
Epigenetic Upregulation of MAGE-A Isoforms Promotes Breast Cancer Cell Aggressiveness

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Small Interfering RNA (siRNA) Transfection
2.3. Virus Infection for the Stable Cell Line
2.4. Quantitative Reverse-Transcription-Polymerase Chain Reaction (qRT-PCR)
2.5. Western Blot Analysis
2.6. Proliferation Assay
2.7. Migration and Invasion Assay
2.8. 3D Culture
2.9. ChIP-qPCR
2.10. RNA-Seq Library
2.11. ChIP-Seq Library Construction
2.12. Data Analysis
2.13. Statistical Analysis
3. Results
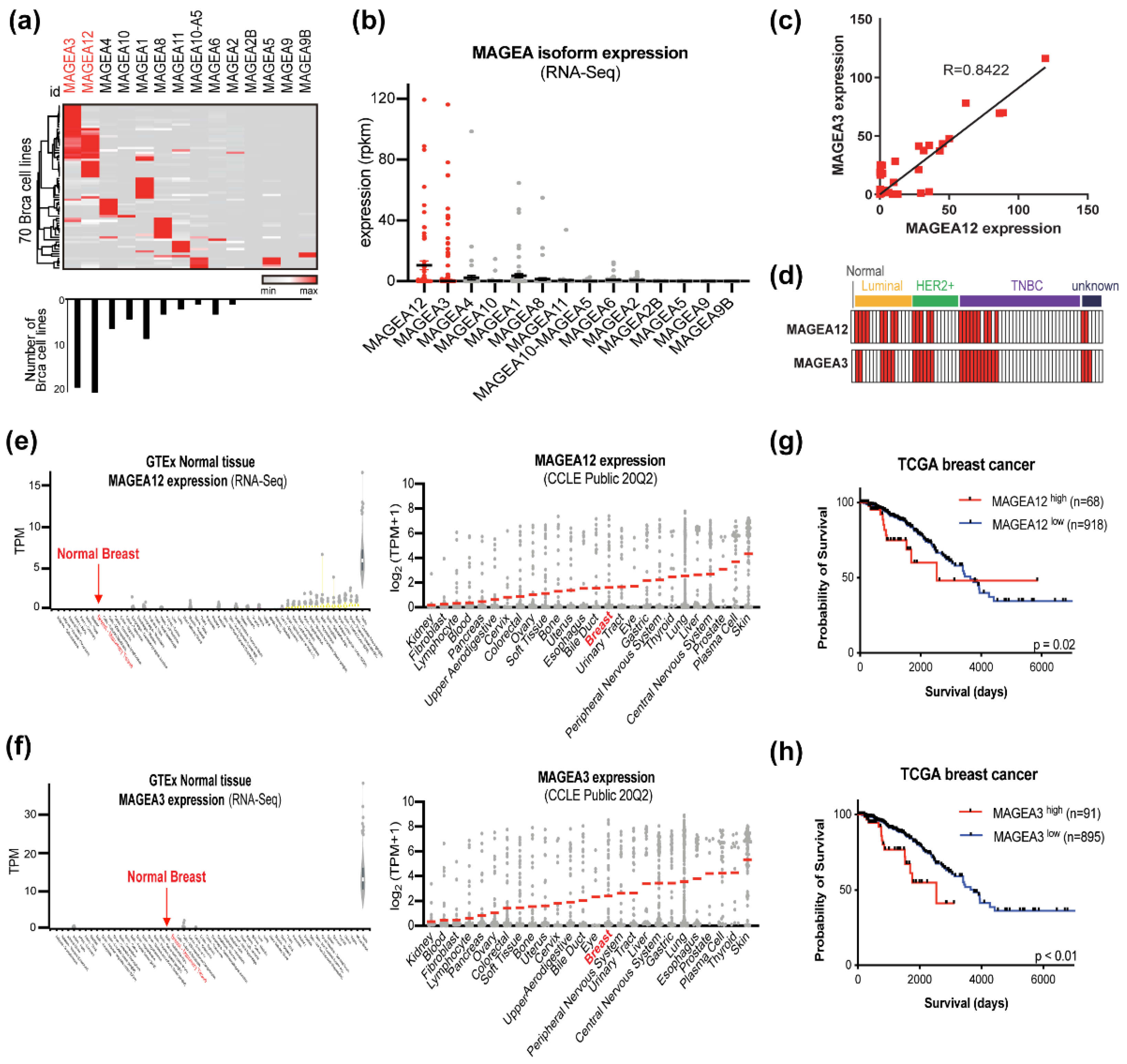
3.1. A Subset of Breast Cancer Cells Expresses Both MAGEA12 and MAGEA3 in a Subtype-Independent Manner
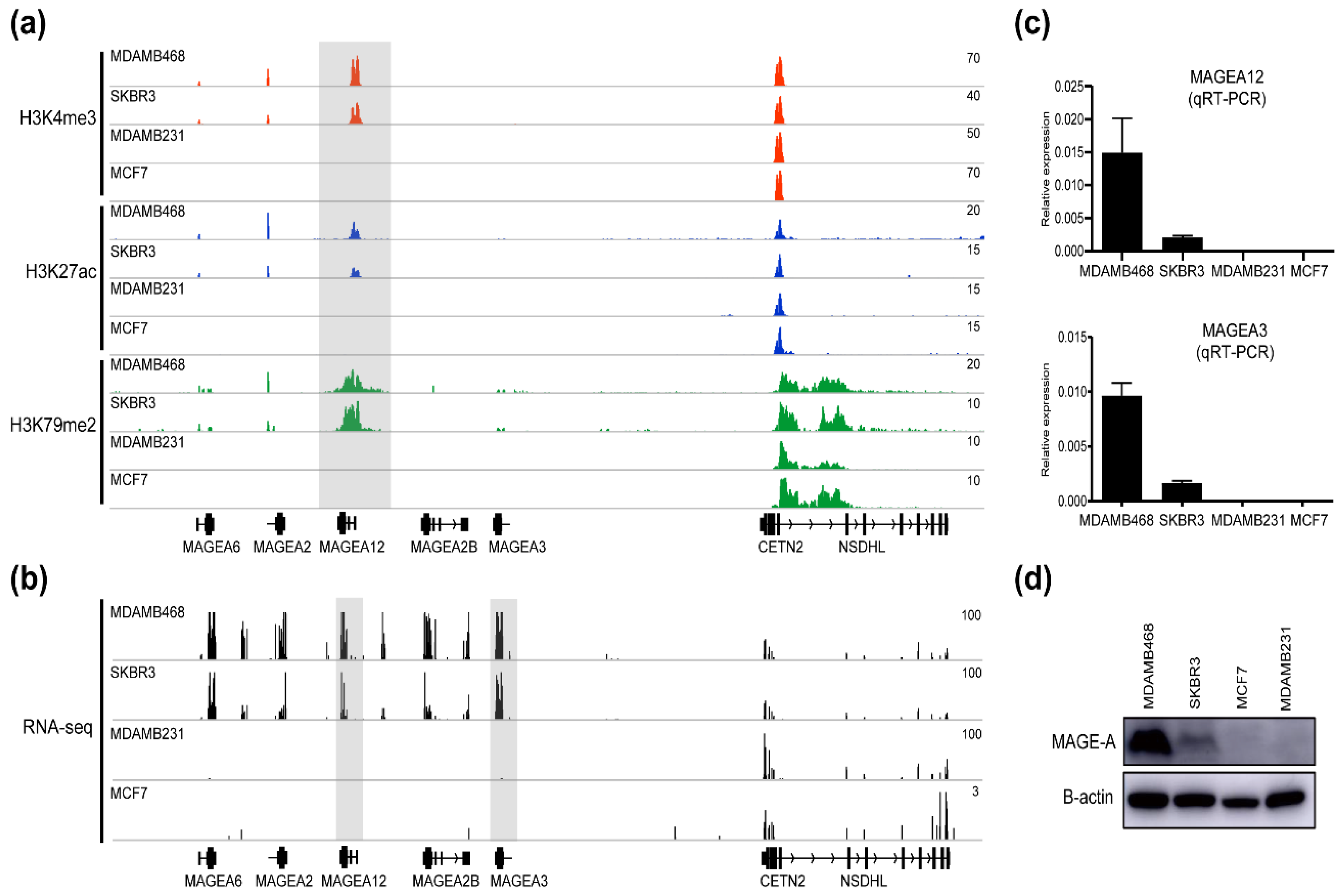
3.2. Chromatin Modifications at MAGE-A Gene Loci in Breast Cancer Cell Lines
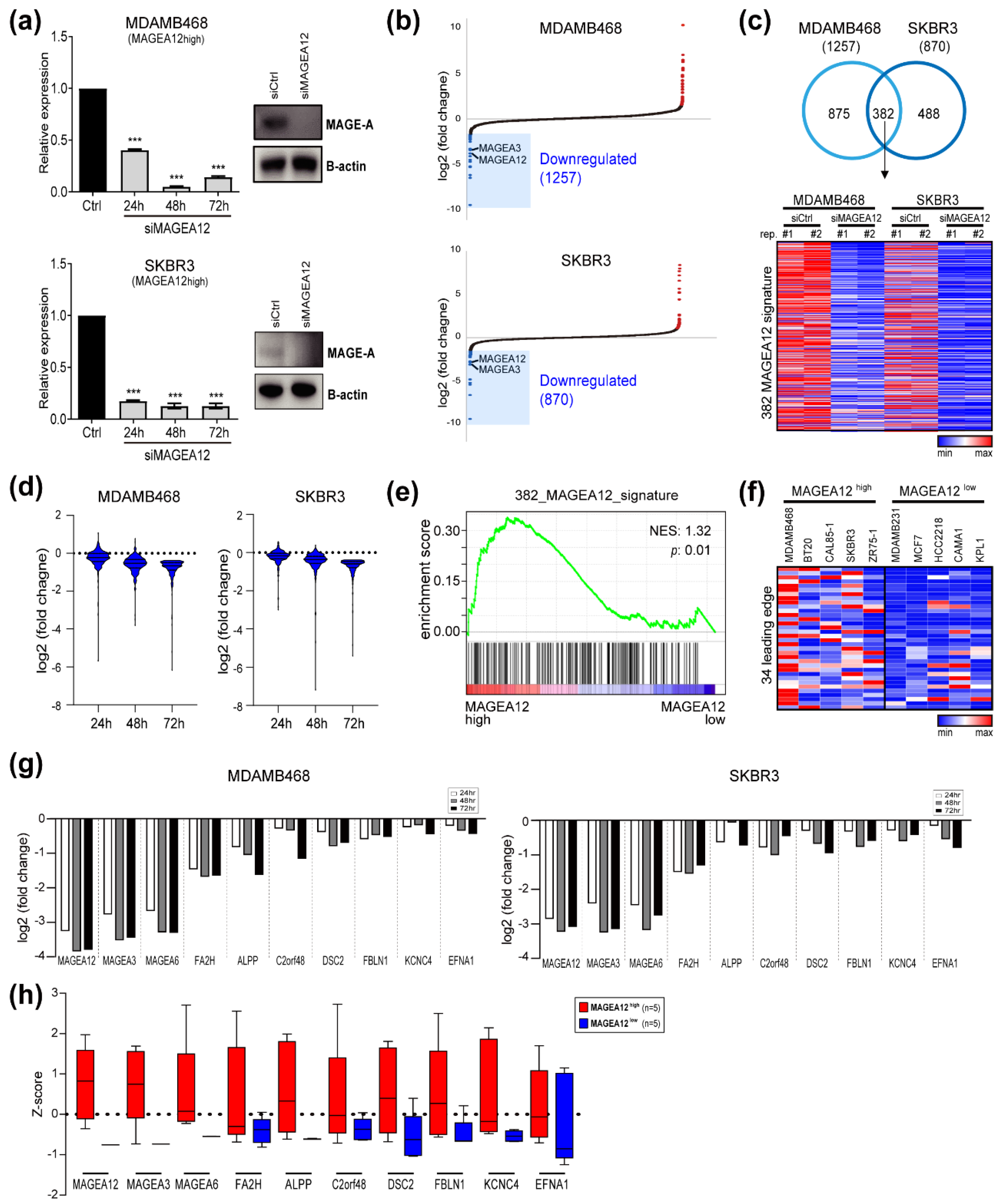
3.3. Identification of MAGEA12 Signature Genes in Breast Cancer
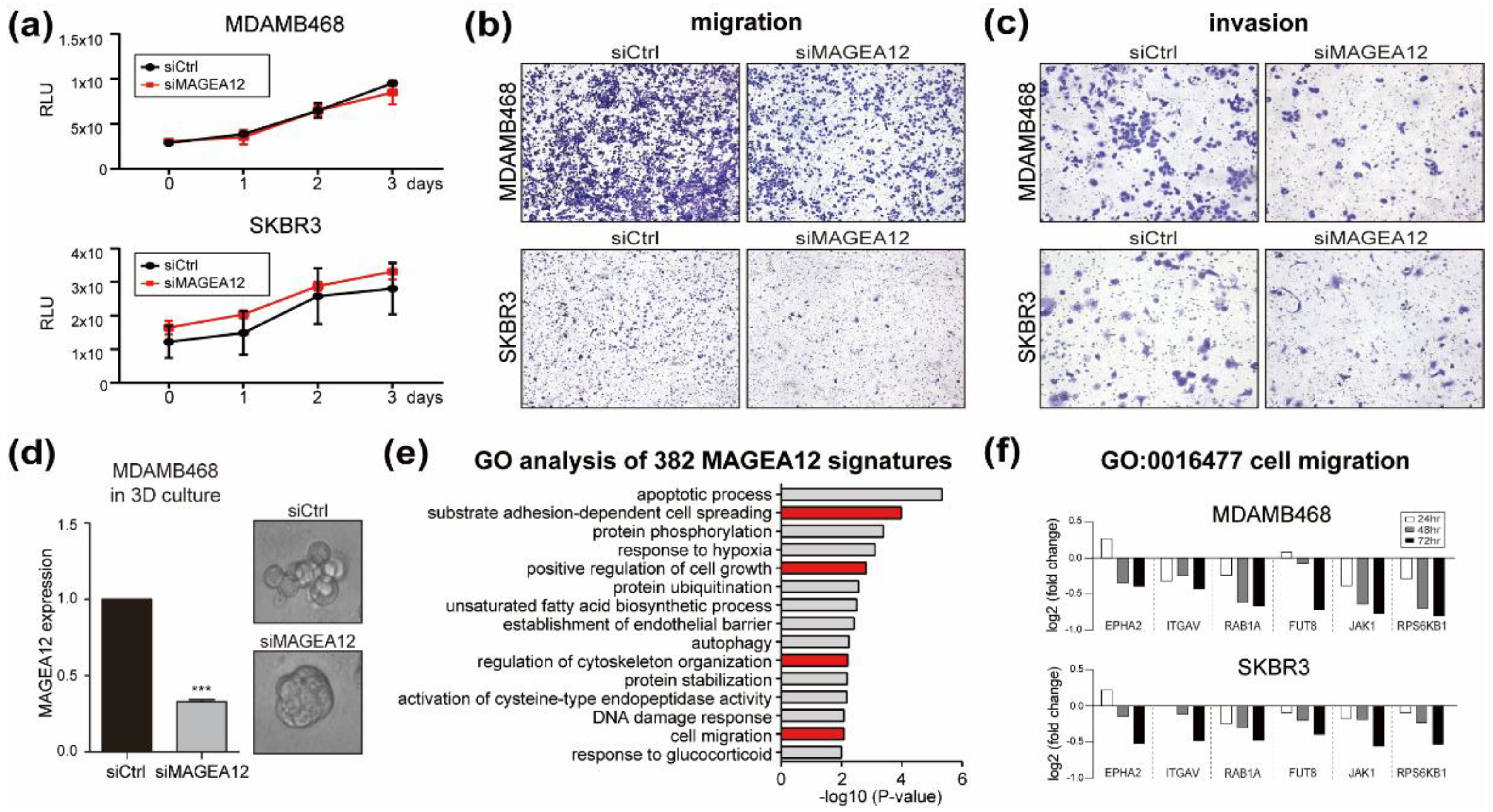
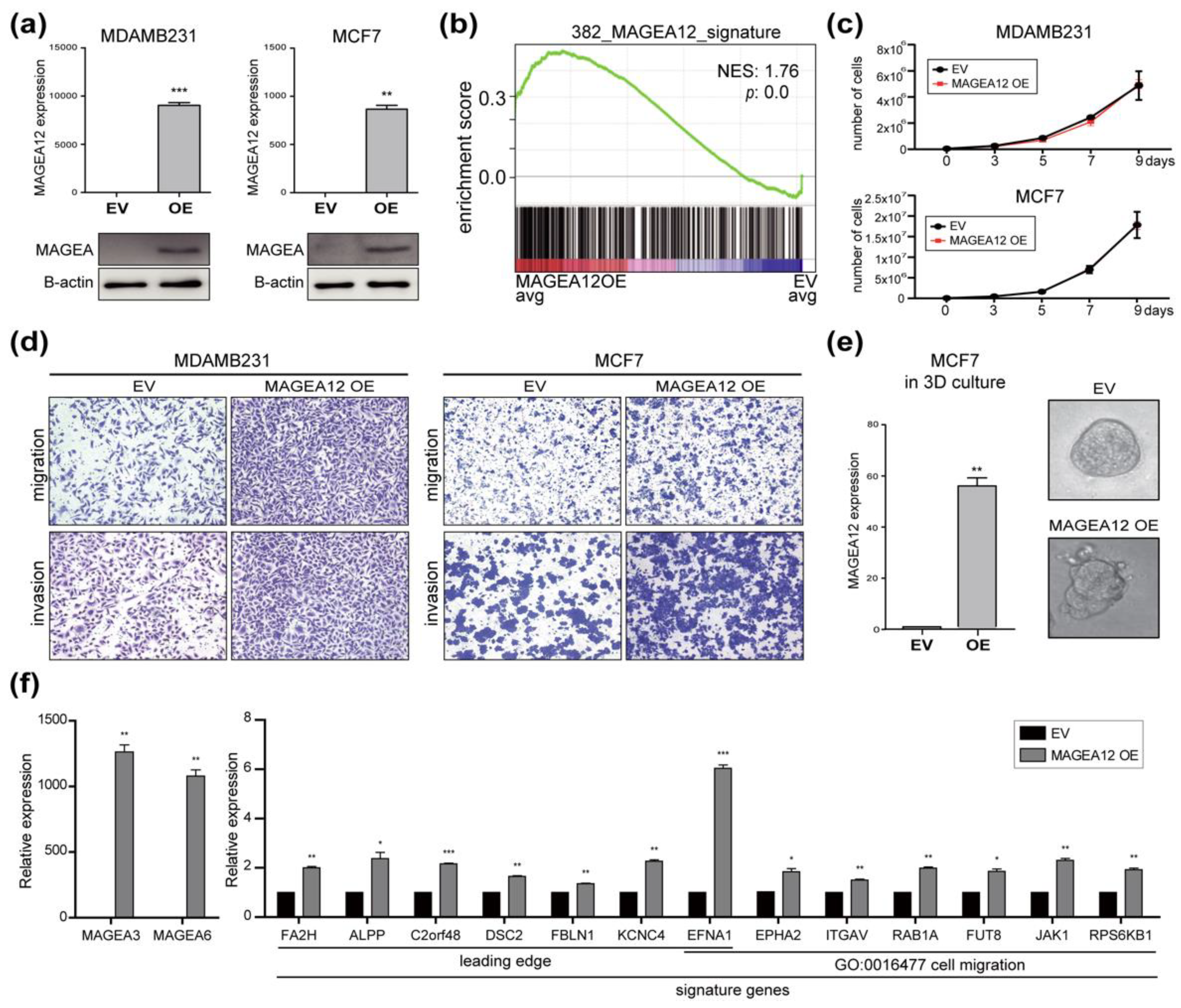
3.4. Effect of MAGEA12 Silencing and Overexpression on Breast Cancer Cell Aggressiveness
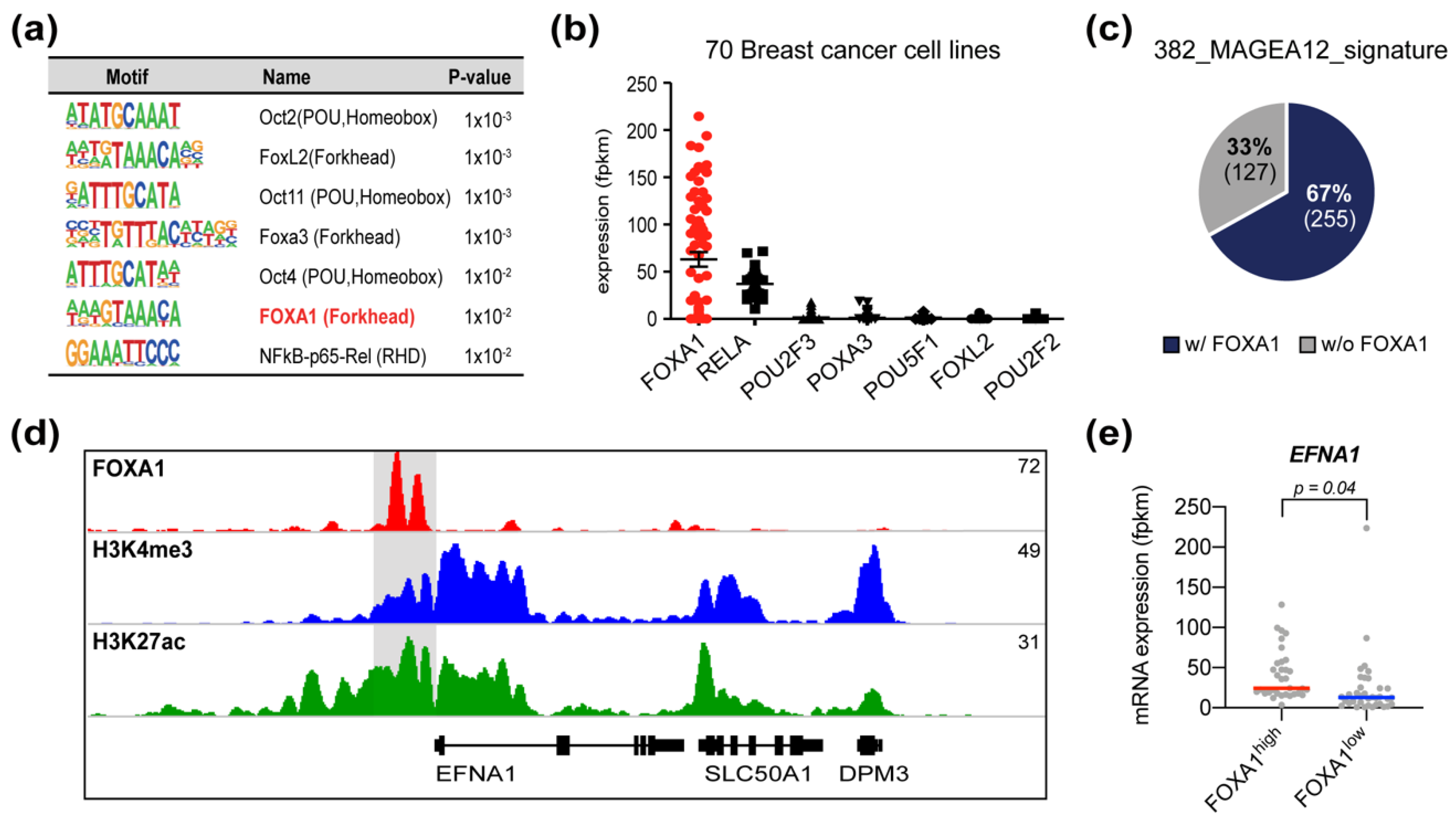
3.5. FOXA1 Is a Candidate Transcription Factor That Regulates MAGEA12 Signature Genes
3.6. Chromatin Modifications in MAGEA12 Signature Genes Parallel MAGEA12 Expression Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.-M.; Kim, H.; Lee, K.-H.; Cho, J.-Y. Analysis of opposing histone modifications H3K4me3 and H3K27me3 reveals candidate diagnostic biomarkers for TNBC and gene set prediction combination. BMB Rep. 2020, 53, 266–271. [Google Scholar] [CrossRef]
- Ji, P.; Gong, Y.; Jin, M.L.; Hu, X.; Di, G.H.; Shao, Z.M. The Burden and Trends of Breast Cancer From 1990 to 2017 at the Global, Regional, and National Levels: Results from the Global Burden of Disease Study 2017. Front. Oncol. 2020, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast cancer in young women: An overview. Updates Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Han, W. Unique features of young age breast cancer and its management. J. Breast Cancer 2014, 17, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Schunkert, E.M.; Zhao, W.; Zanker, K. Breast Cancer Recurrence Risk Assessment: Is Non-Invasive Monitoring an Option? Biomed. Hub 2018, 3. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Oh, S.; Oh, C.; Yoo, K.H. Functional roles of CTCF in breast cancer. BMB Rep. 2017, 50, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Dustin, D.; Fuqua, S.A. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr. Opin. Pharmacol. 2016, 31, 97–103. [Google Scholar] [CrossRef]
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Sang, M.; Lian, Y.; Zhou, X.; Shan, B. MAGE-A family: Attractive targets for cancer immunotherapy. Vaccine 2011, 29, 8496–8500. [Google Scholar] [CrossRef]
- Huang, Z.M.; Jia, Z.C.; Tang, J.; Zhang, Y.; Tian, Y.; Ni, D.J.; Wang, F.; Wu, Y.Z.; Ni, B. Cross-immunizing potential of tumor MAGE-A epitopes recognized by HLA-A*02:01-restricted cytotoxic T lymphocytes. BMB Rep. 2012, 45, 408–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weon, J.L.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florke Gee, R.R.; Chen, H.; Lee, A.K.; Daly, C.A.; Wilander, B.A.; Fon Tacer, K.; Potts, P.R. Emerging roles of the MAGE protein family in stress response pathways. J. Biol. Chem. 2020, 295, 16121–16155. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Meng, L.; Ding, P.; Sang, M. Epigenetic regulation of MAGE family in human cancer progression-DNA methylation, histone modification, and non-coding RNAs. Clin. Epigenetics 2018, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, F.; Huang, W.; Gu, L.; Meng, L.; Ju, Y.; Wu, Y.; Li, J.; Liu, L.; Sang, M. MAGE-A11 is activated through TFCP2/ZEB1 binding sites de-methylation as well as histone modification and facilitates ESCC tumor growth. Oncotarget 2018, 9, 3365–3378. [Google Scholar] [CrossRef] [Green Version]
- Sienel, W.; Varwerk, C.; Linder, A.; Kaiser, D.; Teschner, M.; Delire, M.; Stamatis, G.; Passlick, B. Melanoma associated antigen (MAGE)-A3 expression in Stages I and II non-small cell lung cancer: Results of a multi-center study. Eur. J. Cardio Thorac. Surg. 2004, 25, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Fan, W.; Hu, H.; Zhang, L.; Michel, J.; Wu, Y.; Wang, J.; Jia, L.; Tang, X.; Xu, L.; et al. MAGE-A1 in lung adenocarcinoma as a promising target of chimeric antigen receptor T cells. J. Hematol. Oncol. 2019, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, J.; Ding, N.; Li, Y.; Yang, Y.; Fang, X.; Zhao, H. MAGE-A1 promotes melanoma proliferation and migration through C-JUN activation. Biochem. Biophys. Res. Commun. 2016, 473, 959–965. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshida, K.; Wada, Y.; Hamai, Y.; Sentani, K.; Oue, N.; Yasui, W. Melanoma-associated antigen-A1 expression predicts resistance to docetaxel and paclitaxel in advanced and recurrent gastric cancer. Oncol. Rep. 2007, 18, 329–336. [Google Scholar] [CrossRef]
- Otte, M.; Zafrakas, M.; Riethdorf, L.; Pichlmeier, U.; Loning, T.; Janicke, F.; Pantel, K. MAGE-A gene expression pattern in primary breast cancer. Cancer Res. 2001, 61, 6682–6687. [Google Scholar]
- Wei, Y.; Wang, Y.; Gong, J.; Rao, L.; Wu, Z.; Nie, T.; Shi, D.; Zhang, L. High expression of MAGE-A9 contributes to stemness and malignancy of human hepatocellular carcinoma. Int. J. Oncol. 2018, 52, 219–230. [Google Scholar] [CrossRef]
- Zhan, W.; Zhang, Z.; Zhang, Y.; Ma, J.; Wu, T.; Gu, Y.; Li, Y.; Yang, J. Prognostic value of MAGE-A9 expression in patients with colorectal cancer. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Shiota, H.; Kuroda, K.; Shigematsu, Y.; Ichiki, Y.; Uramoto, H.; Hanagiri, T.; Tanaka, F. Clinical significance of human leukocyte antigen loss and melanoma-associated antigen 4 expression in smokers of non-small cell lung cancer patients. Int. J. Clin. Oncol. 2013, 18, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Brands, R.C.; Kuchler, N.; Fuchs, A.; Linz, C.; Kubler, A.C.; Muller-Richter, U.D. Melanoma-associated antigen expression and the efficacy of tyrosine kinase inhibitors in head and neck cancer. Oncol. Lett. 2015, 10, 1211–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, S.; Kriegebaum, U.; Kuchler, N.; Brands, R.C.; Linz, C.; Kubler, A.C.; Muller-Richter, U.D. Correlation of MAGE-A tumor antigens and the efficacy of various chemotherapeutic agents in head and neck carcinoma cells. Clin. Oral. Investig. 2014, 18, 189–197. [Google Scholar] [CrossRef]
- Bredenbeck, A.; Hollstein, V.M.; Trefzer, U.; Sterry, W.; Walden, P.; Losch, F.O. Coordinated expression of clustered cancer/testis genes encoded in a large inverted repeat DNA structure. Gene 2008, 415, 68–73. [Google Scholar] [CrossRef] [PubMed]
- De Plaen, E.; Arden, K.; Traversari, C.; Gaforio, J.J.; Szikora, J.P.; De Smet, C.; Brasseur, F.; van der Bruggen, P.; Lethe, B.; Lurquin, C.; et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics 1994, 40, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Laiseca, J.E.; Ladelfa, M.F.; Cotignola, J.; Peche, L.Y.; Pascucci, F.A.; Castano, B.A.; Galigniana, M.D.; Schneider, C.; Monte, M. Functional interaction between co-expressed MAGE-A proteins. PLoS ONE 2017, 12, e0178370. [Google Scholar] [CrossRef] [PubMed]
- Trippel, A.; Halling, F.; Heymann, P.; Ayna, M.; Al-Nawas, B.; Ziebart, T. The expression of melanoma-associated antigen A (MAGE-A) in oral squamous cell carcinoma: An evaluation of the significance for tumor prognosis. Oral Maxillofac. Surg. 2019, 23, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Xu, L.; Zhang, S.; Zhu, H.; Mao, G.; Huang, J. High expression levels of MAGE-A9 are correlated with unfavorable survival in lung adenocarcinoma. Oncotarget 2016, 7, 4871–4881. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Fu, M.; Ge, Z.; Zhan, F.; Ding, Y.; Ni, H.; Zhang, W.; Zhu, Y.; Tang, X.; Xiong, L.; et al. High expression of MAGE-A9 correlates with unfavorable survival in hepatocellular carcinoma. Sci. Rep. 2014, 4, 6625. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Luo, G.; Liu, B.; Sun, Y.; Yan, Z. Melanoma-associated antigen A2 is overexpressed in glioma and associated with poor prognosis in glioma patients. Neoplasma 2018, 65, 604–609. [Google Scholar] [CrossRef] [Green Version]
- Lian, Y.; Sang, M.; Ding, C.; Zhou, X.; Fan, X.; Xu, Y.; Lu, W.; Shan, B. Expressions of MAGE-A10 and MAGE-A11 in breast cancers and their prognostic significance: A retrospective clinical study. J. Cancer Res. Clin. Oncol. 2012, 138, 519–527. [Google Scholar] [CrossRef]
- Poojary, M.; Jishnu, P.V.; Kabekkodu, S.P. Prognostic Value of Melanoma-Associated Antigen-A (MAGE-A) Gene Expression in Various Human Cancers: A Systematic Review and Meta-analysis of 7428 Patients and 44 Studies. Mol. Diagn. Ther. 2020, 24, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara-Kuroda, A.; Kato, T.; Abiko, T.; Tsuchikawa, T.; Kyogoku, N.; Ichinokawa, M.; Tanaka, K.; Noji, T.; Hida, Y.; Kaga, K.; et al. Prognostic value of MAGEA4 in primary lung cancer depends on subcellular localization and p53 status. Int. J. Oncol. 2018, 53, 713–724. [Google Scholar] [CrossRef]
- Mei, A.H.; Tung, K.; Han, J.; Perumal, D.; Lagana, A.; Keats, J.; Auclair, D.; Chari, A.; Jagannath, S.; Parekh, S.; et al. MAGE-A inhibit apoptosis and promote proliferation in multiple myeloma through regulation of BIM and p21(Cip1). Oncotarget 2020, 11, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Nardiello, T.; Jungbluth, A.A.; Mei, A.; Diliberto, M.; Huang, X.; Dabrowski, A.; Andrade, V.C.; Wasserstrum, R.; Ely, S.; Niesvizky, R.; et al. MAGE-A inhibits apoptosis in proliferating myeloma cells through repression of Bax and maintenance of survivin. Clin. Cancer Res. 2011, 17, 4309–4319. [Google Scholar] [CrossRef] [Green Version]
- Yanagi, T.; Nagai, K.; Shimizu, H.; Matsuzawa, S.-I. Melanoma antigen A12 regulates cell cycle via tumor suppressor p21 expression. Oncotarget 2017, 8, 68448. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.Y.; Kenny, P.A.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Yoo, K.H.; Oh, S.; Kang, K.; Hensel, T.; Robinson, G.W.; Hennighausen, L. Loss of EZH2 results in precocious mammary gland development and activation of STAT5-dependent genes. Nucleic Acids Res. 2015, 43, 8774–8789. [Google Scholar] [CrossRef] [Green Version]
- Das, P.M.; Ramachandran, K.; vanWert, J.; Singal, R. Chromatin immunoprecipitation assay. Biotechniques 2004, 37, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef]
- Glont, S.E.; Chernukhin, I.; Carroll, J.S. Comprehensive Genomic Analysis Reveals that the Pioneering Function of FOXA1 Is Independent of Hormonal Signaling. Cell Rep. 2019, 26, 2558–2565.e3. [Google Scholar] [CrossRef] [Green Version]
- Badve, S.; Turbin, D.; Thorat, M.A.; Morimiya, A.; Nielsen, T.O.; Perou, C.M.; Dunn, S.; Huntsman, D.G.; Nakshatri, H. FOXA1 expression in breast cancer--correlation with luminal subtype A and survival. Clin. Cancer Res. 2007, 13, 4415–4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Li, G. Role of EFNA1 in tumorigenesis and prospects for cancer therapy. Biomed. Pharmacother. 2020, 130, 110567. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Bai, S.; James, S.R.; Mohler, J.L.; Wilson, E.M. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol. Cancer Res. 2009, 7, 523–535. [Google Scholar] [CrossRef] [Green Version]
- De Smet, C.; Loriot, A.; Boon, T. Promoter-dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE-A1 in tumor cells. Mol. Cell. Biol. 2004, 24, 4781–4790. [Google Scholar] [CrossRef] [Green Version]
- Bensaid, D.; Blondy, T.; Deshayes, S.; Dehame, V.; Bertrand, P.; Gregoire, M.; Errami, M.; Blanquart, C. Assessment of new HDAC inhibitors for immunotherapy of malignant pleural mesothelioma. Clin. Epigenetics 2018, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Liu, F.; Liang, X.; Liu, S.; Huang, X.; Sang, M.; Geng, C. Tumor suppressor let-7a inhibits breast cancer cell proliferation, migration and invasion by targeting MAGE-A1. Neoplasma 2019, 66, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Marcar, L.; Maclaine, N.J.; Hupp, T.R.; Meek, D.W. Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res. 2010, 70, 10362–10370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda, C.T.; Potts, P.R. Oncogenic MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by ubiquitinating and degrading AMPK in cancer. Autophagy 2015, 11, 844–846. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Laganiere, J.; Deblois, G.; Lefebvre, C.; Bataille, A.R.; Robert, F.; Giguere, V. From the Cover: Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc. Natl. Acad. Sci. USA 2005, 102, 11651–11656. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, C.; Kim, H.-R.; Oh, S.; Ko, J.Y.; Kim, Y.; Kang, K.; Yang, Y.; Kim, J.; Park, J.H.; Roe, J.-S.; et al. Epigenetic Upregulation of MAGE-A Isoforms Promotes Breast Cancer Cell Aggressiveness. Cancers 2021, 13, 3176. https://doi.org/10.3390/cancers13133176
Oh C, Kim H-R, Oh S, Ko JY, Kim Y, Kang K, Yang Y, Kim J, Park JH, Roe J-S, et al. Epigenetic Upregulation of MAGE-A Isoforms Promotes Breast Cancer Cell Aggressiveness. Cancers. 2021; 13(13):3176. https://doi.org/10.3390/cancers13133176
Chicago/Turabian StyleOh, Chaeun, Hwa-Ryeon Kim, Sumin Oh, Je Yeong Ko, Yesol Kim, Keunsoo Kang, Young Yang, Jongmin Kim, Jong Hoon Park, Jae-Seok Roe, and et al. 2021. "Epigenetic Upregulation of MAGE-A Isoforms Promotes Breast Cancer Cell Aggressiveness" Cancers 13, no. 13: 3176. https://doi.org/10.3390/cancers13133176
APA StyleOh, C., Kim, H.-R., Oh, S., Ko, J. Y., Kim, Y., Kang, K., Yang, Y., Kim, J., Park, J. H., Roe, J.-S., & Yoo, K. H. (2021). Epigenetic Upregulation of MAGE-A Isoforms Promotes Breast Cancer Cell Aggressiveness. Cancers, 13(13), 3176. https://doi.org/10.3390/cancers13133176