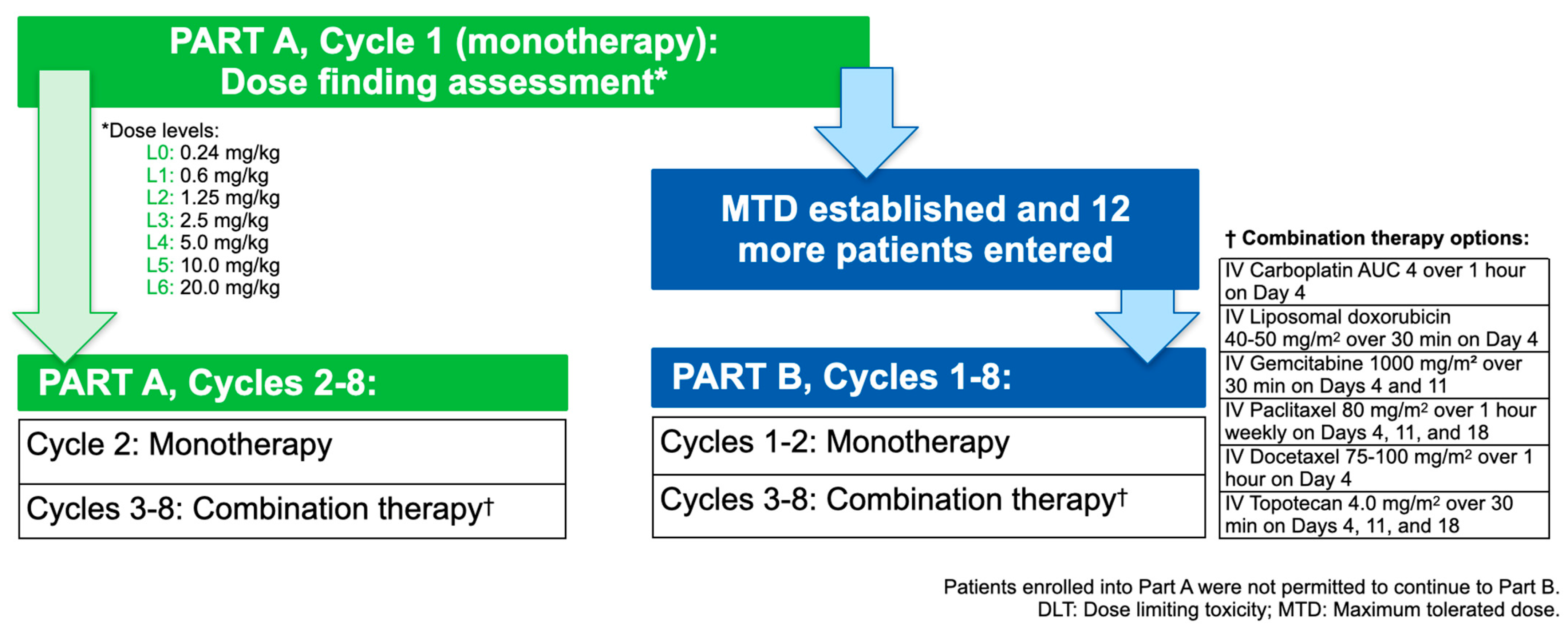
2.1. Study Design
2.2. Patient Population
2.3. Treatments Administered
2.4. Assessments and Evaluations
2.5. Study Endpoints
2.6. Exploratory Analysis of Stem Cell Markers
2.7. Statistical Considerations
3. Results
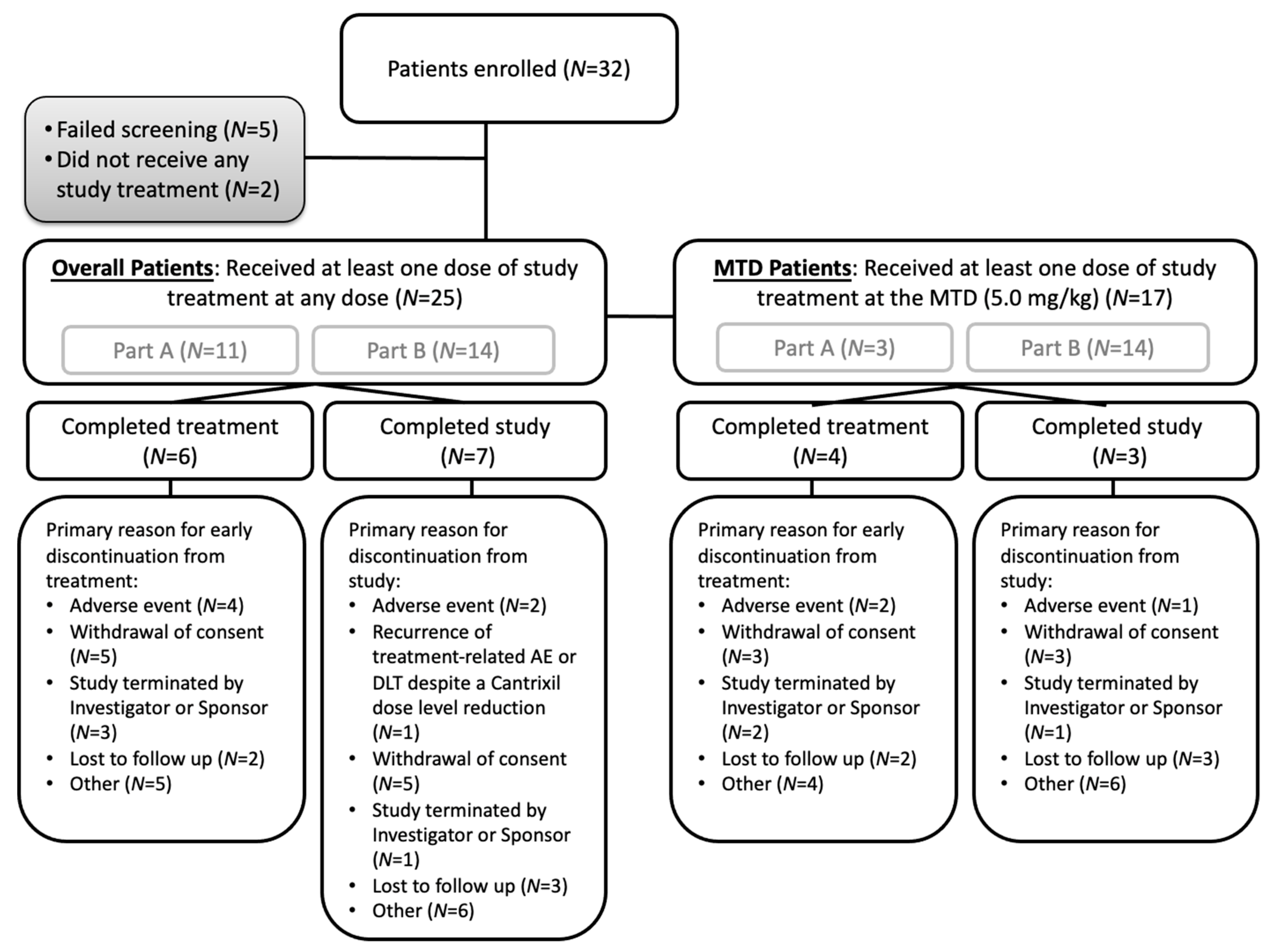
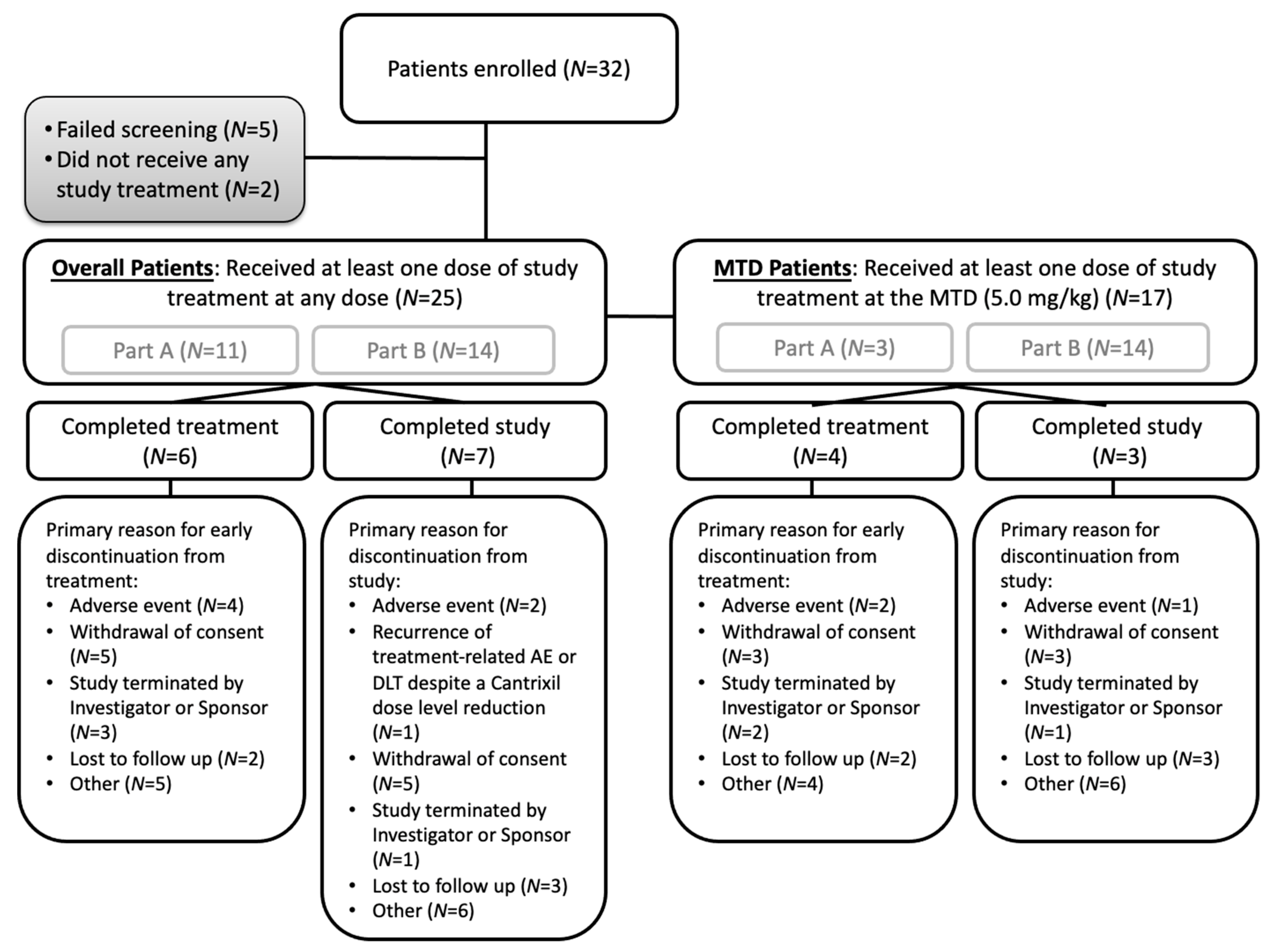
3.1. Patient Population and Baseline Characteristics
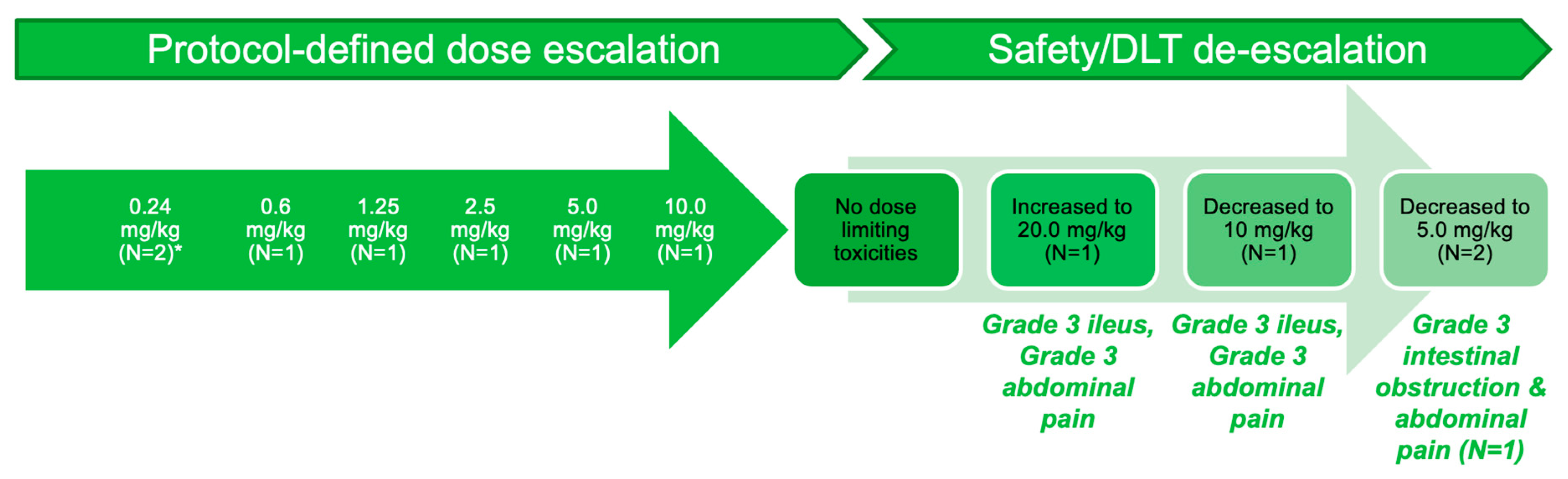
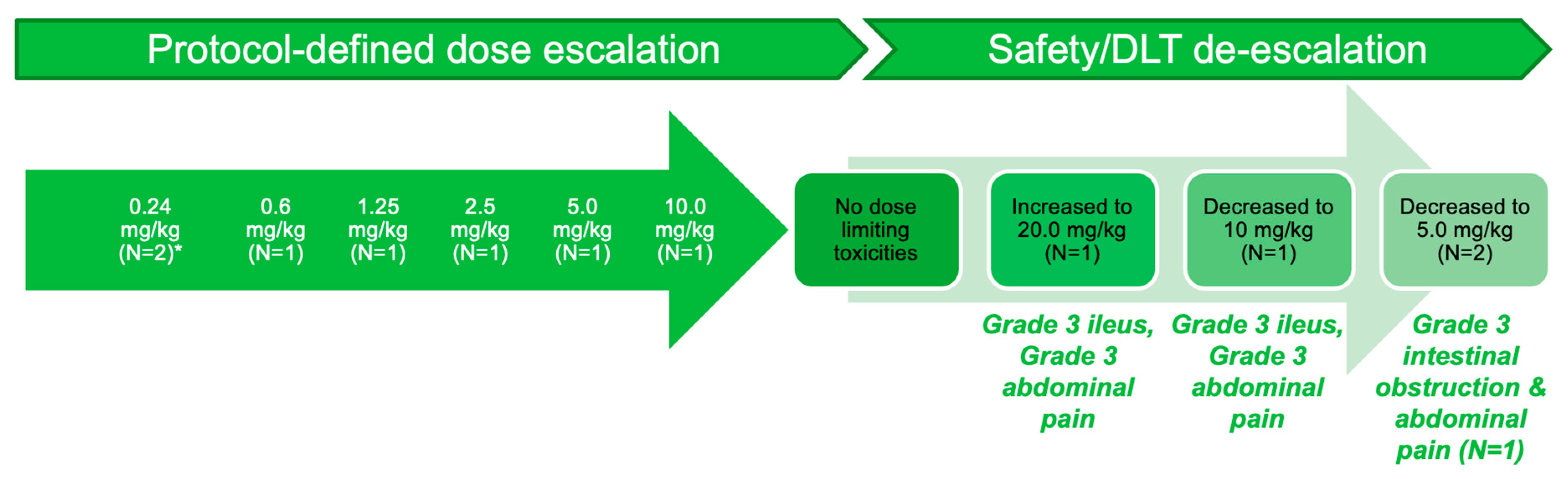
3.2. Part A: Determination of MTD
3.3. Pharmacokinetics
3.4. Safety
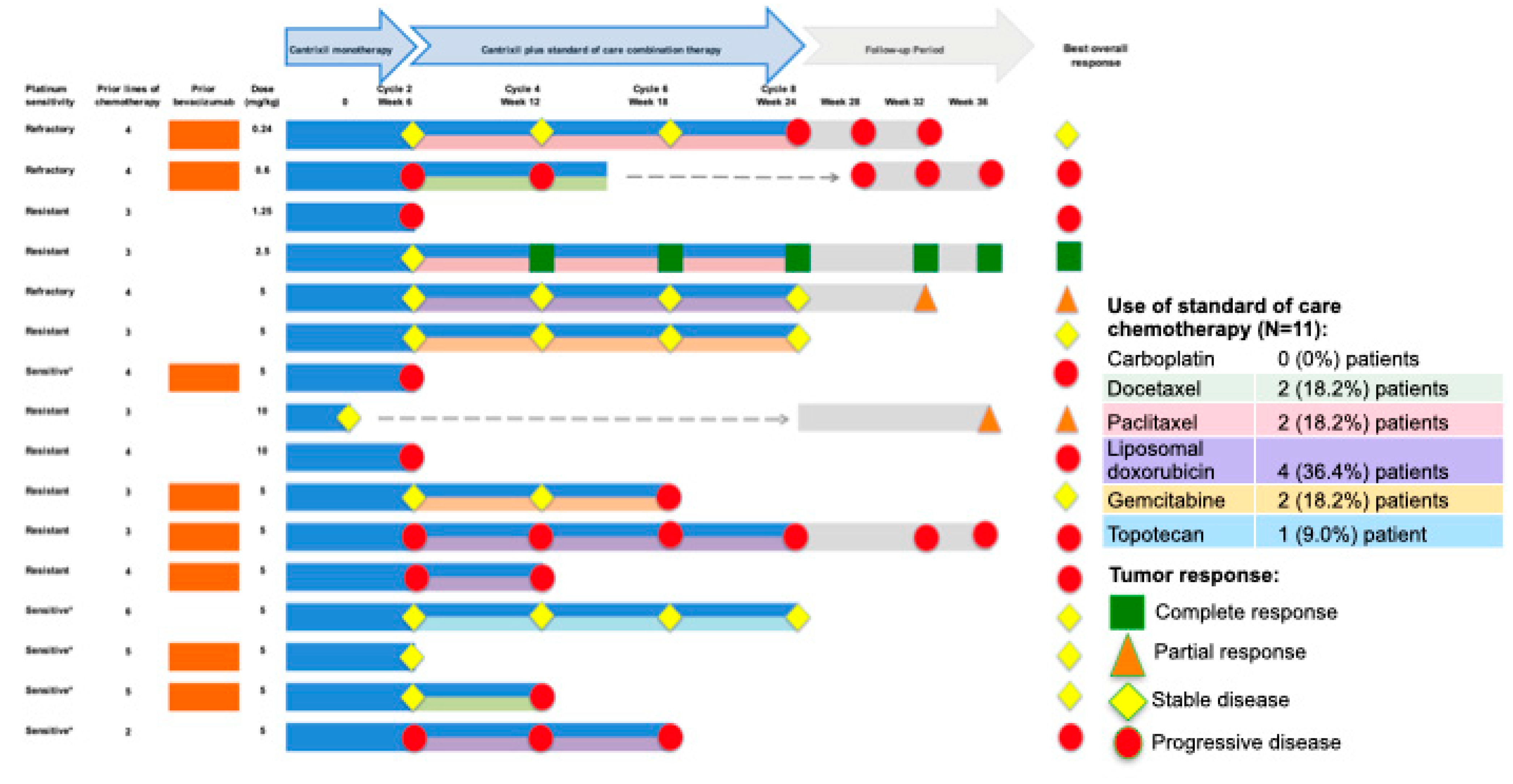
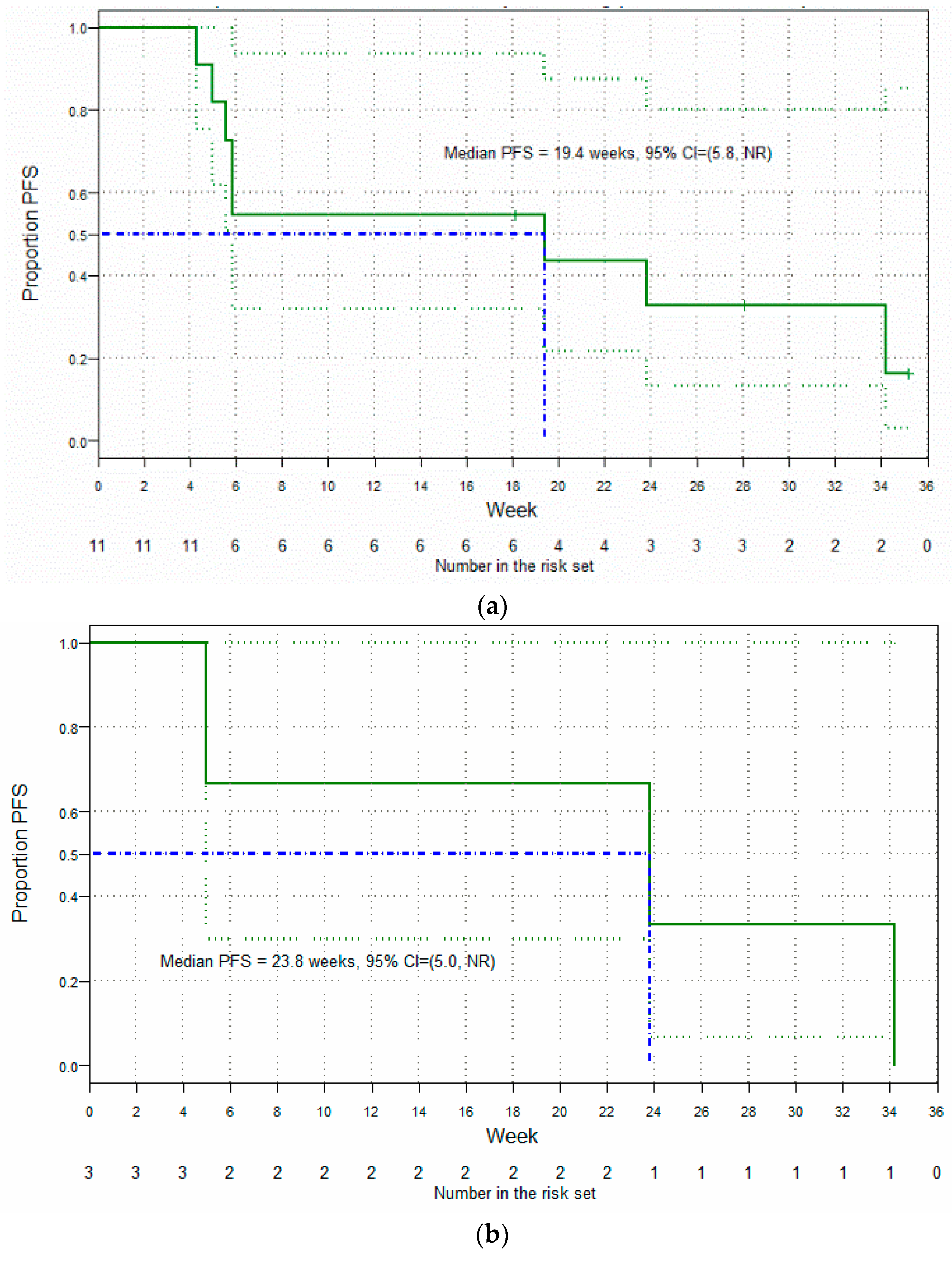
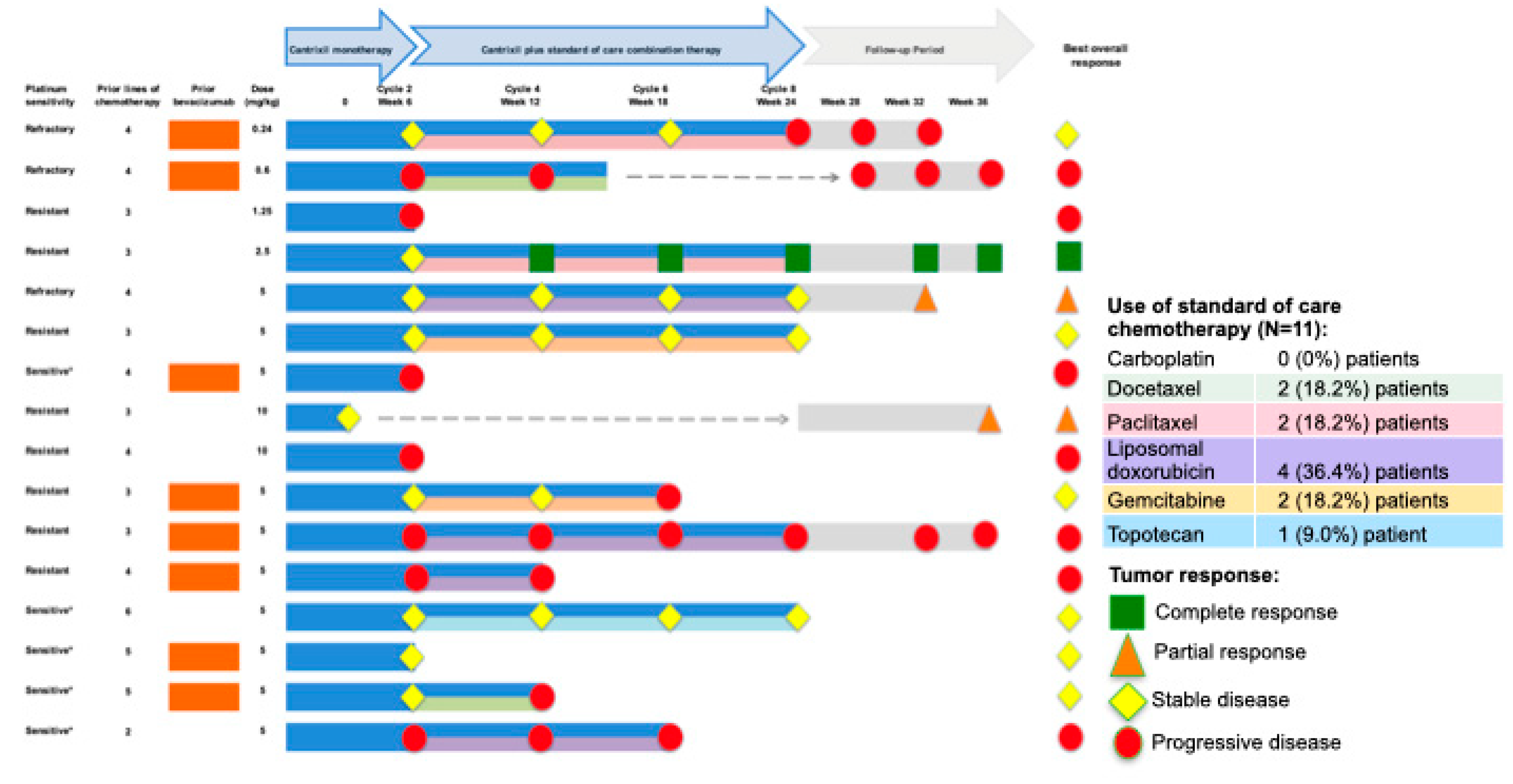
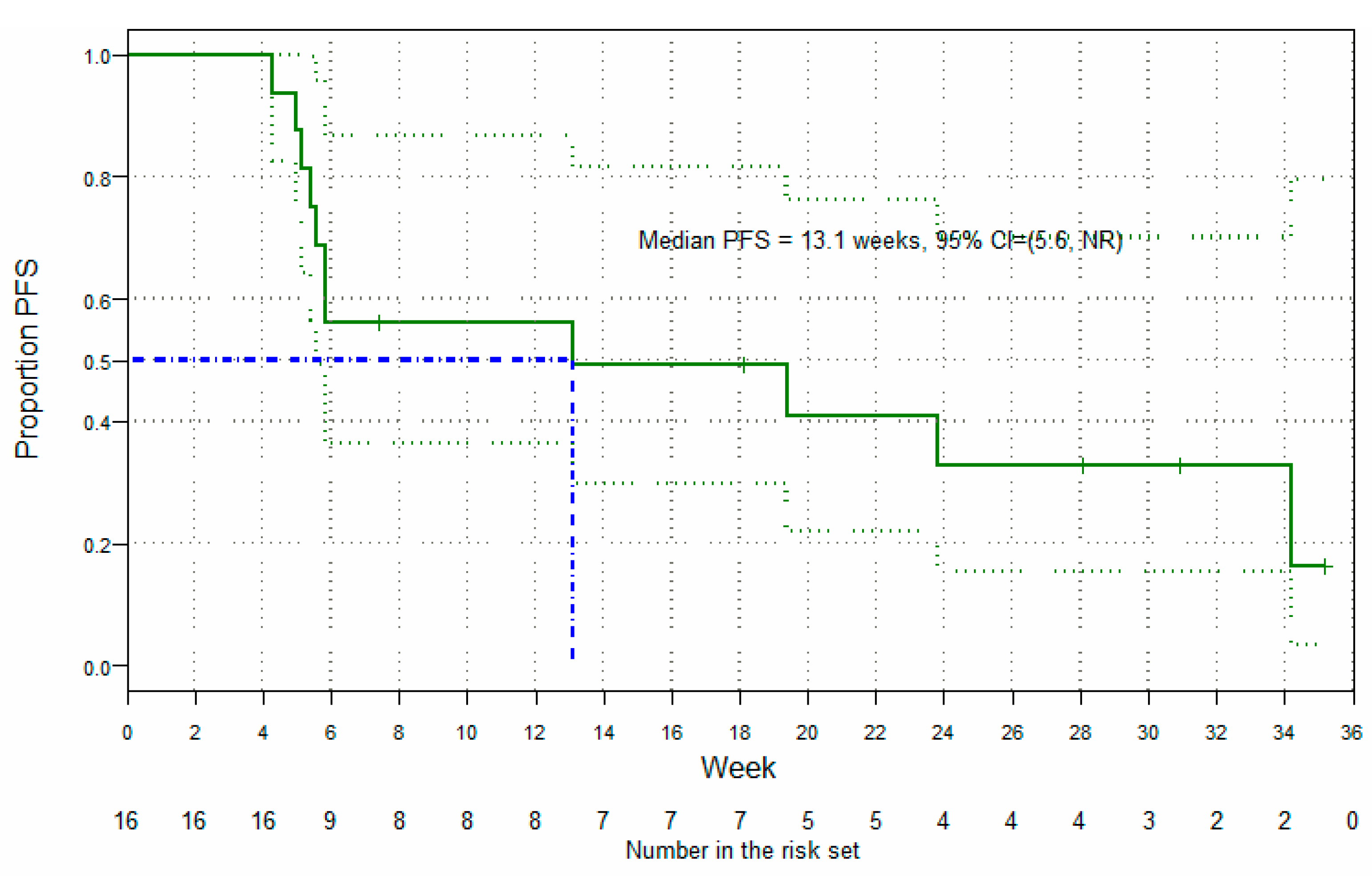
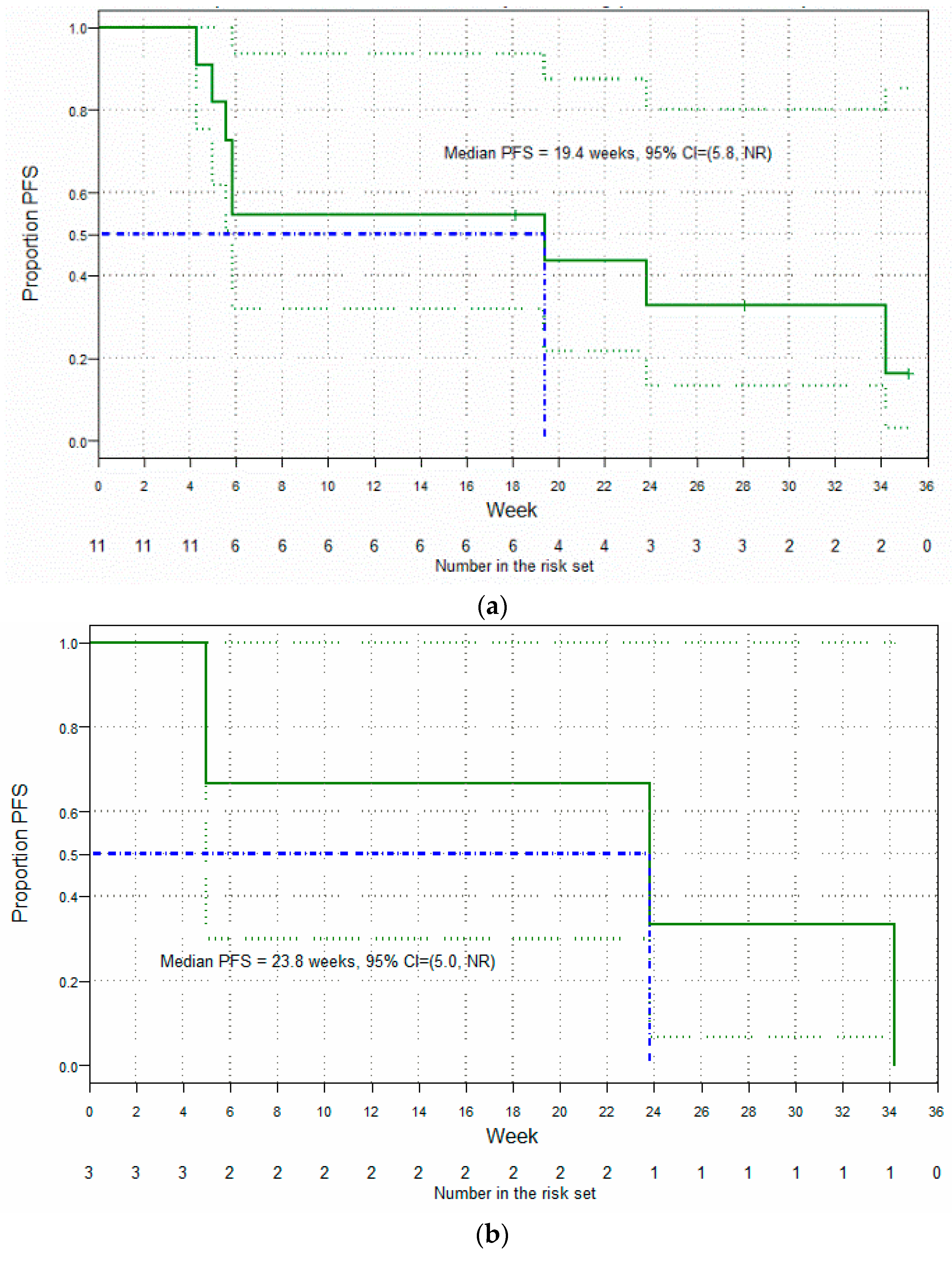
3.5. Clinical Findings: Antitumor Activity
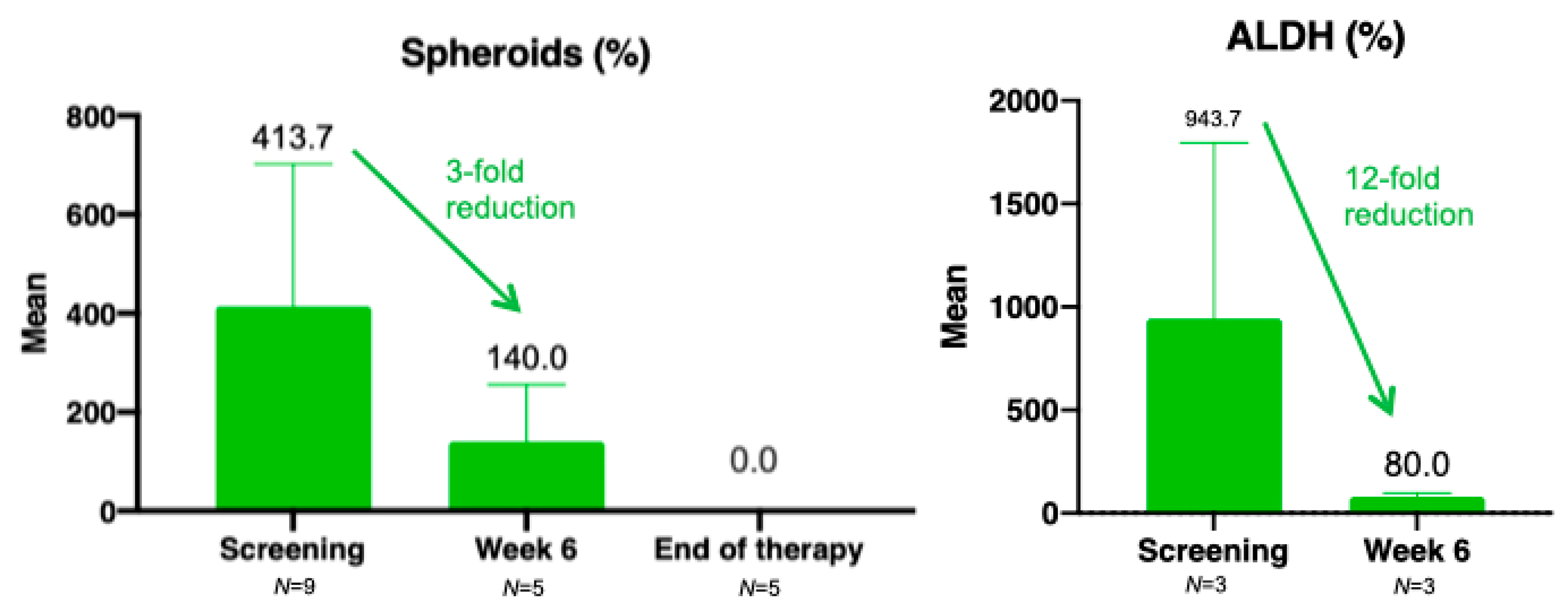
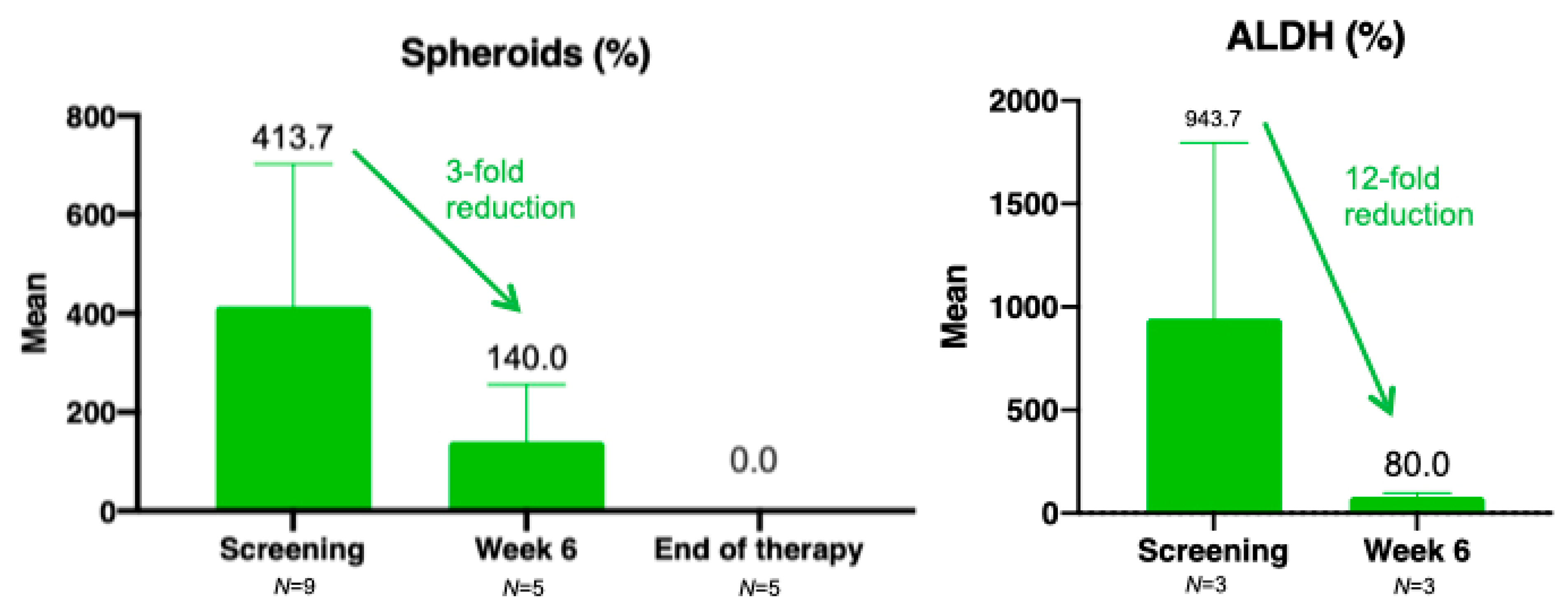
3.6. Translational Research
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021. [Google Scholar] [CrossRef]
- Berek, J.S.; Crum, C.; Friedlander, M. Cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynecol. Obstet. 2015, 131, S111–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitwell, H.J.; Worthington, J.; Blyuss, O.; Gentry-Maharaj, A.; Ryan, A.; Gunu, R.; Kalsi, J.; Menon, U.; Jacobs, I.; Zaikin, A.; et al. Improved early detection of ovarian cancer using longitudinal multimarker models. Br. J. Cancer 2020, 122, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Marth, C.; Reimer, D.; Zeimet, A.G. Front-line therapy of advanced epithelial ovarian cancer: Standard treatment. Ann. Oncol. 2017, 28, viii36–viii39. [Google Scholar] [CrossRef]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.-S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. npj Precis. Oncol. 2018, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips-Chavez, C.; Watson, M.; Coward, J.; Schloss, J. A systematic literature review assessing if genetic biomarkers are predictors for platinum-based chemotherapy response in ovarian cancer patients. Eur. J. Clin. Pharmacol. 2020, 76, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A. First-line treatment of ovarian cancer: Questions and controversies to address. Adv. Med. Oncol. 2018, 10, 1758835918768232. [Google Scholar] [CrossRef]
- Aletti, G.D.; Podratz, K.C.; Cliby, W.A.; Gostout, B.S. Stage IV ovarian cancer: Disease site-specific rationale for postoperative treatment. Gynecol. Oncol. 2009, 112, 22–27. [Google Scholar] [CrossRef]
- Tate, S.; Nishikimi, K.; Matsuoka, A.; Shozu, M. Aggressive surgery for advanced ovarian cancer decreases the risk of intraperitoneal recurrence. Int. J. Clin. Oncol. 2020, 25, 1726–1735. [Google Scholar] [CrossRef]
- Roze, J.F.; Veldhuis, W.B.; Hoogendam, J.P.; Verheijen, R.H.M.; Scholten, R.; Zweemer, R.P. Prognostic value of radiological recurrence patterns in ovarian cancer. Gynecol. Oncol. 2020, 157, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.A.; Cronin, A.; Milne, D.E.; Bookman, M.A.; Burger, R.A.; Cohn, D.E.; Cristea, M.C.; Griggs, J.J.; Keating, N.L.; Levenback, C.F.; et al. Use and Effectiveness of Intraperitoneal Chemotherapy for Treatment of Ovarian Cancer. J. Clin. Oncol. 2015, 33, 2841–2847. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Malekshah, O.M.; Nomani, A.; Patel, N.; Hatefi, A. A novel chemotherapeutic protocol for peritoneal metastasis and inhibition of relapse in drug resistant ovarian cancer. Cancer Med. 2018, 7, 3630–3641. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Morrison, S.; Clarke, M.; Weissman, I. Stem Cells cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef] [Green Version]
- Saif, M.; Ager, E.I.; Field, P.; Lilischkis, K.J. The role of cancer stem cells and the therapeutic potential of TRX-E-002-1 in ovarian cancer. Expert Opin. Orphan Drugs 2018, 6, 465–475. [Google Scholar] [CrossRef]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.-H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Hong, L.; Chefetz, I. How to win the ovarian cancer stem cell battle: Destroying the roots. Cancer Drug Resist. 2020, 4, 1021–1033. [Google Scholar] [CrossRef]
- Kelly, M.G.; Mor, G.; Husband, A.; Malley, D.M.; Baker, L.; Azodi, M.; Schwartz, P.E.; Rutherford, T.J. Phase II Evaluation of Phenoxodiol in Combination with Cisplatin or Paclitaxel in Women with Platinum/Taxane-Refractory/Resistant Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancers. Int. J. Gynecol. Cancer 2011, 21, 633. [Google Scholar] [CrossRef]
- Bristow, R.E.; Tomacruz, R.S.; Armstrong, D.K.; Trimble, E.L.; Montz, F.J. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: A meta-analysis. J. Clin. Oncol. 2002, 20, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Silasi, D.A.; Alvero, A.B.; Rutherford, T.J.; Brown, D.; Mor, G. Phenoxodiol: Pharmacology and clinical experience in cancer monotherapy and in combination with chemotherapeutic drugs. Expert Opin. Pharm. 2009, 10, 1059–1067. [Google Scholar] [CrossRef]
- Stevenson, A.J.; Ager, E.I.; Proctor, M.A.; Skalamera, D.; Heaton, A.; Brown, D.; Gabrielli, B.G. Mechanism of action of the third generation benzopyrans and evaluation of their broad anti-cancer activity in vitro and in vivo. Sci. Rep. 2018, 8, 5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvero, A.B.; Heaton, A.; Lima, E.; Pitruzzello, M.; Sumi, N.; Yang-Hartwich, Y.; Cardenas, C.; Steinmacher, S.; Silasi, D.A.; Brown, D.; et al. TRX-E-002-1 Induces c-Jun-Dependent Apoptosis in Ovarian Cancer Stem Cells and Prevents Recurrence In Vivo. Mol. Cancer Ther. 2016, 15, 1279–1290. [Google Scholar] [CrossRef] [Green Version]
- Alter, R.; Turaga, K.; Lengyel, E. Are We Ready for Hyperthermic Intraperitoneal Chemotherapy in the Upfront Treatment of Ovarian Cancer? JAMA Netw. Open 2020, 3, e2014184. [Google Scholar] [CrossRef] [PubMed]
- Rustin, G.J.; Vergote, I.; Eisenhauer, E.; Pujade-Lauraine, E.; Quinn, M.; Thigpen, T.; du Bois, A.; Kristensen, G.; Jakobsen, A.; Sagae, S.; et al. Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int. J. Gynecol. Cancer 2011, 21, 419–423. [Google Scholar] [CrossRef]
- Winner, K.R.K.; Steinkamp, M.P.; Lee, R.J.; Swat, M.; Muller, C.Y.; Moses, M.E.; Jiang, Y.; Wilson, B.S. Spatial Modeling of Drug Delivery Routes for Treatment of Disseminated Ovarian Cancer. Cancer Res. 2016, 76, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saif, M.W.; Heaton, A.; Lilischkis, K.; Garner, J.; Brown, D.M. Pharmacology and toxicology of the novel investigational agent Cantrixil (TRX-E-002-1). Cancer Chemother. Pharmacol. 2017, 79, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Hoskins, P.J. Intraperitoneal Therapy for British Columbia; BC Cancer Agency Gynecological Tumour Group: Vancouver, BC, Canada, 2006; Available online: http://www.bccancer.bc.ca/books/Documents/Gynecology/GyneovaryIntraperitonealChemotherapyforBC_June06.pdf. (accessed on 27 May 2021).
- Alberts, D.S.; Liu, P.Y.; Hannigan, E.V.; O’Toole, R.; Williams, S.D.; Young, J.A.; Franklin, E.W.; Clarke-Pearson, D.L.; Malviya, V.K.; DuBeshter, B. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N. Engl. J. Med. 1996, 335, 1950–1955. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Markman, M.; Bundy, B.N.; Alberts, D.S.; Fowler, J.M.; Clark-Pearson, D.L.; Carson, L.F.; Wadler, S.; Sickel, J. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: An intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J. Clin. Oncol. 2001, 19, 1001–1007. [Google Scholar] [CrossRef]
- Landrum, L.M.; Java, J.; Mathews, C.A.; Lanneau, G.S., Jr.; Copeland, L.J.; Armstrong, D.K.; Walker, J.L. Prognostic factors for stage III epithelial ovarian cancer treated with intraperitoneal chemotherapy: A Gynecologic Oncology Group study. Gynecol. Oncol. 2013, 130, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Coward, J.I.; Middleton, K.; Murphy, F. New perspectives on targeted therapy in ovarian cancer. Int. J. Women’s Health 2015, 7, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, X.; Chen, D.; Cai, J.; Fu, Y.; Kang, H.; Gao, J.; Gao, K.; Du, N. Intraperitoneal administration of cisplatin plus bevacizumab for the management of malignant ascites in ovarian epithelial cancer: Results of a phase III clinical trial. Med. Oncol. (NorthwoodLond. Engl.) 2015, 32, 292. [Google Scholar] [CrossRef]
- El-Shami, K.; Elsaid, A.; El-Kerm, Y. Open-label safety and efficacy pilot trial of intraperitoneal bevacizumab as palliative treatment in refractory malignant ascites. J. Clin. Oncol. 2007, 25, 9043. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Maxwell, G.L.; Chernofsky, M.R.; Bernstein, S.A.; Farley, J.H.; Rose, G.S. Intraperitoneal bevacizumab for the palliation of malignant ascites in refractory ovarian cancer. Gynecol. Oncol. 2008, 111, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Qiu, J.T.; Lai, C.H.; Huang, H.J.; Lin, C.T.; Chen, M.Y.; Chou, H.H.; Huang, K.G.; Chang, T.C. Outcomes and prognoses of patients with ovarian cancer using bevacizumab: 6-year experience in a tertiary care hospital of northern Taiwan. PLoS ONE 2017, 12, e0175703. [Google Scholar] [CrossRef]
- Sjoquist, K.M.; Espinoza, D.; Mileshkin, L.; Ananda, S.; Shannon, C.M.; Yip, S.; Goh, J.C.; Bowtell, D.; Harrison, M.; Friedlander, M.L. REZOLVE (ANZGOG-1101): A phase 2 trial of intraperitoneal bevacizumab to treat symptomatic ascites in patients with chemotherapy-resistant, epithelial ovarian cancer. Gynecol. Oncol. 2021, 161, 374–381. [Google Scholar] [CrossRef]
- Bellati, F.; Napoletano, C.; Ruscito, I.; Pastore, M.; Pernice, M.; Antonilli, M.; Nuti, M.; Benedetti Panici, P. Complete remission of ovarian cancer induced intractable malignant ascites with intraperitoneal bevacizumab. Immunological observations and a literature review. Invest. New Drugs 2010, 28, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Napoletano, C.; Ruscito, I.; Bellati, F.; Zizzari, I.G.; Rahimi, H.; Gasparri, M.L.; Antonilli, M.; Panici, P.B.; Rughetti, A.; Nuti, M. Bevacizumab-Based Chemotherapy Triggers Immunological Effects in Responding Multi-Treated Recurrent Ovarian Cancer Patients by Favoring the Recruitment of Effector T Cell Subsets. J. Clin. Med. 2019, 8, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandial, D.D.; Farshchi-Heydari, S.; Larson, C.A.; Elliott, G.I.; Wrasidlo, W.J.; Howell, S.B. Enhanced delivery of cisplatin to intraperitoneal ovarian carcinomas mediated by the effects of bortezomib on the human copper transporter 1. Clin. Cancer Res. 2009, 15, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Jandial, D.A.; Brady, W.E.; Howell, S.B.; Lankes, H.A.; Schilder, R.J.; Beumer, J.H.; Christner, S.M.; Strychor, S.; Powell, M.A.; Hagemann, A.R.; et al. A phase I pharmacokinetic study of intraperitoneal bortezomib and carboplatin in patients with persistent or recurrent ovarian cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huehls, A.M.; Wagner, J.M.; Huntoon, C.J.; Geng, L.; Erlichman, C.; Patel, A.G.; Kaufmann, S.H.; Karnitz, L.M. Poly(ADP-Ribose) polymerase inhibition synergizes with 5-fluorodeoxyuridine but not 5-fluorouracil in ovarian cancer cells. Cancer Res. 2011, 71, 4944–4954. [Google Scholar] [CrossRef] [Green Version]
- Muggia, F.; Bonetti, A. History of intraperitoneal platinum drug delivery for ovarian cancer and its future applications. Cancer Drug Resist. 2021, 4. [Google Scholar] [CrossRef]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2016, 140, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian Cancer Spheroid Cells with Stem Cell-Like Properties Contribute to Tumor Generation, Metastasis and Chemotherapy Resistance through Hypoxia-Resistant Metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Hwang, S.; Jeong, J.-Y.; Jung, S.G.; Choi, M.C.; Joo, W.D.; Song, S.H.; Lee, C.; An, H.J. Integrative analysis of transcription factors and microRNAs in ovarian cancer cell spheroids. J. Ovarian Res. 2020, 13, 16. [Google Scholar] [CrossRef]
- Muñoz-Galván, S.; Carnero, A. Targeting Cancer Stem Cells to Overcome Therapy Resistance in Ovarian Cancer. Cells 2020, 9, 1402. [Google Scholar] [CrossRef] [PubMed]
- Vassalli, G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, C.N.; Goodman, B.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting Aldehyde Dehydrogenase Cancer Stem Cells in Ovarian Cancer. Mol. Cancer Ther. 2010, 9, 3186. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Part A | Patients Receiving MTD a | All Patients |
|---|---|---|---|
| n = 11 | n = 17 | n = 25 | |
| Age, median (range), y | 66 (42–79) | 61 (46–79) | 62 (42–79) |
| BMI, median (range), kg/m2 | 28.2 (20.1–38.3) | 29.2 (18.8–36.6) | 28.2 (18.8–38.3) |
| Time since diagnosis, median (range), m | 52.7 (17.2–87.8) | 62.3 (14.0–255.8) | 48.5 (14.0–255.8) |
| ECOG performance status, n (%) | |||
| 0 | 8 (72.7) | 6 (35.3) | 12 (48.0) |
| 1 | 3 (27.3) | 11 (64.7) | 13 (52.0) |
| Tumor stage at study entry, n (%) | |||
| III | 4 (36.4) | 6 (35.3) | 10 (40.0) |
| IV | 7 (63.6) | 11 (64.7) | 15 (60.0) |
| Tumor type, n (%) | |||
| Epithelial ovarian cancer | 9 (81.8) | 11 (64.7) | 18 (72.0) |
| Fallopian tube cancer | 1 (9.1) | 2 (11.8) | 3 (12.0) |
| Primary peritoneal cancer | 1 (9.1) | 4 (23.5) | 4 (16.0) |
| Histological subtype, n (%) | |||
| Serous Carcinoma | 8 (72.7) | 11 (64.7) | 17 (68.0) |
| Endometrioid Carcinoma | 1 (9.1) | 1 (5.9) | 1 (4.0) |
| Clear Cell Carcinoma | 0 (0.0) | 1 (5.9) | 1 (4.0) |
| Other | 2 (18.2) | 4 (23.5) | 6 (24.0) |
| Prior lines of therapy, median (range) | 4 (3–6) | 5 (2–11) | 5 (2–11) |
| Prior chemotherapy, n (%) | |||
| Platinum | 11 (100.0) | 17 (100.0) | 25 (100.0) |
| Taxane | 11 (100.0) | 17 (100.0) | 25 (100.0) |
| Anthracycline | 11 (100.0) | 12 (70.6) | 19 (76.0) |
| Gemcitabine | 7 (63.6) | 11 (64.7) | 15 (60.0) |
| Melphalan | 0 (0.0) | 3 (17.6) | 3 (12.0) |
| Topotecan | 0 (0.0) | 3 (17.6) | 3 (12.0) |
| Cyclophosphamide | 0 (0.0) | 1 (5.9) | 1 (4.0) |
| Prior bevacizumab, n (%) | 4 (36.4) | 10 (58.8) | 13 (52.0) |
| Platinum status, n (%) | |||
| Refractory (Pt-Rf) | 3 (27.3) | 1 (5.9) | 3 (12.0) |
| Resistant (Pt-R) | 7 (63.6) | 11 (64.7) | 17 (68.0) |
| Sensitive, but intolerant b (Pt-S) | 1 (9.1) | 5 (29.4) | 5 (20.0) |
| BRCA status, n (%) | |||
| BRCA1 mutation | 1 (9.1) | 3 (17.6) | 3 (12.0) |
| BRCA2 mutation | 0 (0.0) | 1 (5.9) | 1 (4.0) |
| Negative | 7 (63.6) | 12 (70.6) | 17 (68.0) |
| Unknown | 3 (27.3) | 1 (5.9) | 4 (16.0) |
| Number (%) of Patients with Adverse Event, MedDRA Preferred Term | Part A | Patients Receiving MTD a | All Patients | |||
|---|---|---|---|---|---|---|
| n = 11 | n = 17 | n = 25 | ||||
| Monotherapy | Grade 1–2 | Grade 3 b | Grade 1–2 | Grade 3 b | Grade 1–2 | Grade 3 b |
| Any adverse event | 4 (36.4) | 5 (45.5) | 5 (29.4) | 7 (41.2) | 8 (32.0) | 10 (40.0) |
| Abdominal pain | 4 (36.4) | 3 (27.3) | 1 (5.9) | 4 (23.5) | 5 (20.0) | 5 (20.0) |
| Vomiting | 4 (36.4) | 1 (9.1) | 6 (35.3) | 1 (5.9) | 8 (32.0) | 2 (8.0) |
| Nausea | 4 (36.4) | 1 (9.1) | 4 (23.5) | 0 (0.0) | 6 (24.0) | 1 (4.0) |
| Diarrhea | 3 (27.3) | 0 (0.0) | 2 (11.8) | 0 (0.0) | 4 (16.0) | 0 (0.0) |
| Abdominal distension | 2 (18.2) | 0 (0.0) | 3 (17.6) | 0 (0.0) | 4 (16.0) | 0 (0.0) |
| Abdominal discomfort | 1 (9.1) | 0 (0.0) | 1 (5.9) | 0 (0.0) | 2 (8.0) | 0 (0.0) |
| Ileus | 0 (0.0) | 2 (18.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (8.0) |
| Fatigue | 5 (45.5) | 0 (0.0) | 3 (17.6) | 0 (0.0) | 8 (32.0) | 0 (0.0) |
| Combination therapy | ||||||
| Any adverse event | 2 (18.2) | 1 (9.1) | 2(11.8) | 2(11.8) | 3 (12.0) | 2 (8.0) |
| Abdominal pain | 2 (18.2) | 0 (0.0) | 2 (11.8) | 0 (0.0) | 3 (12.0) | 0 (0.0) |
| Neutropenia | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.9) | 0 (0.0) | 2 (8.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coward, J.I.; Barve, M.A.; Kichenadasse, G.; Moore, K.N.; Harnett, P.R.; Berg, D.; Garner, J.S.; Dizon, D.S. Maximum Tolerated Dose and Anti-Tumor Activity of Intraperitoneal Cantrixil (TRX-E-002-1) in Patients with Persistent or Recurrent Ovarian Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer: Phase I Study Results. Cancers 2021, 13, 3196. https://doi.org/10.3390/cancers13133196
Coward JI, Barve MA, Kichenadasse G, Moore KN, Harnett PR, Berg D, Garner JS, Dizon DS. Maximum Tolerated Dose and Anti-Tumor Activity of Intraperitoneal Cantrixil (TRX-E-002-1) in Patients with Persistent or Recurrent Ovarian Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer: Phase I Study Results. Cancers. 2021; 13(13):3196. https://doi.org/10.3390/cancers13133196
Chicago/Turabian StyleCoward, Jermaine I., Minal A. Barve, Ganessan Kichenadasse, Kathleen N. Moore, Paul R. Harnett, Daniel Berg, James S. Garner, and Don S. Dizon. 2021. "Maximum Tolerated Dose and Anti-Tumor Activity of Intraperitoneal Cantrixil (TRX-E-002-1) in Patients with Persistent or Recurrent Ovarian Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer: Phase I Study Results" Cancers 13, no. 13: 3196. https://doi.org/10.3390/cancers13133196
APA StyleCoward, J. I., Barve, M. A., Kichenadasse, G., Moore, K. N., Harnett, P. R., Berg, D., Garner, J. S., & Dizon, D. S. (2021). Maximum Tolerated Dose and Anti-Tumor Activity of Intraperitoneal Cantrixil (TRX-E-002-1) in Patients with Persistent or Recurrent Ovarian Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer: Phase I Study Results. Cancers, 13(13), 3196. https://doi.org/10.3390/cancers13133196