
Transcript-Level Dysregulation of BCL2 Family Genes in Acute Myeloblastic Leukemia


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
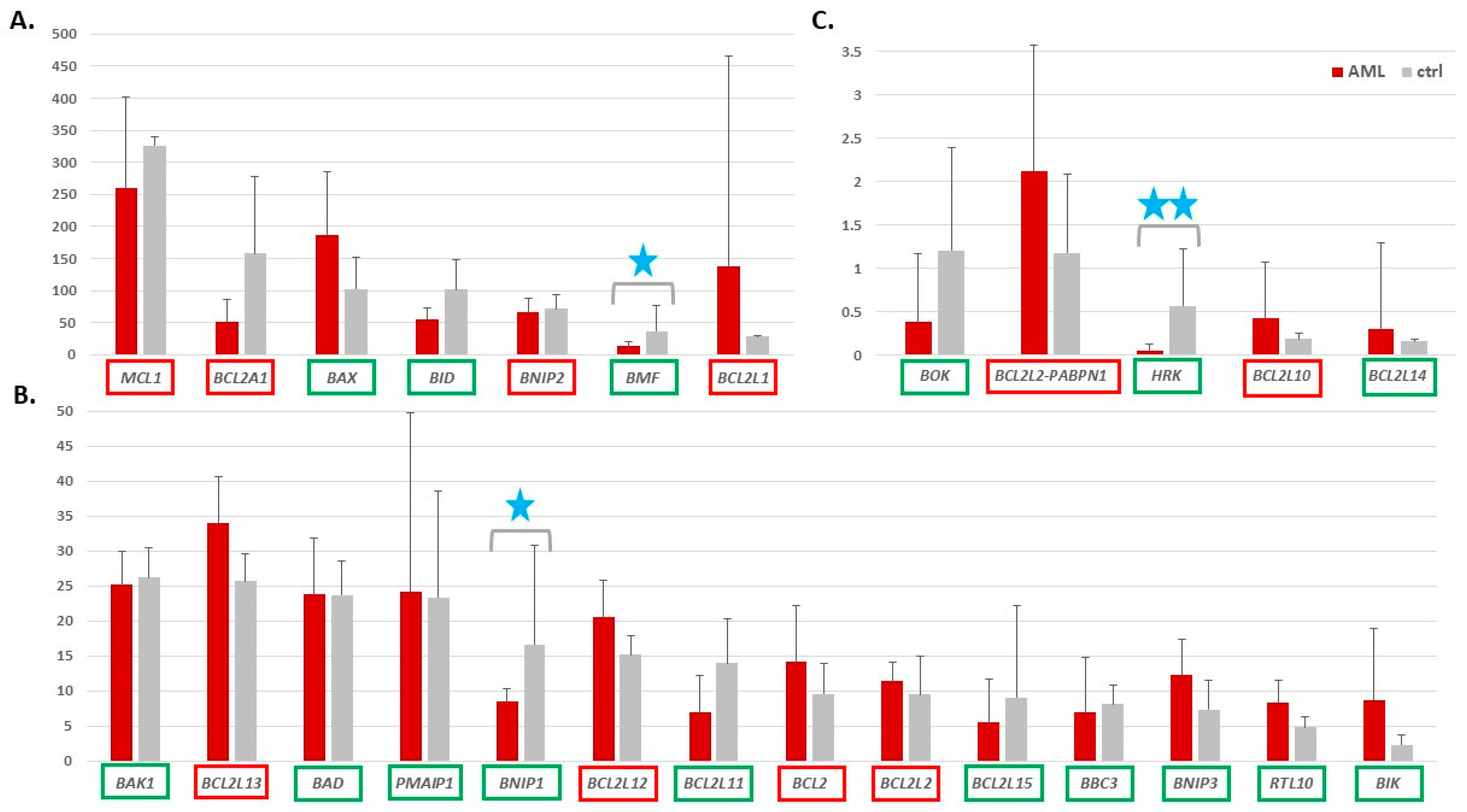
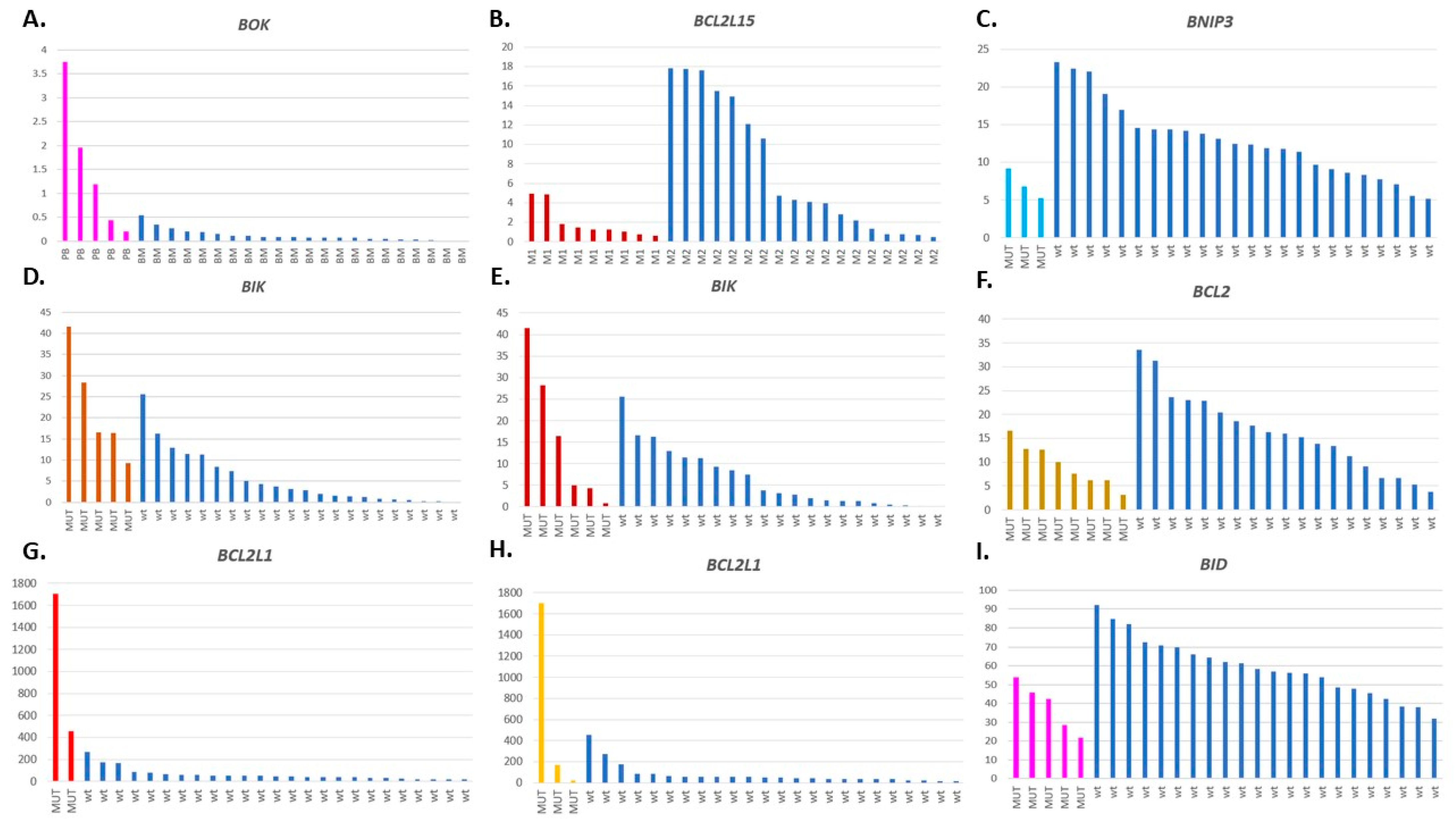
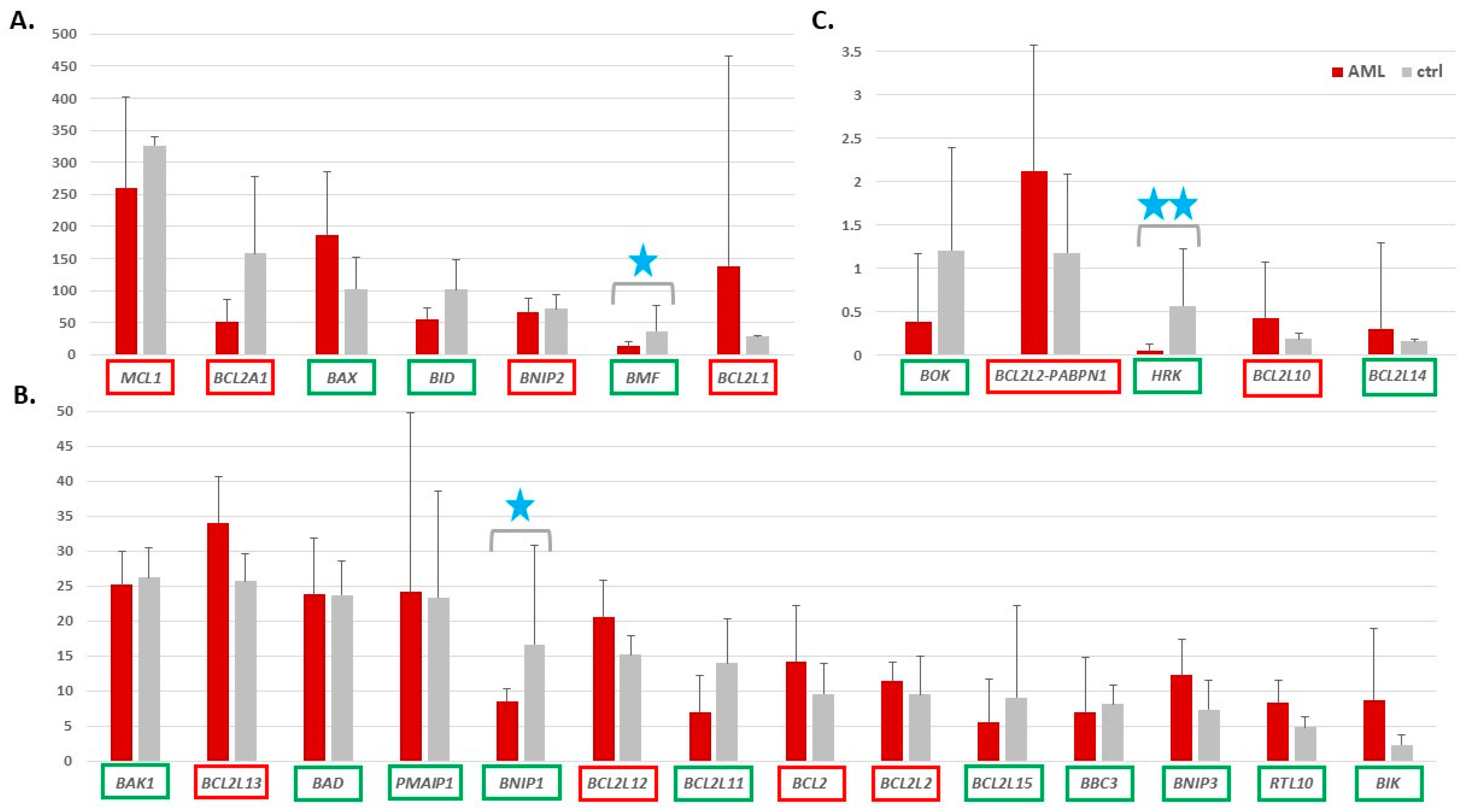
3.1. Expression of Genes Encoding Proteins from the BCL2 Family
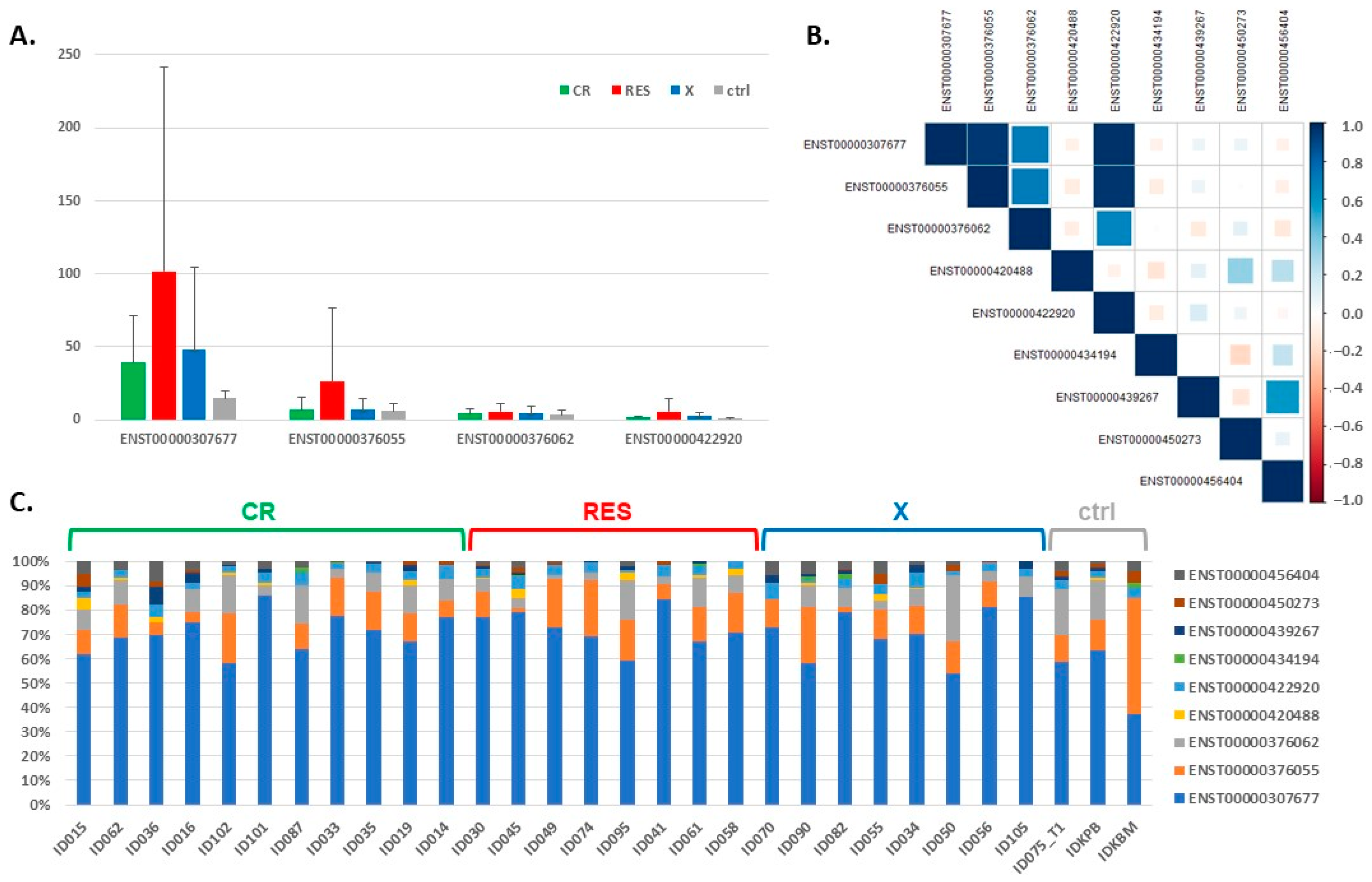
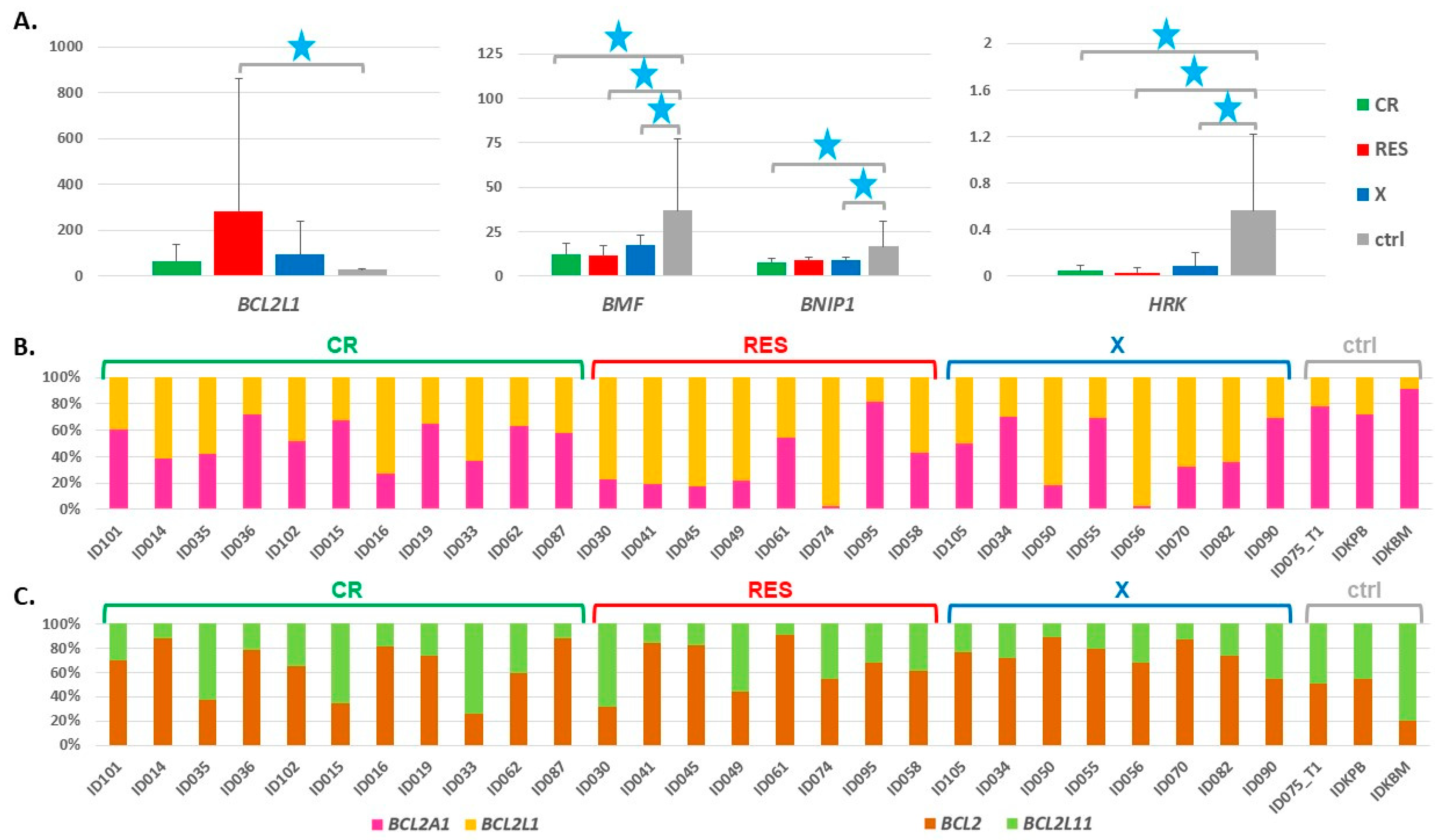
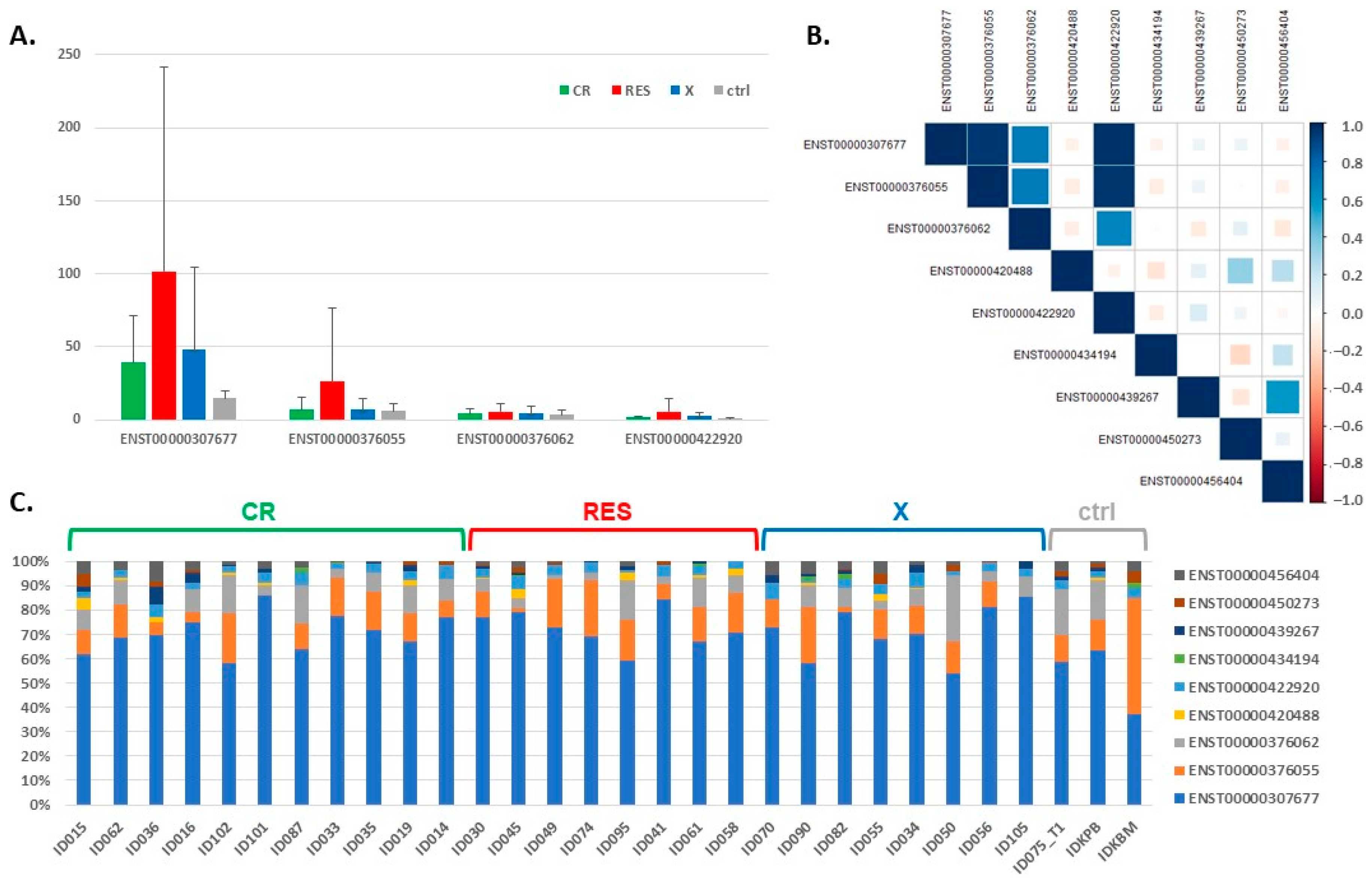
3.2. Expression of the BCL2L1 Gene Isoforms
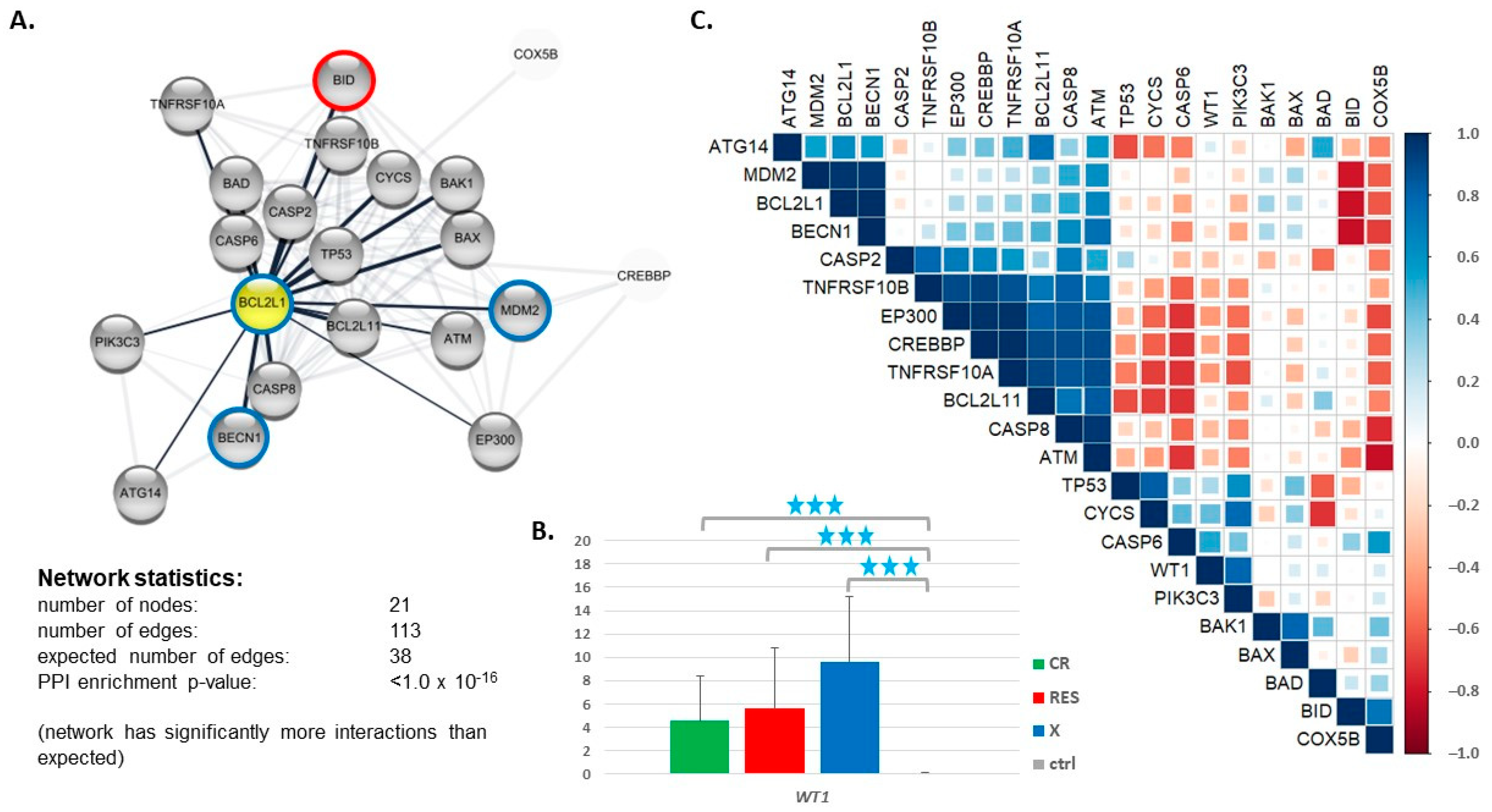
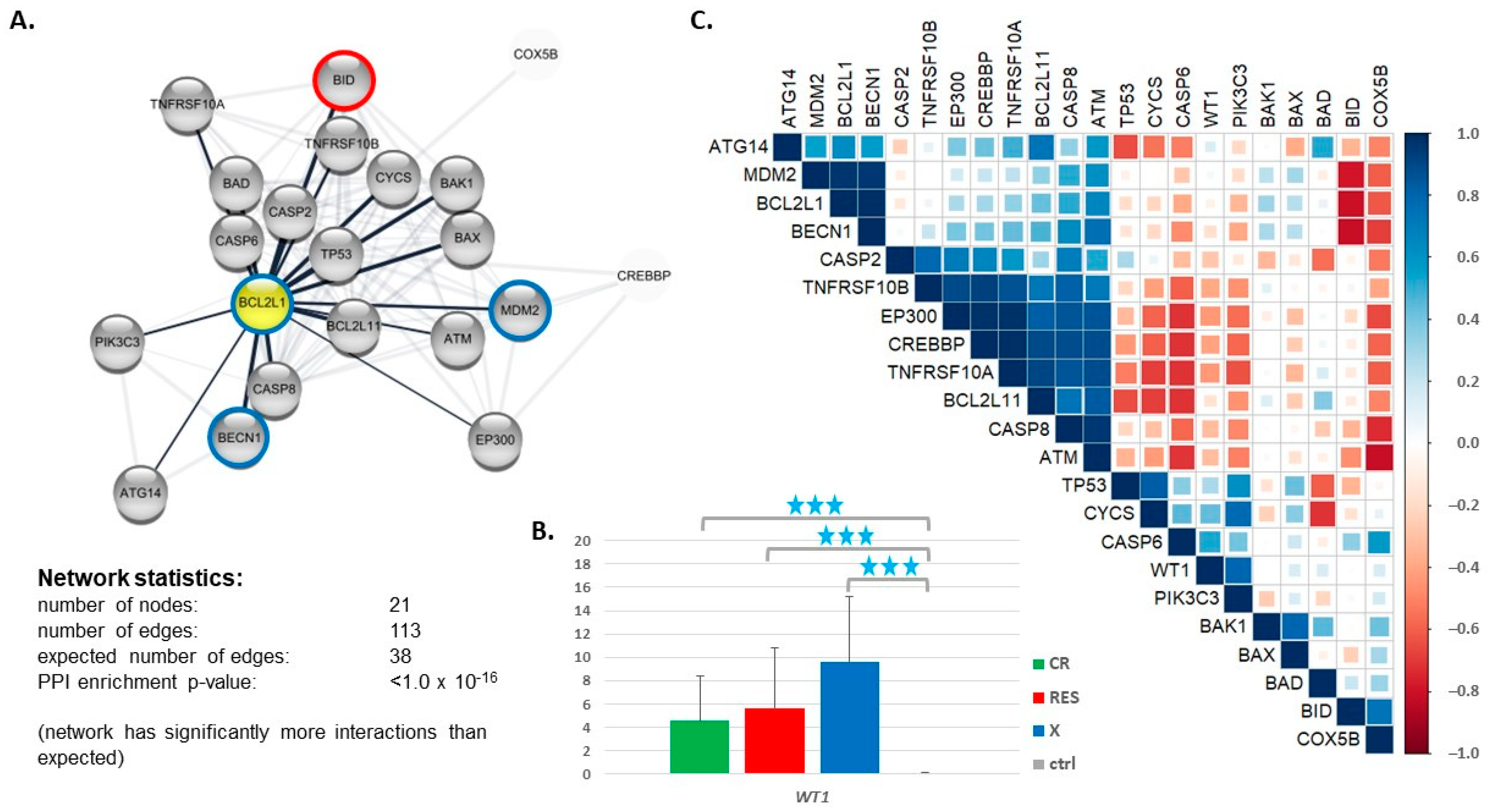
3.3. Expression of Genes Encoding Proteins Interacting with BCL2L1
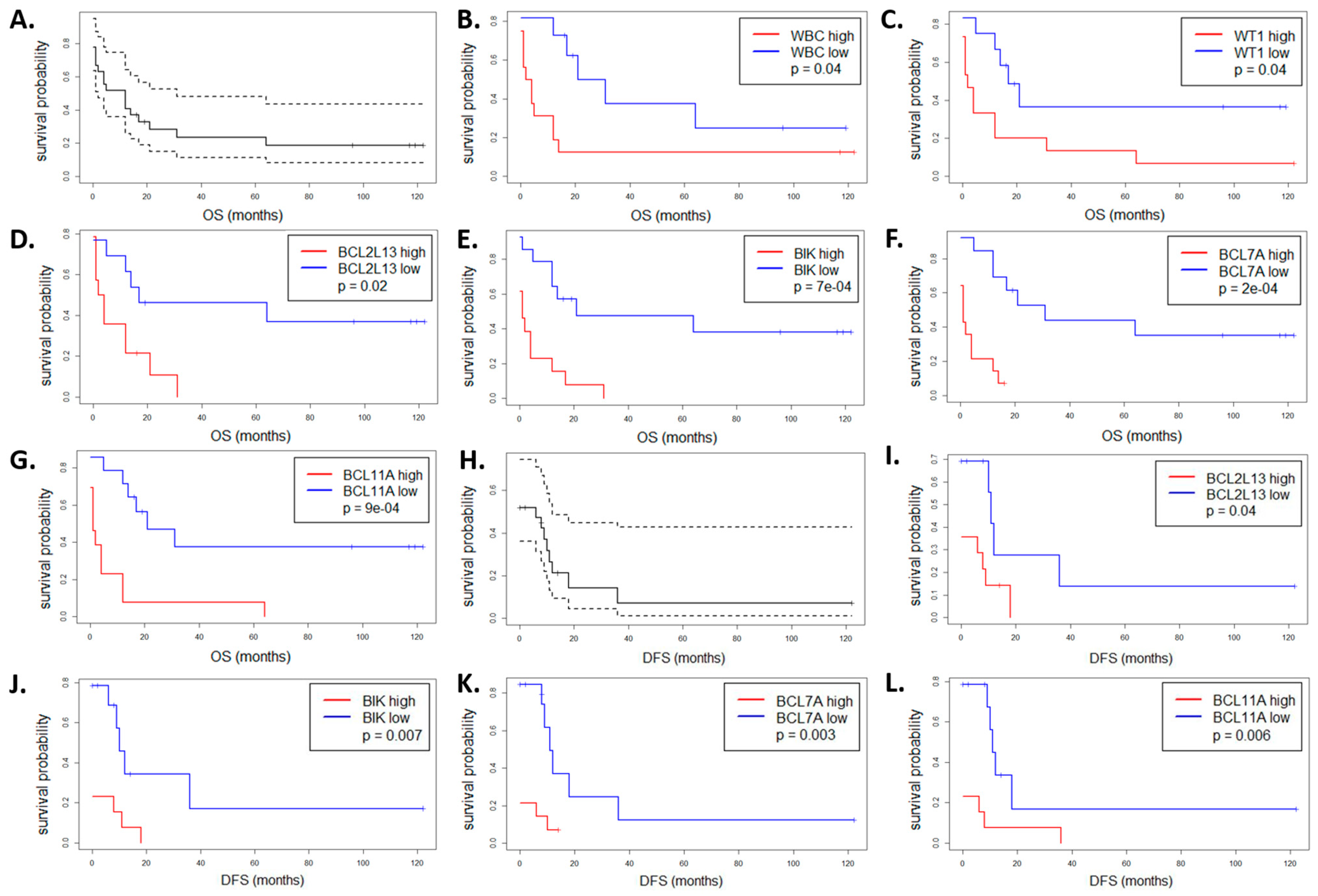
3.4. Impact of the BCL2 Gene Family Expression Levels on the Patient Outcome
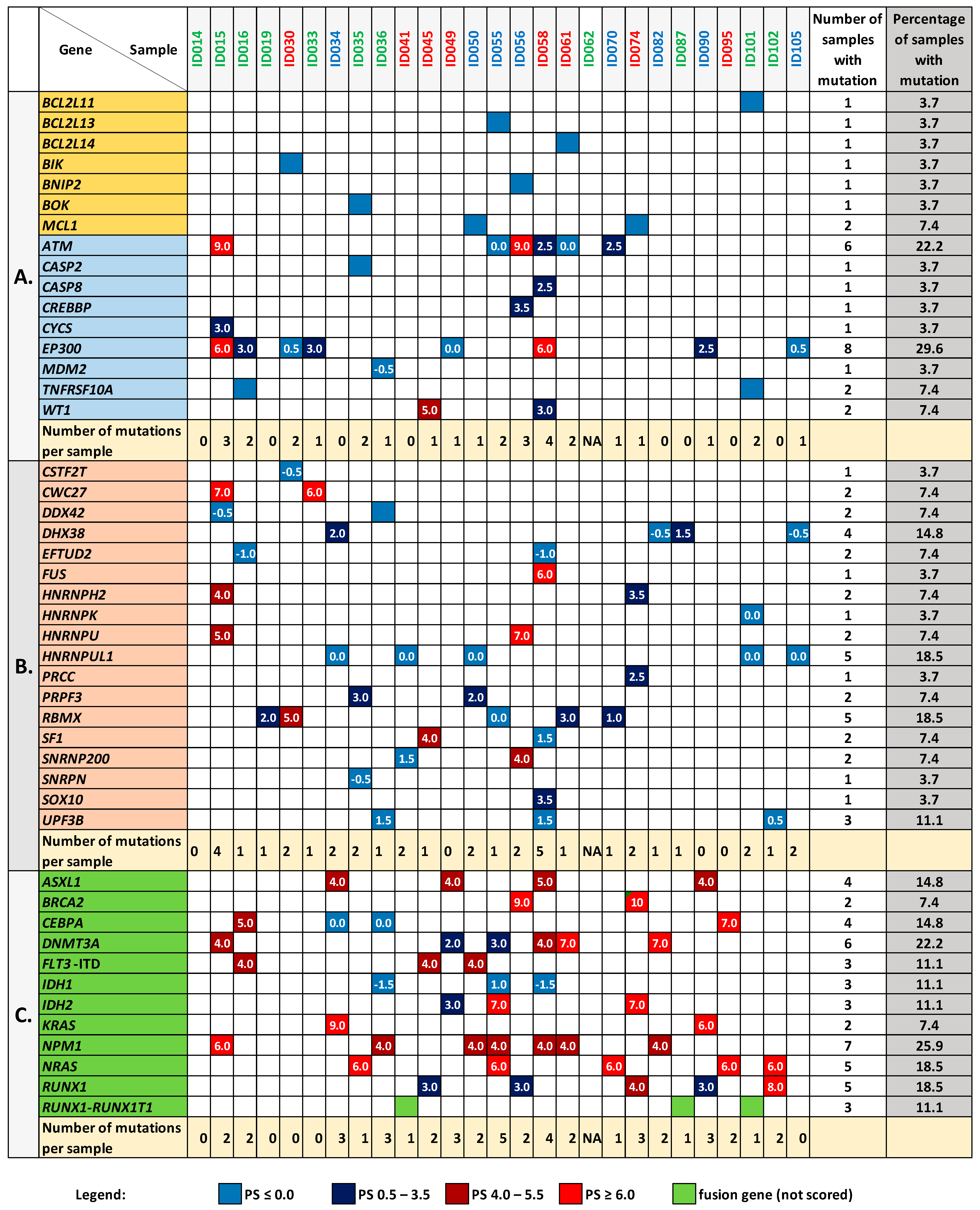
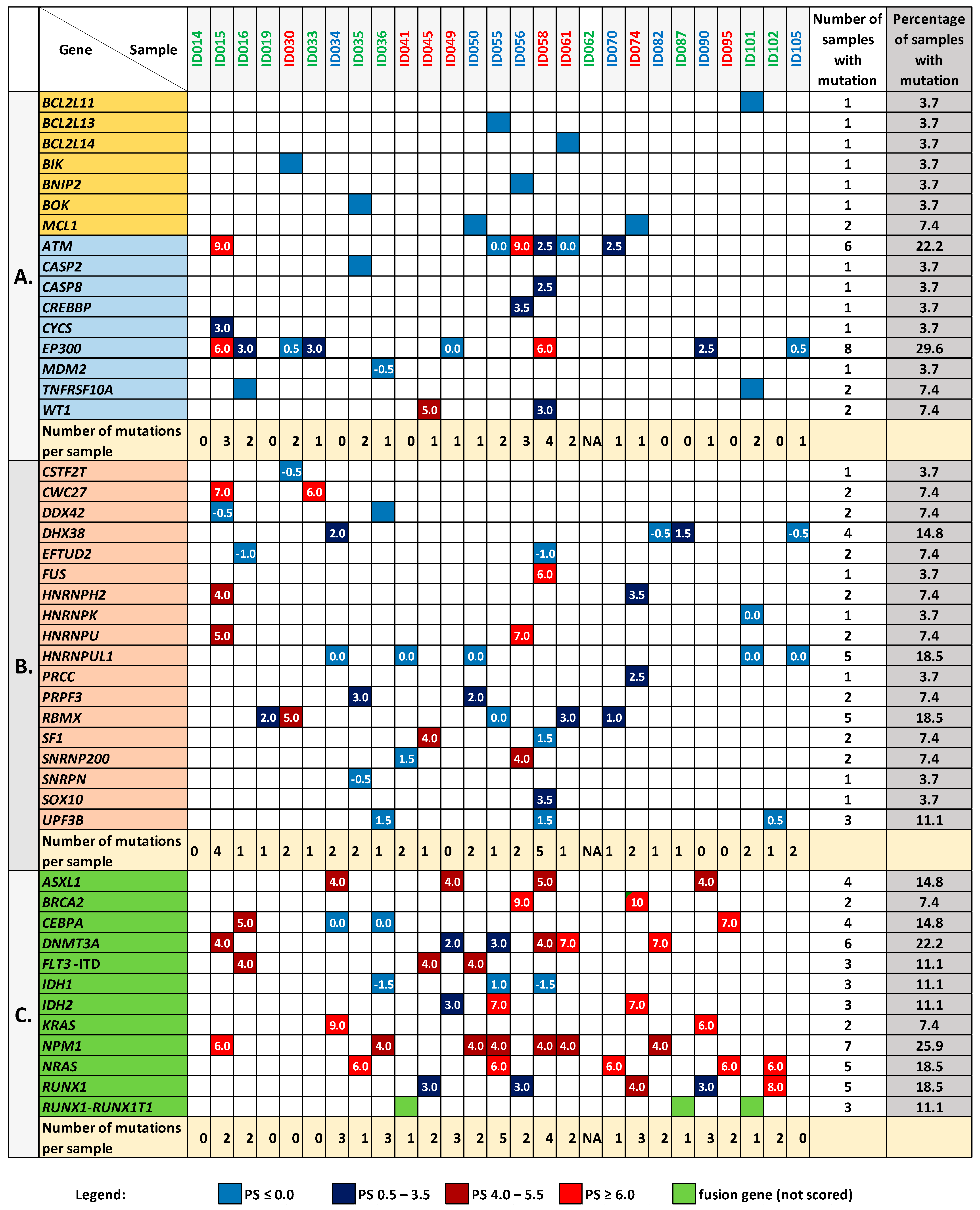
3.5. Mutation Analysis of the BCL2 Family Genes and Genes Encoding Proteins Interacting with BCL2L1
3.6. Mutation Analysis in the Genes Encoding Splicing-Associated Proteins
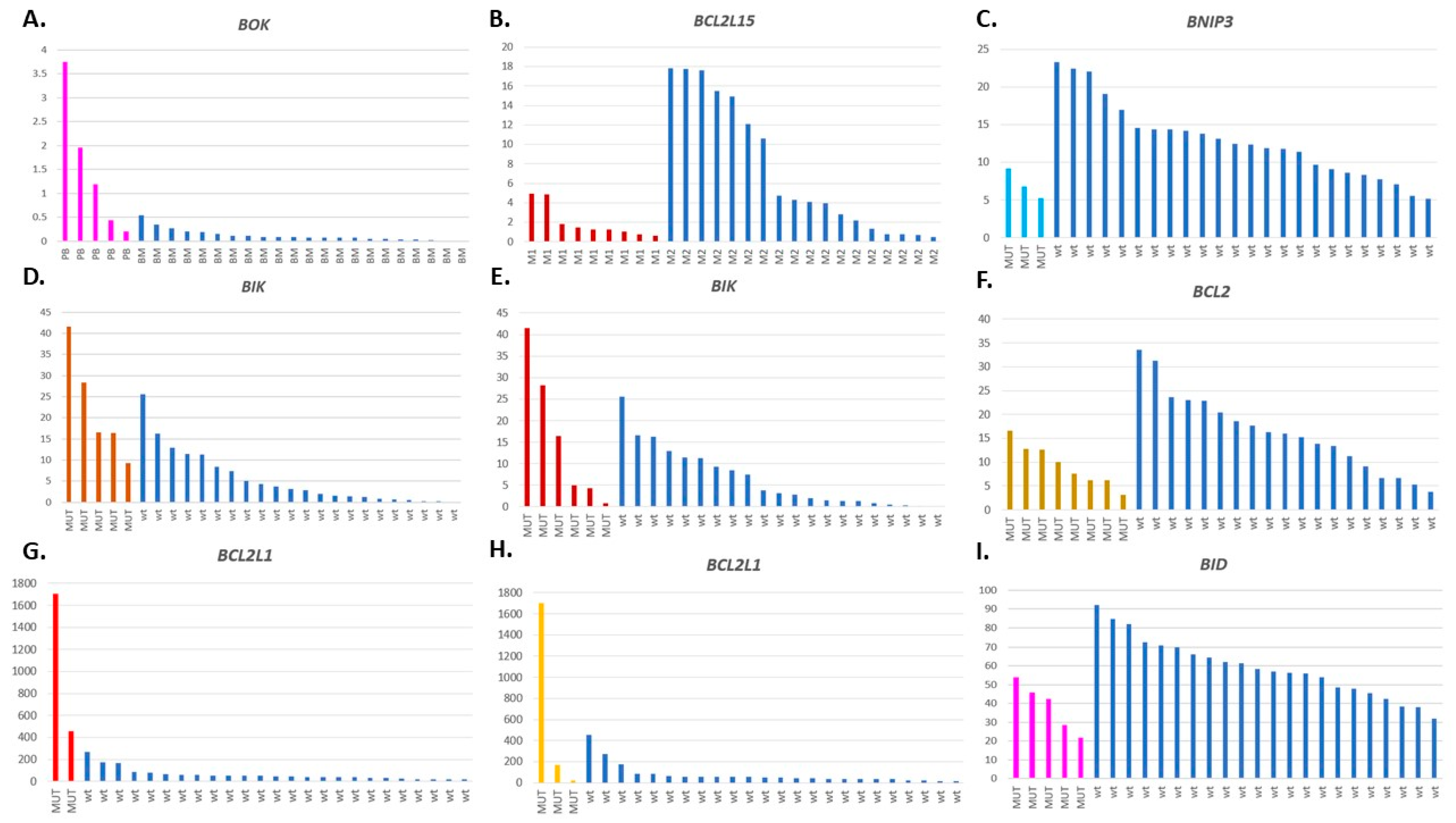
3.7. Relations between the BCL2 Family Gene Expression, Mutation Status, and Other Clinical Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Deschler, B.; Lübbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [CrossRef] [Green Version]
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Handschuh, L. Not Only Mutations Matter: Molecular Picture of Acute Myeloid Leukemia Emerging from Transcriptome Studies. J. Oncol. 2019, 2019, 1–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, S.L.; Cotter, T.G. Functional Aspects of Apoptosis in Hematopoiesis and Consequences of Failure. Adv. Cancer Res. 1997, 71, 121–164. [Google Scholar] [PubMed]
- Reed, J.C.; Pellecchia, M. Apoptosis-based therapies for hematologic malignancies. Blood 2005, 106, 408–418. [Google Scholar] [CrossRef]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatok, J.; Racay, P. Bcl-2 family proteins: Master regulators of cell survival. Biomol. Concepts 2016, 7, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardwick, J.M.; Soane, L. Multiple Functions of BCL-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risberg, K.; Redalen, K.R.; Sønstevold, L.; Bjørnetrø, T.; Sølvernes, J.; Ree, A.H. Pro-survival responses to the dual inhibition of anti-apoptotic Bcl-2 family proteins and mTOR-mediated signaling in hypoxic colorectal carcinoma cells. BMC Cancer 2016, 16, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Akgul, C.; Moulding, D.A.; Edwards, S.W. Alternative splicing of Bcl-2-related genes: Functional consequences and potential therapeutic applications. Cell. Mol. Life Sci. 2004, 61, 2189–2199. [Google Scholar] [CrossRef]
- Reed, J.C. Bcl-2 family proteins. Oncogene 1998, 17, 3225–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Popgeorgiev, N.; Jabbour, L.; Gillet, G. Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front. Cell Dev. Biol. 2018, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juin, P.; Geneste, O.; Gautier, F.; Depil, S.; Campone, M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat. Rev. Cancer 2013, 13, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 is a Mechanism of Intrinsic Resistance to ABT-199 which can be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensi, L.; Longo, R.; Vecchi, A.; Messora, C.; Garagnani, L.; Bernardi, S.; Tamassia, M.G.; Sacchi, S. Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica 1995, 80, 98–102. [Google Scholar] [PubMed]
- Lauria, F.; Raspadori, D.; Rondelli, D.; Ventura, M.; Fiacchini, M.; Visani, G.; Forconi, F.; Tura, S. High bcl-2 expression in acute myeloid leukemia cells correlates with CD34 positivity and complete remission rate. Leukemia 1997, 11, 2075–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, T.; Xu, Z.; Gu, Y.; Ma, J.; Li, X.; Guo, H.; Wen, X.; Zhang, W.; Yang, L.; et al. BCL2 overexpression: Clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 2019, 14, 68. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- Lessene, G.; Czabotar, P.E.; Colman, P.M. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 989–1000. [Google Scholar] [CrossRef]
- Malpas, J.; Mathé, G.; Hayat, M. Comparison of the effects of cytosine arabinoside, cytosine arabinoside and thioguanine, and cytosine arabinoside and daunorubicin in acute myeloid leukemia. Bull. Cancer 1974, 61, 411–418. [Google Scholar]
- Lichtman, M.A. A historical perspective on the development of the cytarabine (7days) and daunorubicin (3days) treatment regimen for acute myelogenous leukemia: 2013 the 40th anniversary of 7+3. Blood Cells. Mol. Dis. 2013, 50, 119–130. [Google Scholar] [CrossRef]
- Preliminary results of a trial on acute myeloid leukemia comparing the effect of cytosine arabinoside, the combination of cytosine arabinoside and thioguanine and the combination of cytosine arabinoside and daunorubicine. Recent Results Cancer Res. Fortschritte Krebsforsch. Progres Dans Rech. Sur Cancer 1973, 43, 165–168.
- Bose, P.; Vachhani, P.; Cortes, J.E. Treatment of Relapsed/Refractory Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2017, 18, 17. [Google Scholar] [CrossRef]
- Rowe, J.M.; Tallman, M.S. How I treat acute myeloid leukemia. Blood 2010, 116, 3147–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, J.M. Will new agents impact survival in AML? Best Pract. Res. Clin. Haematol. 2019, 32, 101094. [Google Scholar] [CrossRef]
- Dlamini, Z.; Shoba, B.; Hull, R. Splicing machinery genomics events in acute myeloid leukaemia (AML): In search for therapeutic targets, diagnostic and prognostic biomarkers. Am. J. Cancer Res. 2020, 10, 2690–2704. [Google Scholar] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3 -Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Bogenberger, J.M.; Tibes, R. Targeting Apoptosis in Acute Myeloid Leukemia: Current Status and Future Directions of BCL-2 Inhibition with Venetoclax and Beyond. Target. Oncol. 2020, 15, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, L.; Kaźmierczak, M.; Milewski, M.; Goralski, M.; Łuczak, M.; Wojtaszewska, M.; Uszczyńska-Ratajczak, B.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Gene expression profiling of acute myeloid leukemia samples from adult patients with AML-M1 and -M2 through boutique microarrays, real-time PCR and droplet digital PCR. Int. J. Oncol. 2018, 52, 656–678. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, L.; Wojciechowski, P.; Kazmierczak, M.; Marcinkowska-Swojak, M.; Luczak, M.; Lewandowski, K.; Komarnicki, M.; Blazewicz, J.; Figlerowicz, M.; Kozlowski, P. NPM1 alternative transcripts are upregulated in acute myeloid and lymphoblastic leukemia and their expression level affects patient outcome. J. Transl. Med. 2018, 16, 232. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Cheson, B.D.; Bennett, J.M.; Kopecky, K.J.; Büchner, T.; Willman, C.L.; Estey, E.H.; Schiffer, C.A.; Doehner, H.; Tallman, M.S.; Lister, T.A.; et al. Revised Recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J. Clin. Oncol. 2003, 21, 4642–4649. [Google Scholar] [CrossRef]
- de Greef, G.E.; van Putten, W.L.J.; Boogaerts, M.; Huijgens, P.C.; Verdonck, L.F.; Vellenga, E.; Theobald, M.; Jacky, E.; Lowenberg, B.; The Dutch-Belgian Hemato-Oncology Co-operative Group HOVON; et al. Criteria for defining a complete remission in acute myeloid leukaemia revisited. An analysis of patients treated in HOVON-SAKK co-operative group studies. Br. J. Haematol. 2005, 128, 184–191. [Google Scholar] [CrossRef]
- Bloomfield, C.D.; Estey, E.; Pleyer, L.; Schuh, A.C.; Stein, E.M.; Tallman, M.S.; Wei, A. Time to repeal and replace response criteria for acute myeloid leukemia? Blood Rev. 2018, 32, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Singh, S.; Chen, X.; Zheng, Z.; Huang, J.; Lin, T.; Li, H. The landscape of chimeric RNAs in bladder urothelial carcinoma. Int. J. Biochem. Cell Biol. 2019, 110, 50–58. [Google Scholar] [CrossRef]
- Del Gaizo Moore, V.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Invest. 2007, 117, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Boise, L.H.; González-García, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nuñez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Bansal, H.; Seifert, T.; Bachier, C.; Rao, M.; Tomlinson, G.; Iyer, S.P.; Bansal, S. The Transcription Factor Wilms Tumor 1 Confers Resistance in Myeloid Leukemia Cells against the Proapoptotic Therapeutic Agent TRAIL (Tumor Necrosis Factor α-related Apoptosis-inducing Ligand) by Regulating the Antiapoptotic Protein Bcl-xL. J. Biol. Chem. 2012, 287, 32875–32880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domen, J. The Role of Apoptosis in Regulating Hematopoiesis and Hematopoietic Stem Cells. Immunol. Res. 2000, 22, 83–94. [Google Scholar] [CrossRef]
- Lin, J.-C.; Tsao, M.-F.; Lin, Y.-J. Differential Impacts of Alternative Splicing Networks on Apoptosis. Int. J. Mol. Sci. 2016, 17, 2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cory, S. Regulation of Lymphocyte Survival by the BCL-2 Gene Family. Annu. Rev. Immunol. 1995, 13, 513–543. [Google Scholar] [CrossRef]
- Vikström, I.B.; Slomp, A.; Carrington, E.M.; Moesbergen, L.M.; Chang, C.; Kelly, G.L.; Glaser, S.P.; Jansen, J.H.M.; Leusen, J.H.W.; Strasser, A.; et al. MCL-1 is required throughout B-cell development and its loss sensitizes specific B-cell subsets to inhibition of BCL-2 or BCL-XL. Cell Death Dis. 2016, 7, e2345. [Google Scholar] [CrossRef] [Green Version]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Overexpression of the human BCL-2 gene product results in growth enhancement of Epstein-Barr virus-immortalized B cells. Proc. Natl. Acad. Sci. USA 1989, 86, 1958–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairbairn, L.J.; Cowling, G.J.; Reipert, B.M.; Dexter, T.M. Suppression of apoptosis allows differentiation and development of a multipotent hemopoietic cell line in the absence of added growth factors. Cell 1993, 74, 823–832. [Google Scholar] [CrossRef]
- Zhou, P.; Qian, L.; Bieszczad, C.K.; Noelle, R.; Binder, M.; Levy, N.B.; Craig, R.W. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood 1998, 92, 3226–3239. [Google Scholar] [CrossRef] [PubMed]
- Maung, Z.T.; MacLean, F.R.; Reid, M.M.; Pearson, A.D.J.; Proctor, S.J.; Hamilton, P.J.; Hall, A.G. The relationship between bcl-2 expression and response to chemotherapy in acute leukaemia. Br. J. Haematol. 1994, 88, 105–109. [Google Scholar] [CrossRef]
- Bincolettu, C.; Saad, S.T.O.; Silva, E.S.; Queiroz, M.L.S. Haematopoietic response and bcl-2 expression in patients with acute myeloid leukaemia. Eur. J. Haematol. 2009, 62, 38–42. [Google Scholar] [CrossRef]
- Nagy, B.; Lundán, T.; Larramendy, M.L.; Aalto, Y.; Zhu, Y.; Niini, T.; Edgren, H.; Ferrer, A.; Vilpo, J.; Elonen, E.; et al. Abnormal expression of apoptosis-related genes in haematological malignancies: Overexpression of MYC is poor prognostic sign in mantle cell lymphoma. Br. J. Haematol. 2003, 120, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillet, P.; Strasser, A. BH3-only proteins—Evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 2002, 115, 1567–1574. [Google Scholar] [CrossRef]
- Willis, S.N. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallis, M.; Zhu, Y.-M.; Russell, N. Bcl-xL is heterogeneously expressed by acute myeloblastic leukaemia cells and is associated with autonomous growth in vitro and with P-glycoprotein expression. Leukemia 1997, 11, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.M.; Mitra, R.; Vogel, A.N.; Liu, J.; Gong, J.Z.; Eischen, C.M. Targeting BCL-W and BCL-XL as a therapeutic strategy for Hodgkin lymphoma. Leukemia 2020, 34, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Leiter, U.; Schmid, R.M.; Kaskel, P.; Peter, R.U.; Krähn, G. Antiapoptotic bcl-2 and bcl-xL in advanced malignant melanoma. Arch. Dermatol. Res. 2000, 292, 225–232. [Google Scholar] [CrossRef]
- Liu, J.R.; Fletcher, B.; Page, C.; Hu, C.; Nunez, G.; Baker, V. Bcl-xLIs Expressed in Ovarian Carcinoma and Modulates Chemotherapy-Induced Apoptosis. Gynecol. Oncol. 1998, 70, 398–403. [Google Scholar] [CrossRef]
- España, L.; Fernández, Y.; Rubio, N.; Torregrosa, A.; Blanco, J.; Sierra, A. Overexpression of Bcl-xL in Human Breast Cancer Cells Enhances Organ-Selective Lymph Node Metastasis. Breast Cancer Res. Treat. 2004, 87, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Castilla, C.; Congregado, B.; Chinchón, D.; Torrubia, F.J.; Japón, M.A.; Sáez, C. Bcl-xL Is Overexpressed in Hormone-Resistant Prostate Cancer and Promotes Survival of LNCaP Cells via Interaction with Proapoptotic Bak. Endocrinology 2006, 147, 4960–4967. [Google Scholar] [CrossRef] [PubMed]
- Scherr, A.-L.; Mock, A.; Gdynia, G.; Schmitt, N.; Heilig, C.E.; Korell, F.; Rhadakrishnan, P.; Hoffmeister, P.; Metzeler, K.H.; Schulze-Osthoff, K.; et al. Identification of BCL-XL as highly active survival factor and promising therapeutic target in colorectal cancer. Cell Death Dis. 2020, 11, 875. [Google Scholar] [CrossRef]
- Sochalska, M.; Tuzlak, S.; Egle, A.; Villunger, A. Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: Implications for targeted therapy. FEBS J. 2015, 282, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Rinkenberger, J.L.; Horning, S.; Klocke, B.; Roth, K.; Korsmeyer, S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000, 14, 23–27. [Google Scholar]
- Vogler, M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.; Schneider, P.A.; Dai, H.; Dogan, A.; Maurer, M.J.; Church, A.K.; Novak, A.J.; Feldman, A.L.; Wu, X.; Ding, H.; et al. BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood 2015, 125, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Inokuchi, K.; Tarusawa, M.; Dan, K. Mutation of bcl-x gene in non-Hodgkin’s lymphoma. Am. J. Hematol. 2002, 69, 74–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Cho, S.-Y.; Kim, H.; Na, D.; Han, J.Y.; Chae, J.; Park, C.; Park, O.-K.; Min, S.; Kang, J.; et al. Genomic alterations in BCL2L1 and DLC1 contribute to drug sensitivity in gastric cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 12492–12497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Nam, S.W.; Kim, C.J.; Cho, Y.G.; Lee, J.H.; Kim, H.S.; Park, W.S.; Kim, S.H.; et al. Inactivating mutations of proapoptotic Bad gene in human colon cancers. Carcinogenesis 2004, 25, 1371–1376. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Briggs, J.M. Functional Implications of the spectrum of BCL2 mutations in Lymphoma. Mutat. Res. Mutat. Res. 2016, 769, 1–18. [Google Scholar] [CrossRef]
- Fresquet, V.; Rieger, M.; Carolis, C.; García-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef] [Green Version]
- Bilbao-Sieyro, C.; Rodríguez-Medina, C.; Florido, Y.; Stuckey, R.; Sáez, M.N.; Sánchez-Sosa, S.; González Martín, J.M.; Santana, G.; González-Pérez, E.; Cruz-Cruz, N.; et al. BCL2 Expression at Post-Induction and Complete Remission Impact Outcome in Acute Myeloid Leukemia. Diagnostics 2020, 10, 1048. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Röck, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1364–1372. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, A.Y.; Salmon-Divon, M.; Zahavi, T.; Barash, Y.; Levy-Drummer, R.S.; Jacob-Hirsch, J.; Peretz, T. Determination of Molecular Markers for BRCA1 and BRCA2 Heterozygosity Using Gene Expression Profiling. Cancer Prev. Res. 2013, 6, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsen, K.; Sørensen, K.D.; Brems-Eskildsen, A.S.; Modin, C.; Gaustadnes, M.; Hein, A.-M.K.; Kruhøffer, M.; Laurberg, S.; Borre, M.; Wang, K.; et al. Alternative Splicing in Colon, Bladder, and Prostate Cancer Identified by Exon Array Analysis. Mol. Cell. Proteom. 2008, 7, 1214–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamia, S.; Haibe-Kains, B.; Pilarski, P.M.; Bar-Natan, M.; Pevzner, S.; Avet-Loiseau, H.; Lode, L.; Verselis, S.; Fox, E.A.; Burke, J.; et al. A Genome-Wide Aberrant RNA Splicing in Patients with Acute Myeloid Leukemia Identifies Novel Potential Disease Markers and Therapeutic Targets. Clin. Cancer Res. 2014, 20, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An Alternative Splicing Network Links Cell-Cycle Control to Apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Jeromin, S.; Bacher, U.; Bayer, K.; Dicker, F.; Eder, C.; Fasan, A.; Grossmann, V.; Kohlmann, A.; Kern, W.; Haferlach, C.; et al. SF3B1 Mutations Are Detectable in 48.9% of Acute Myeloid Leukemia with Normal Karyotype (AML-NK) and ≥15% Ring Sideroblasts and Are Closely Related to FLT3-ITD and RUNX1 Mutations. Blood 2012, 120, 406. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Jeromin, S.; Perglerová, K.; Haferlach, C.; Kern, W.; Haferlach, T. SF3B1 Mutations in AML, MDS and MDS/MPN-RS-T Are Accompanied By Different Other Gene Mutations: Impact for Targeted Treatment Studies. Blood 2016, 128, 1976. [Google Scholar] [CrossRef]
- Bamopoulos, S.A.; Batcha, A.M.N.; Jurinovic, V.; Rothenberg-Thurley, M.; Janke, H.; Ksienzyk, B.; Philippou-Massier, J.; Graf, A.; Krebs, S.; Blum, H.; et al. Clinical presentation and differential splicing of SRSF2, U2AF1 and SF3B1 mutations in patients with acute myeloid leukemia. Leukemia 2020, 34, 2621–2634. [Google Scholar] [CrossRef]
- Luczak, M.; Kaźmierczak, M.; Handschuh, L.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Comparative proteome analysis of acute myeloid leukemia with and without maturation. J. Proteom. 2012, 75, 5734–5748. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Fiebig, A.A.; Zhu, W.; Hollerbach, C.; Leber, B.; Andrews, D.W. Bcl-XL is qualitatively different from and ten times more effective than Bcl-2 when expressed in a breast cancer cell line. BMC Cancer 2006, 6, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Kline, M.P.; Rajkumar, S.V.; Timm, M.M.; Kimlinger, T.K.; Haug, J.L.; Lust, J.A.; Greipp, P.R.; Kumar, S. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia 2007, 21, 1549–1560. [Google Scholar] [CrossRef]
- Karakas, T.; Maurer, U.; Weidmann, E.; Miething, C.C.; Hoelzer, D.; Bergmann, L. High expression of bcl-2 mRNA as a determinant of poor prognosis in acute myeloid leukemia. Ann. Oncol. 1998, 9, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Karakas, T.; Miething, C.; Maurer, U.; Weidmann, E.; Ackermann, H.; Hoelzer, D.; Bergmann, L. The coexpression of the apoptosis-related genes bcl-2 and wt1 in predicting survival in adult acute myeloid leukemia. Leukemia 2002, 16, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Del Poeta, G.; Venditti, A.; Del Principe, M.I.; Maurillo, L.; Buccisano, F.; Tamburini, A.; Cox, M.C.; Franchi, A.; Bruno, A.; Mazzone, C.; et al. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood 2003, 101, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- De haes, I.; Dendooven, A.; Mercier, M.L.; Puylaert, P.; Vermeulen, K.; Kockx, M.; Deiteren, K.; Maes, M.-B.; Berneman, Z.; Anguille, S. Absence of BCL-2 Expression Identifies a Subgroup of AML with Distinct Phenotypic, Molecular, and Clinical Characteristics. J. Clin. Med. 2020, 9, 3090. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Jiang, S.; Zhang, X.; Konopleva, M.; Estrov, Z.; Snell, V.; Xie, Z.; Okcu, M.; Sanchez-Williams, G.; Dong, J.; et al. Expression of Bcl-2-related genes in normal and AML progenitors: Changes induced by chemotherapy and retinoic acid. Leukemia 1999, 13, 1881–1892. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Patients (F/M) | FAB Type (Number) | Age Median (Range) | WBC Count [×109/L] Mean (Range) | NPM1 mut. | FLT3 mut. | RUNX1/RUNX1T1 | Karyotype |
|---|---|---|---|---|---|---|---|---|
| CR | 11 (6/5) | M1 (3) M2 (8) | 52 (19–64) | 36.3 (3.6–111) | 2 | 2 | 2 | normal (1) aberrant (4) NA (6) |
| RES | 8 (3/5) | M1 (2) M2 (6) | 53.5 (21–64) | 62.17 (1.3–146) | 2 | 1 | 1 | normal (1) aberrant (4) NA (3) |
| X | 8 (4/4) | M1 (4) M2 (4) | 51 (25–65) | 90.43 (19.9–233) | 3 | 1 | - | normal (1) aberrant (2) NA (5) |
| All patients | 27 (13/14) | M1 (9) M2 (18) | 52 (19–65) | 60.0 (1.3–233) | 7 | 4 | 3 | normal (3) aberrant (10) NA (14) |
| Group | BCL2A1/BCL2L1 Ratio | BCL2/BCL2L11 Ratio |
|---|---|---|
| AML—mean ratio (st. dev.) CR—mean ratio (st. dev.) RES—mean ratio (st. dev.) | 1.15 (1.02) | 3.26 (2.53) |
| 1.31 (0.7) | 2.93 (2.43) | |
| 0.92 (1.44) | 3.16 (3.05) | |
| X—mean ratio (st. dev.) ctrl—mean ratio (st. dev.) | 1.15 (0.99) | 3.81 (2.3) |
| 5.67 (4.56) | 0.83 (0.50) | |
| AML vs. ctrl * CR vs. ctrl* | 0.0034 | 0.033 |
| 0.0110 | 0.1264 | |
| RES vs. ctrl * X vs. ctrl * | 0.0485 | 0.1333 |
| 0.0121 | 0.0121 | |
| RES vs. X * | 0.5054 | 0.3823 |
| RES vs. CR * | 0.0506 | 0.9678 |
| CR vs. X * | 0.5448 | 0.3950 |
| BCL2L1 Gene Isoform | Length of the Gene (Protein) | CR—Mean TPM (st. dev.) | RES—Mean TPM (st. dev.) | X—Mean TPM (st. dev.) | Ctrl—Mean TPM (st. dev.) |
|---|---|---|---|---|---|
| ENST00000307677 | 2574 bp (233 aa) | 39.14 (32.37) | 101.71 (140.27) | 48.01 (56.36) | 14.90 (4.68) |
| ENST00000376055 | 2496 bp (170 aa) | 7.05 (8.13) | 26.29 (50.13) | 6.89 (7.52) | 6.32 (5.18) |
| ENST00000376062 | 2578 bp (233 aa) | 4.49 (3.26) | 5.55 (5.80) | 4.67 (4.93) | 3.39 (2.95) |
| ENST00000420488 | 2444 bp (233 aa) | 0.41 (0.45) | 0.75 (0.88) | 0.22 (0.26) | 0.11 (0.19) |
| ENST00000422920 | 2227 bp (170 aa) | 1.82 (0.91) | 5.89 (8.84) | 2.44 (2.62) | 0.97 (0.15) |
| ENST00000434194 | 2441 bp (233 aa) | 0.10 (0.25) | 0.11 (0.19) | 0.23 (0.45) | 0.16 (0.28) |
| ENST00000439267 | 2426 bp (233 aa) | 0.48 (0.53) | 0.48 (0.51) | 0.63 (0.67) | 0.25 (0.22) |
| ENST00000450273 | 1292 bp (307 aa) | 0.35 (0.50) | 0.51 (0.52) | 0.48 (0.59) | 0.76 (0.40) |
| ENST00000456404 | 2562 bp (233 aa) | 0.97 (0.63) | 0.72 (0.83) | 1.20 (0.95) | 0.87 (0.47) |
| Expression of Genes | Associated Factor | p Value |
|---|---|---|
| BCL2L1, BID, BOK, HRK | BRCA2_status, RUNX1_status | 4.145 × 10−10 *** 0.04287 * |
| BCL2L1, BID, BOK, HRK | BRCA2 status | 2.106 × 10−9 *** |
| BCL2L1, BID, HRK | RUNX1 status | 0.0002187 *** |
| BCL2L10, BCL2L2, HRK | HNRNPU status | 8.038 × 10−7 *** |
| BCL2L1, BBC3, BOK, BCL2L10, BCL2L11 | HNRNPH2 status | 1.129 × 10−5 *** |
| BCL2L11, BCL2L13, BID, BIK, BNIP1 | WBC count | 6.459 × 10−5 *** |
| HRK, BAK1, BCL2A1, BID, BNIP1 | SNRNP200 status | 0.002168 ** |
| BCL2L10, BNIP2 | DDX42 status | 0.000545 *** |
| BCL2, BCL2A1, BCL2L10, BID, BNIP1, MCL1 | CWC27 status | 0.007744 ** |
| BMF, RTL10, BAX, BCL2L11, BCL2L2 | CEBPA status | 0.00383 ** |
| BCL2L13, BIK, PMAIP1 | RBMX status | 0.0004931 *** |
| MCL1, BCL2L11 | HNRNPUL1 status | 0.05703 |
| BCL2L2.PABPN1, BNIP2 | DHX38 status | 0.009772 ** |
| BCL2, BCL2L11, BCL2L12, BCL2L15 | EP300 status | 0.04793 * |
| BCL2L13, BCL2L2, BIK | ATM status | 0.003997 ** |
| BAX, BCL2A1, BNIP2 | KRAS status | 0.01254 * |
| BCL2L13, BCL2L2 | DNMT3A status | 0.02401 * |
| BNIP1, BCL2L2.PABPN1 | IDH1 status | 0.01978 * |
| BBC3, BCL2L1, BCL2L11, BOK, MCL1 | IDH2 status | 0.02641 * |
| BCL2L1, BOK | IDH2 status, MCL1 status | 0.001286 ** 0.007325 ** |
| BCL2L1, BOK | IDH2 status, MCL1 status, BRCA2 status | 1.755 × 10−5 *** 4.508 × 10−5 *** 4.624 × 10−6 *** |
| BCL2L1, BOK | MCL1 status | 0.001242 ** |
| BAD, BAK1, BCL2L11, BID, BNIP2, BNIP3 | RUNX1-RUNX1T1 status | 0.002101 ** |
| BCL2L13, BID, BIK, BNIP1 | NPM1 status | 0.02555 * |
| BAD, BAX, BCL2L1, BCL2L11, BCL2L13, BCL2L2.PABPN1, BID, BIK, BNIP1, BOK | Blast percentage in PB | 0.003099 ** |
| BCL2L1, BCL2L11, BCL2L15, BCL2L2.PABPN1, BID, BIK, BNIP1, BOK, HRK | Blast percentage in BM | 0.000261 *** |
| BAK1, BBC3, BCL2L1, BCL2L11, BCL2L12, BOK | Tissue type (PB, BM) | 0.0004833 *** |
| BCL2L15, BCL2L2, BNIP1, BNIP3 | FAB type (M1, M2) | 0.03326 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Handschuh, L.; Wojciechowski, P.; Kazmierczak, M.; Lewandowski, K. Transcript-Level Dysregulation of BCL2 Family Genes in Acute Myeloblastic Leukemia. Cancers 2021, 13, 3175. https://doi.org/10.3390/cancers13133175
Handschuh L, Wojciechowski P, Kazmierczak M, Lewandowski K. Transcript-Level Dysregulation of BCL2 Family Genes in Acute Myeloblastic Leukemia. Cancers. 2021; 13(13):3175. https://doi.org/10.3390/cancers13133175
Chicago/Turabian StyleHandschuh, Luiza, Pawel Wojciechowski, Maciej Kazmierczak, and Krzysztof Lewandowski. 2021. "Transcript-Level Dysregulation of BCL2 Family Genes in Acute Myeloblastic Leukemia" Cancers 13, no. 13: 3175. https://doi.org/10.3390/cancers13133175
APA StyleHandschuh, L., Wojciechowski, P., Kazmierczak, M., & Lewandowski, K. (2021). Transcript-Level Dysregulation of BCL2 Family Genes in Acute Myeloblastic Leukemia. Cancers, 13(13), 3175. https://doi.org/10.3390/cancers13133175