
Cancer-Associated Fibroblasts in Breast Cancer Treatment Response and Metastasis

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
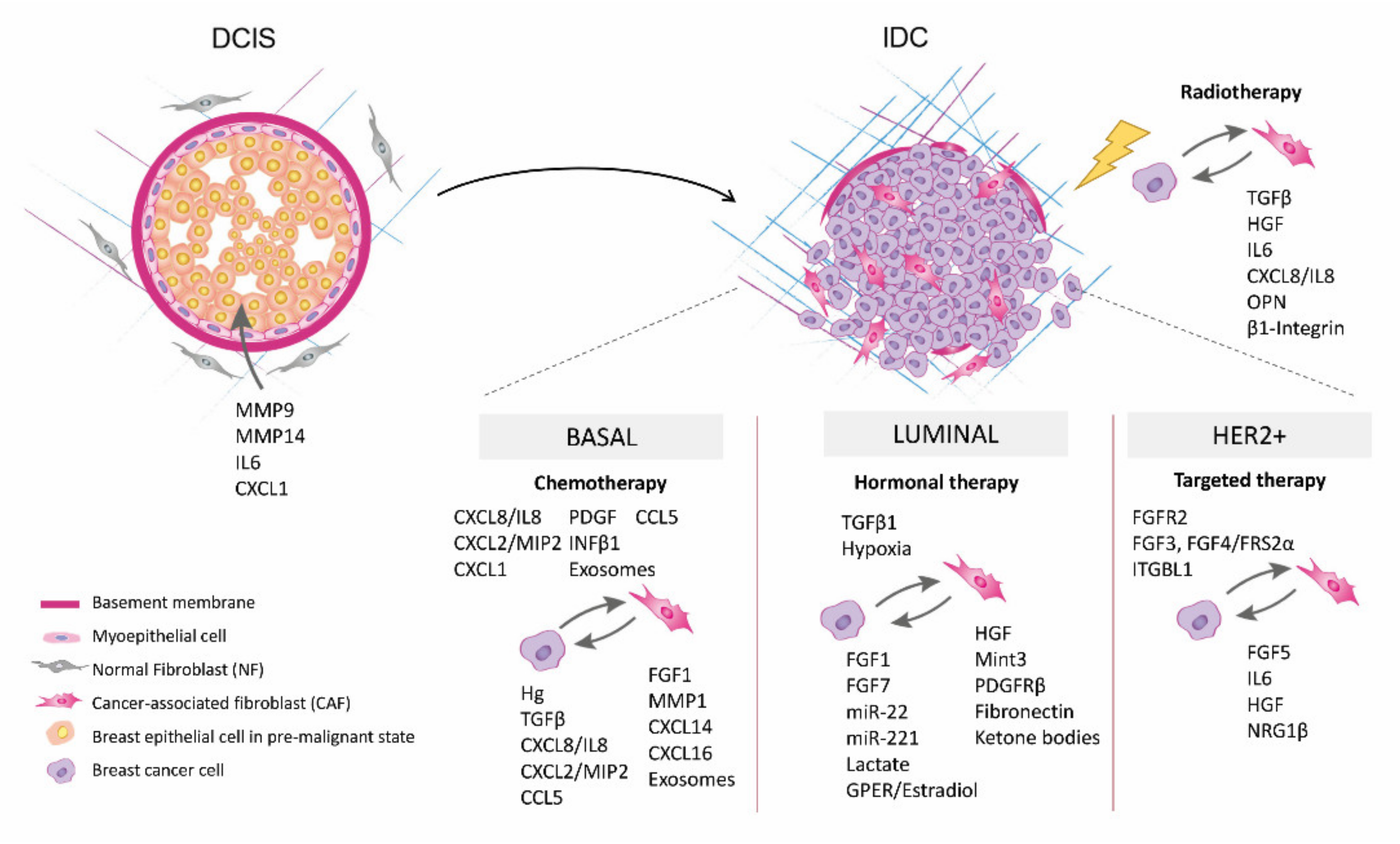
2. Role of Fibroblasts on Ductal Carcinoma In Situ Progression
3. Role of CAFs in BrCa Therapy Resistance
3.1. Radiotherapy Resistance and CAFs
3.2. CAFs and Chemotherapy Resistance in Triple Negative BrCa
3.3. CAFs and Hormonal Therapy Resistance in Luminal BrCa
3.4. CAFs- and HER2-Targeted Therapy Resistance in HER2-Enriched BrCa
4. CAFs and BrCa Metastasis
4.1. Contribution of CAFs from Primary Tumor Stroma to Metastasis Progression
4.2. Contribution of Fibroblasts from Metastatic Sites to Metastasis Formation
5. Targeting CAFs to Prevent BrCa Progression
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Robson, M.; Goessl, C.; Domchek, S. Olaparib for metastatic germline BRCA-mutated breast cancer. N. Engl. J. Med. 2017, 377, 1792–1793. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Imparato, V.; Masone, S.; Masullo, M.; Nasso, R.; Montagnani, S.; Arcucci, A. Involvement of breast cancer-associated fibroblasts in tumor development, therapy resistance and evaluation of potential therapeutic strategies. Curr. Med. Chem. 2018, 25, 3414–3434. [Google Scholar] [CrossRef] [PubMed]
- Arcucci, A.; Ruocco, M.R.; Granato, G.; Sacco, A.M.; Montagnani, S. Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. BioMed Res. Int. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchsbaum, R.J.; Oh, S.Y. Breast cancer-associated fibroblasts: Where we are and where we need to go. Cancers 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target Ther. 2021, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Iii, H.; et al. Cancer-associated fibroblasts: Versatile players in the tumor microenvironment. Cancers 2020, 12, 2652. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic reprogramming of cancer associated fibroblasts: The slavery of stromal fibroblasts. BioMed Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteran, L.; Erez, N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Cancer Facts & Figures 2020; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Pareja, F.; Brown, D.N.; Lee, J.Y.; Paula, A.D.C.; Selenica, P.; Bi, R.; Geyer, F.C.; Gazzo, A.; da Silva, E.M.; Vahdatinia, M.; et al. Whole-exome sequencing analysis of the progression from non–low-grade ductal carcinoma in situto invasive ductal carcinoma. Clin. Cancer Res. 2020, 26, 3682–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.C.; Machado, H.L.; Schwertfeger, K.L. Breaking through to the other side: Microenvironment contributions to DCIS initiation and progression. J. Mammary Gland. Biol. Neoplasia 2018, 23, 207–221. [Google Scholar] [CrossRef]
- Ma, X.-J.; Dahiya, S.; Richardson, E.; Erlander, M.; Sgroi, D.C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009, 11, R7. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Beck, A.H.; Webster, J.A.; Espinosa, I.; Montgomery, K.; Varma, S.; van de Rijn, M.; Jensen, K.C.; West, R.B. Analysis of stromal signatures in the tumor microenvironment of ductal carcinoma in situ. Breast Cancer Res. Treat. 2010, 123, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Nogueira, P.; Mancino, M.; Fuster, G.; Bragado, P.; de Puig, M.P.; Gascón, P.; Casado, F.J.; Carbó, N. Breast mammographic density: Stromal implications on breast cancer detection and therapy. J. Clin. Med. 2020, 9, 776. [Google Scholar] [CrossRef] [Green Version]
- Lisanti, M.P.; Reeves, K.; Peiris-Pagès, M.; Chadwick, A.L.; Sanchez-Alvarez, R.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Tsirigos, A.; Pavlides, S.; et al. JNK1 stress signaling is hyper-activated in high breast density and the tumor stroma: Connecting fibrosis, inflammation, and stemness for cancer prevention. Cell Cycle 2013, 13, 580–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Peluffo, G.; Chen, H.; Gelman, R.; Schnitt, S.; Polyak, K. Role of COX-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc. Natl. Acad. Sci. USA 2009, 106, 3372–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, K.E.; Yang, N.; Pehlke, C.; Keely, P.J.; Eliceiri, K.W.; Friedl, A.; Beebe, D.J. Transition to invasion in breast cancer: A microfluidic in vitro model enables examination of spatial and temporal effects. Integr. Biol. 2011, 3, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuala, K.O.; Sameni, M.; Shah, S.; Aggarwal, N.; Simonait, M.L.; Franco, O.E.; Hong, Y.; Hayward, S.W.; Behbod, F.; Mattingly, R.R.; et al. Il-6 signaling between ductal carcinoma in situ cells and carcinoma-associated fibroblasts mediates tumor cell growth and migration. BMC Cancer 2015, 15, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, T.T.; Prechtl, A.M.; Pearson, G.W. Breast cancer subtype-specific interactions with the microenvironment dictate mechanisms of invasion. Cancer Res. 2011, 71, 6857–6866. [Google Scholar] [CrossRef] [Green Version]
- Conklin, M.W.; Gangnon, R.E.; Sprague, B.L.; van Germert, L.; Hampton, J.M.; Eliceiri, K.W.; Bredfeldt, J.S.; Liu, Y.; Surachaicharn, N.; Newcomb, P.A.; et al. Collagen alignment as a predictor of recurrence after ductal carcinoma in situ. Cancer Epidemiol. Biomark. Prev. 2017, 27, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Sprague, B.L.; Vacek, P.M.; Mulrow, S.E.; Evans, M.F.; Trentham-Dietz, A.; Herschorn, S.D.; James, T.A.; Surachaicharn, N.; Keikhosravi, A.; Eliceiri, K.W.; et al. Collagen organization in relation to ductal carcinoma in situ pathology and outcomes. Cancer Epidemiol. Biomark. Prev. 2021, 30, 80–88. [Google Scholar] [CrossRef]
- Bernard, S.; Myers, M.; Bin Fang, W.; Zinda, B.; Smart, C.; Lambert, D.; Zou, A.; Fan, F.; Cheng, N. CXCL1 derived from mammary fibroblasts promotes progression of mammary lesions to invasive carcinoma through CXCR2 dependent mechanisms. J. Mammary Gland. Biol. Neoplasia 2018, 23, 249–267. [Google Scholar] [CrossRef]
- Shaker, H.; Bundred, N.; Albadry, H.; Nicholson, S.; Castle, J.; Lumsden, L.; Pritchard, S.; Landberg, G.; Kirwan, C. PO-21—Stromal fibroblasts in preinvasive breast cancer (ductal carcinoma in situ, DCIS) demonstrate a cancer-like procoagulant phenotypic switch that may facilitate invasion. Thromb. Res. 2016, 140, S184. [Google Scholar] [CrossRef]
- Sameni, M.; Cavallo-Medved, D.; Franco, O.E.; Chalasani, A.; Ji, K.; Aggarwal, N.; Anbalagan, A.; Chen, X.; Mattingly, R.R.; Hayward, S.W.; et al. Pathomimetic avatars reveal divergent roles of microenvironment in invasive transition of ductal carcinoma in situ. Breast Cancer Res. 2017, 19, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaker, H.; Bundred, N.J.; Landberg, G.; Pritchard, S.A.; Albadry, H.; Nicholson, S.L.; Harries, L.J.; Heah, J.Y.E.; Castle, J.; Kirwan, C.C. Breast cancer stromal clotting activation (Tissue Factor and thrombin): A pre-invasive phenomena that is prognostic in invasion. Cancer Med. 2020, 9, 1768–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.-N.; Liu, Z.; Tian, Y.; Zhao, P.-P.; Hua, X. FAP-a and GOLPH3 are hallmarks of DCIS progression to invasive breast cancer. Front. Oncol. 2019, 9, 1424. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Dasgupta, A.; Nguyen, K.H.; Liu, C.; Kovatich, A.J.; Schwartz, G.F.; Pestell, R.G.; Sotgia, F.; Rui, H.; Lisanti, M.P. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol. Ther. 2009, 8, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Krisnawan, V.E.; Stanley, J.A.; Schwarz, J.K.; DeNardo, D.G. Tumor microenvironment as a regulator of radiation therapy: New insights into stromal-mediated radioresistance. Cancers 2020, 12, 2916. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, Y.; Tan, Y.; Wei, Q.; Yu, W. Cancer-associated fibroblasts in radiotherapy: Challenges and new opportunities. Cell Commun. Signal. 2019, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Barker, H.E.; Paget, J.T.E.; Khan, A.; Harrington, K. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Steer, A.; Cordes, N.; Jendrossek, V.; Klein, D. Impact of cancer-associated fibroblast on the radiation-response of solid xenograft tumors. Front. Mol. Biosci. 2019, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Barcellos-Hoff, M.H.; Derynck, R.; Tsang, M.L.; A Weatherbee, J. Transforming growth factor-beta activation in irradiated murine mammary gland. J. Clin. Investig. 1994, 93, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Bouquet, F.; Pal, A.; Pilones, K.A.; Demaria, S.; Hann, B.; Akhurst, R.J.; Babb, J.; Lonning, S.M.; Dewyngaert, J.K.; Formenti, S.C.; et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 2011, 17, 6754–6765. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; Arteaga, C.L. Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [Green Version]
- Park, C.C.; Zhang, H.J.; Yao, E.S.; Park, C.J.; Bissell, M.J. β1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res. 2008, 68, 4398–4405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Victor, C.T.-S.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.K.C.; Stuart, J.; Chuang, Y.-Y.E.; Little, J.B.; Yuan, Z.-M. Low-dose radiation-induced senescent stromal fibroblasts render nearby breast cancer cells radioresistant. Radiat. Res. 2009, 172, 306–313. [Google Scholar] [CrossRef]
- Pareja, F.; Geyer, F.C.; Marchiò, C.; Burke, K.A.; Weigelt, B.; Reis-Filho, J.S. Triple-negative breast cancer: The importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer 2016, 2, 16036. [Google Scholar] [CrossRef]
- Rong, G.; Kang, H.; Wang, Y.; Hai, T.; Sun, H. Candidate markers that associate with chemotherapy resistance in breast cancer through the study on taxotere-induced damage to tumor microenvironment and gene expression profiling of Carcinoma-Associated Fibroblasts (CAFs). PLoS ONE 2013, 8, e70960. [Google Scholar] [CrossRef] [Green Version]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.; Wu, S.Z.; Roden, D.; Chan, C.-L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, M.; Zheng, K.; Tang, S.; Ma, S. Expression pattern analysis and drug differential sensitivity of cancer-associated fibroblasts in triple-negative breast cancer. Transl. Oncol. 2021, 14, 100891. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, X.; Ran, W.; Meng, J.; Zhai, Y.; Zhang, P.; Yin, Q.; Yu, H.; Zhang, Z.; Li, Y. Regulating cancer associated fibroblasts with losartan-loaded injectable peptide hydrogel to potentiate chemotherapy in inhibiting growth and lung metastasis of triple negative breast cancer. Biomaterials 2017, 144, 60–67. [Google Scholar] [CrossRef]
- Maia, A.; Gu, Z.; Koch, A.; Berdiel-Acer, M.; Will, R.; Schlesner, M.; Wiemann, S. IFNβ1 secreted by breast cancer cells undergoing chemotherapy reprograms stromal fibroblasts to support tumour growth after treatment. Mol. Oncol. 2021, 15, 1308–1329. [Google Scholar] [CrossRef]
- Roswall, P.; Bocci, M.; Bartoschek, M.; Li, H.; Kristiansen, G.; Jansson, S.; Lehn, S.; Sjolund, J.; Reid, S.; Larsson, C.; et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat. Med. 2018, 24, 463–473. [Google Scholar] [CrossRef]
- Wee, Z.N.; Yatim, S.M.J.M.; Kohlbauer, V.K.; Feng, M.; Goh, J.Y.; Bao, Y.; Lee, P.L.; Zhang, S.; Wang, P.P.; Lim, E.; et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat. Commun. 2015, 6, 8746. [Google Scholar] [CrossRef] [PubMed]
- Karagoz, K.; Sinha, R.; Arga, K.Y. Triple negative breast cancer: A multi-omics network discovery strategy for candidate targets and driving pathways. OMICS: A J. Integr. Biol. 2015, 19, 115–130. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Wakabayashi, H.; Matsumine, A.; Sudo, A.; Uchida, A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem. Biophys. Res. Commun. 2011, 407, 525–530. [Google Scholar] [CrossRef]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.-H. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal. Cell. Pathol. 2010, 33, 61–79. [Google Scholar] [CrossRef]
- Pinilla, S.; Alt, E.; Khalek, F.A.; Jotzu, C.; Muehlberg, F.; Beckmann, C.; Song, Y.-H. Tissue resident stem cells produce CCL5 under the influence of cancer cells and thereby promote breast cancer cell invasion. Cancer Lett. 2009, 284, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Allaoui, R.; Bergenfelz, C.; Mohlin, S.; Hagerling, C.; Salari, K.; Werb, Z.; Anderson, R.; Ethier, S.P.; Jirström, K.; Påhlman, S.; et al. Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat. Commun. 2016, 7, 13050. [Google Scholar] [CrossRef]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Morein, D.; Rubinstein-Achiasaf, L.; Sprinzak, D.; Wiemann, S.; Körner, C.; Ehrlich, M.; Ben-Baruch, A. Notch-mediated tumor-stroma-inflammation networks promote invasive properties and CXCL8 expression in triple-negative breast cancer. Front. Immunol. 2019, 10, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-stroma-inflammation networks promote pro-metastatic chemokines and aggressiveness characteristics in triple-negative breast cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [Green Version]
- Sjöberg, E.; Augsten, M.; Bergh, J.; Jirström, K.; Östman, A. Expression of the chemokine CXCL14 in the tumour stroma is an independent marker of survival in breast cancer. Br. J. Cancer 2016, 114, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, K.; Ishida, M.; Yanai, H.; Tsuta, K.; Sekimoto, M.; Sugie, T. Prognostic significance of PD-L1-positive cancer-associated fibroblasts in patients with triple-negative breast cancer. BMC Cancer 2021, 21, 1–10. [Google Scholar] [CrossRef]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [Green Version]
- Coates, A.S.; Winer, E.P.; Goldhirsch, A.; Gelber, R.D.; Gnant, M.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J.; Andre, F.; Baselga, J.; et al. Tailoring therapies—improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann. Oncol. 2015, 26, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Liu, M.C.; Bouker, K.B.; Gu, Z.; Lee, R.Y.; Zhu, Y.; Skaar, T.C.; Gomez, B.; O’Brien, K.; Wang, Y.; et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003, 22, 7316–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG); Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Yang, G.; Yu, T.; Luo, S.; Wu, C.; Sun, Y.; Liu, M.; Tu, G. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr.-Relat. Cancer 2014, 21, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Liu, M.; Yang, L.; Tu, G.; Zhu, Q.; Chen, M.; Cheng, H.; Luo, H.; Fu, W.; Li, Z.; et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: A new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res. 2015, 17, 1–18. [Google Scholar]
- Jena, B.C.; Das, C.K.; Banerjee, I.; Das, S.; Bharadwaj, D.; Majumder, R.; Mandal, M. Paracrine TGF-β1 from breast cancer contributes to chemoresistance in cancer associated fibroblasts via upregulation of the p44/42 MAPK signaling pathway. Biochem. Pharmacol. 2021, 186, 114474. [Google Scholar] [CrossRef]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.-A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.-E.; et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Goldberg, A.F.; Lin, Z.; Ko, Y.-H.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol. Ther. 2011, 12, 924–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Cantarin, M.P.M.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Dasgupta, A.; Sotgia, F.; Mercier, I.; Pestell, R.G.; Sabel, M.; Kleer, C.G.; Brody, J.R.; Lisanti, M.P. An absence of Stromal Caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am. J. Pathol. 2009, 174, 2023–2034. [Google Scholar] [CrossRef] [Green Version]
- Hiscox, S.; Jordan, N.J.; Jiang, W.; Harper, M.; McClelland, R.; Smith, C.; I Nicholson, R. Chronic exposure to fulvestrant promotes overexpression of the c-Met receptor in breast cancer cells: Implications for tumour–stroma interactions. Endocrine-Related Cancer 2006, 13, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.; Knowles, M.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 2011, 130, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Turczyk, L.; Kitowska, K.; Mieszkowska, M.; Mieczkowski, K.; Czaplinska, D.; Piasecka, D.; Kordek, R.; Skladanowski, A.C.; Potemski, P.; Romanska, H.M.; et al. FGFR2-driven signaling counteracts tamoxifen effect on ERα-positive breast cancer cells. Neoplasia 2017, 19, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.; Santner, S.; Carolin, K.A.; Tait, L. Direct involvement of breast tumor fibroblasts in the modulation of tamoxifen sensitivity. Am. J. Pathol. 2007, 170, 1546–1560. [Google Scholar] [CrossRef] [Green Version]
- Nakaoka, H.J.; Tanei, Z.; Hara, T.; Weng, J.S.; Kanamori, A.; Hayashi, T.; Sato, H.; Orimo, A.; Otsuji, K.; Tada, K.; et al. Mint3-mediated L1CAM expression in fibroblasts promotes cancer cell proliferation via integrin α5β1 and tumour growth. Oncogenesis 2017, 6, e334. [Google Scholar] [CrossRef]
- Busch, S.; Rydén, L.; Stål, O.; Jirstrom, K.; Landberg, G. Low ERK phosphorylation in cancer-associated fibroblasts is associated with tamoxifen resistance in pre-menopausal breast cancer. PLoS ONE 2012, 7, e45669. [Google Scholar] [CrossRef]
- Pontiggia, O.; Sampayo, R.; Raffo, D.; Motter, A.; Xu, R.; Bissell, M.J.; Joffé, E.B.D.K.; Simian, M. The tumor microenvironment modulates tamoxifen resistance in breast cancer: A role for soluble stromal factors and fibronectin through β1 integrin. Breast Cancer Res. Treat. 2011, 133, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyh, B.; Dittmer, A.; Lange, T.; Martens, J.W.M.; Dittmer, J. Stromal cells promote anti-estrogen resistance of breast cancer cells through an insulin-like growth factor binding protein 5 (IGFBP5)/B-cell leukemia/lymphoma 3 (Bcl-3) axis. Oncotarget 2015, 6, 39307–39328. [Google Scholar] [CrossRef] [Green Version]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Li, X.; Zeng, C.; Liu, C.; Hao, Q.; Li, W.; Zhang, K.; Zhang, W.; Wang, S.; Zhao, H.; et al. CD63 + Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Adv. Sci. 2020, 7, 2002518. [Google Scholar] [CrossRef]
- Sansone, P.; Berishaj, M.; Rajasekhar, V.K.; Ceccarelli, C.; Chang, Q.; Strillacci, A.; Savini, C.; Shapiro, L.; Bowman, R.L.; Mastroleo, C.; et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. 2017, 77, 1927–1941. [Google Scholar] [CrossRef] [Green Version]
- Carr, J.A.; Havstad, S.; Zarbo, R.J.; Divine, G.; Mackowiak, P.; Velanovich, V. The association of HER-2/neu amplification with breast cancer recurrence. Arch. Surg. 2000, 135, 1469–1474. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Campone, M.; Berton-Rigaud, D.; Bourbouloux, E.; Sophie, S.; Zanetti, A.; Frenel, J.-S. Her2 positive breast cancer: Practices. Bull. Cancer 2011, 98, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H.L.; Doval, D.C.; Chavez, M.A.; Ang, P.C.-S.; Aziz, Z.; Nag, S.; Ng, C.; Franco, S.X.; Chow, L.W.; Arbushites, M.C.; et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J. Clin. Oncol. 2008, 26, 2999–3005. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.C.; Ades, F.; de Azambuja, E.; Piccart-Gebhart, M. Trastuzumab for patients with HER2 positive breast cancer: Delivery, duration and combination therapies. Breast 2013, 22, S152–S155. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Wan, C.; Yang, H.; Xu, L.; Dong, Q.; Du, C.; Du, J.; Li, F. Her2-targeted multifunctional nano-theranostic platform mediates tumor microenvironment remodeling and immune activation for breast cancer treatment. Int. J. Nanomed. 2020, 15, 10007–10028. [Google Scholar] [CrossRef]
- Nguyen, M.; de Ninno, A.; Mencattini, A.; Mermet-Meillon, F.; Fornabaio, G.; Evans, S.S.; Cossutta, M.; Khira, Y.; Han, W.; Sirven, P.; et al. Dissecting effects of anti-cancer drugs and cancer-associated fibroblasts by on-chip reconstitution of immunocompetent tumor microenvironments. Cell Rep. 2018, 25, 3884–3893.e3. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Tabassum, D.P.; Janiszewska, M.; Place, A.E.; Trinh, A.; Rozhok, A.I.; Pyne, S.; Guerriero, J.L.; Shu, S.; Ekram, M.; et al. Spatial proximity to fibroblasts impacts molecular features and therapeutic sensitivity of breast cancer cells influencing clinical outcomes. Cancer Res. 2016, 76, 6495–6506. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-H.; Pelissier, F.A.; Zhang, H.; Lakins, J.; Weaver, V.M.; Park, C.; LaBarge, M.A. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol. Biol. Cell 2015, 26, 3946–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Korn, R.L.; Rosen, L.S.; Lorusso, P.; Dychter, S.S.; Zhu, J.; Maneval, D.C.; Jiang, P.; Shepard, H.M.; Frost, G.; et al. Phase 1 trials of PEGylated recombinant human hyaluronidase PH20 in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Nogueira, P.; Mancino, M.; Fuster, G.; Lopez-Plana, A.; Jauregui, P.; Almendro, V.; Enreig, E.; Menéndez, S.; Rojo, F.; Noguera-Castells, A.; et al. Tumor-associated fibroblasts promote HER2-targeted therapy resistance through FGFR2 Activation. Clin. Cancer Res. 2019, 26, 1432–1448. [Google Scholar] [CrossRef] [Green Version]
- Akhand, S.S.; Chen, H.; Purdy, S.C.; Liu, Z.; Anderson, J.C.; Willey, C.D.; Wendt, M.K. Fibroblast growth factor receptor facilitates recurrence of minimal residual disease following trastuzumab emtansine therapy. NPJ Breast Cancer 2021, 7, 1–11. [Google Scholar] [CrossRef]
- Hanker, A.; Garrett, J.J.; Estrada, M.V.; Moore, P.P.; Ericsson, P.G.P.; Koch, J.J.; Langley, E.E.; Singh, S.S.; Kim, P.P.; Frampton, G.G.; et al. HER2-overexpressing breast cancers amplify FGFR signaling upon acquisition of resistance to dual therapeutic blockade of HER2. Clin. Cancer Res. 2017, 23, 4323–4334. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-H.; Lee, J.K.; LaBarge, M.A. Fabrication and Use of MicroEnvironment microArrays (MEArrays). J. Vis. Exp. 2012, 2012, e4152. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.S.; Dane, M.; Chin, K.; Tatarova, Z.; Liu, M.; Liby, T.; Thompson, W.; Smith, R.; Nederlof, M.; Bucher, E.; et al. Microenvironment-mediated mechanisms of resistance to HER2 inhibitors differ between HER2+ breast cancer subtypes. Cell Syst. 2018, 6, 329–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nat. Cell Biol. 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Zhang, Y.; Zhao, M.; Shen, K.; Qu, Q.; Lou, Y.; Liu, J.; Huang, O.; Chen, X.; Wu, J. Cancer-associated fibroblasts induce trastuzumab resistance in HER2 positive breast cancer cells. Mol. BioSyst. 2015, 11, 1029–1040. [Google Scholar] [CrossRef]
- Zervantonakis, I.K.; Poskus, M.D.; Scott, A.L.; Selfors, L.M.; Lin, J.-R.; Dillon, D.A.; Pathania, S.; Sorger, P.K.; Mills, G.B.; Brugge, J.S. Fibroblast–tumor cell signaling limits HER2 kinase therapy response via activation of MTOR and antiapoptotic pathways. Proc. Natl. Acad. Sci. USA 2020, 117, 16500–16508. [Google Scholar] [CrossRef]
- Qiao, A.; Gu, F.; Guo, X.; Zhang, X.; Fu, L. Breast cancer-associated fibroblasts: Their roles in tumor initiation, progression and clinical applications. Front. Med. 2016, 10, 33–40. [Google Scholar] [CrossRef]
- Hasebe, T. Tumor–stromal interactions in breast tumor progression—Significance of histological heterogeneity of tumor—Stromal fibroblasts. Expert Opin. Ther. Targets 2013, 17, 449–460. [Google Scholar] [CrossRef]
- Suh, J.; Kim, D.; Lee, Y.; Jang, J.; Surh, Y. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol. Carcinog. 2020, 59, 1028–1040. [Google Scholar] [CrossRef]
- Matà, R.; Palladino, C.; Nicolosi, M.L.; Presti, A.R.L.; Malaguarnera, R.; Ragusa, M.; Sciortino, D.; Morrione, A.; Maggiolini, M.; Vella, V.; et al. IGF-I induces upregulation of DDR1 collagen receptor in breast cancer cells by suppressing MIR-199a-5p through the PI3K/AKT pathway. Oncotarget 2015, 7, 7683–7700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, K.M.; Pettee, K.M.; Rubinic-Minotti, K.; Su, R.; Nestor-Kalinoski, A.; Eisenmann, K.M. Carcinoma associated fibroblasts (CAFs) promote breast cancer motility by suppressing mammalian Diaphanous-related formin-2 (mDia2). PLoS ONE 2018, 13, e0195278. [Google Scholar] [CrossRef] [PubMed]
- Farmaki, E.; Chatzistamou, I.; Kaza, V.; Kiaris, H. A CCL8 gradient drives breast cancer cell dissemination. Oncogene 2016, 35, 6309–6318. [Google Scholar] [CrossRef]
- Erez, N.; Glanz, S.; Raz, Y.; Avivi, C.; Barshack, I. Cancer Associated Fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013, 437, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Lee, K.W.; Cho, J.A.; Park, H.; Lim, E.H. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2011, 40, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3–p38 MAPK signalling. Cancer Lett. 2019, 442, 320–333. [Google Scholar] [CrossRef]
- Giorello, M.B.; Borzone, F.R.; Labovsky, V.; Piccioni, F.V.; Chasseing, N.A. Cancer-associated fibroblasts in the breast tumor microenvironment. J. Mammary Gland. Biol. Neoplasia 2021, 1–21. [Google Scholar] [CrossRef]
- Zhang, X.H.-F.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massagué, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 154, 1060–1073. [Google Scholar] [CrossRef] [Green Version]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 signaling plays a critical role in the epithelial–mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, J.-S.; Kang, M.-J.; Suchar, A.M.; Shimamura, T.; Kohn, E.A.; Michalowska, A.M.; Jordan, V.C.; Hirohashi, S.; Wakefield, L.M. Chemokine (C-C Motif) Ligand 2 mediates the prometastatic effect of dysadherin in human breast cancer cells. Cancer Res. 2006, 66, 7176–7184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuhara, R.; Irié, T.; Suzuki, K.; Sawada, T.; Miwa, N.; Sasaki, A.; Tsunoda, Y.; Nakamura, S.; Mishima, K. The β-catenin signaling pathway induces aggressive potential in breast cancer by up-regulating the chemokine CCL5. Exp. Cell Res. 2015, 338, 22–31. [Google Scholar] [CrossRef]
- Chen, J.-Q.; Russo, J. ERα-negative and triple negative breast cancer: Molecular features and potential therapeutic approaches. Biochim. Biophys. Acta Bioenerg. 2009, 1796, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Hartman, Z.C.; Poage, G.M.; Den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory Cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [Green Version]
- Lv, D.; Zhang, Y.; Kim, H.-J.; Zhang, L.; Ma, X. CCL5 as a potential immunotherapeutic target in triple-negative breast cancer. Cell. Mol. Immunol. 2013, 10, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andò, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2011, 8, 263–275. [Google Scholar] [CrossRef]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2015, 7, 1262–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, C.; Barone, I.; Vircillo, V.; Panza, S.; Malivindi, R.; Gelsomino, L.; Pellegrino, M.; Rago, V.; Mauro, L.; Lanzino, M.; et al. Activated FXR inhibits leptin signaling and counteracts tumor-promoting activities of cancer-associated fibroblasts in breast malignancy. Sci. Rep. 2016, 6, 21782. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Li, K.; Pang, X.; Guo, B.; Su, M.; Huang, Y.; Wang, N.; Ji, F.; Zhong, C.; Yang, J.; et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 2016, 35, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luga, V.; Wrana, J.L. Tumor–stroma interaction: Revealing fibroblast-secreted exosomes as potent regulators of wnt-planar cell polarity signaling in cancer metastasis. Cancer Res. 2013, 73, 6843–6847. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-S.; Ma, S.; Dou, H.; Liu, F.; Zhang, S.-Y.; Jiang, C.; Xiao, M.; Huang, Y.-X. Breast cancer-derived exosomes regulate cell invasion and metastasis in breast cancer via miR-146a to activate cancer associated fibroblasts in tumor microenvironment. Exp. Cell Res. 2020, 391, 111983. [Google Scholar] [CrossRef]
- Chen, B.; Sang, Y.; Song, X.; Zhang, D.; Wang, L.; Zhao, W.; Liang, Y.; Zhang, N.; Yang, Q. Exosomal miR-500a-5p derived from cancer-associated fibroblasts promotes breast cancer cell proliferation and metastasis through targeting USP28. Theranostics 2021, 11, 3932–3947. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Liu, B.; DeFilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 2013, 15, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Jia, H.-H.; Xu, Y.-Q.; Zhou, X.; Zhao, X.-H.; Wang, Y.; Song, X.; Zhu, Z.-Y.; Sun, T.; Dou, Y.; et al. Paracrine and epigenetic control of CAF-induced metastasis: The role of HOTAIR stimulated by TGF-ß1 secretion. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xuejun, T.; Tian, X.; Mohammad, M.; Movassaghi, M.; Naber, S.P.; Kuperwasser, C.; Buchsbaum, R.J. The fibroblast Tiam1-osteopontin pathway modulates breast cancer invasion and metastasis. Breast Cancer Res. 2016, 18, 1–15. [Google Scholar]
- Wang, M.; Zhang, J.; Huang, Y.; Ji, S.; Shao, G.; Feng, S.; Chen, D.; Zhao, K.; Wang, Z.; Wu, A. Cancer-associated fibroblasts autophagy enhances progression of triple-negative breast cancer cells. Med Sci. Monit. 2017, 23, 3904–3912. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Fu, M.; Wang, J.; Xia, C.; Zhang, H.; Xiong, Y.; He, J.; Liu, J.; Liu, B.; Pan, S.; et al. TGF-β1-activated cancer-associated fibroblasts promote breast cancer invasion, metastasis and epithelial-mesenchymal transition by autophagy or overexpression of FAP-α. Biochem. Pharmacol. 2021, 188, 114527. [Google Scholar] [CrossRef]
- Gagliano, T.; Shah, K.; Gargani, S.; Lao, L.; Alsaleem, M.; Chen, J.; Ntafis, V.; Huang, P.; Ditsiou, A.; Vella, V.; et al. PIK3Cδ expression by fibroblasts promotes triple-negative breast cancer progression. J. Clin. Investig. 2020, 130, 3188–3204. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial–mesenchymal plasticity. Life Sci. Alliance 2019, 2, e201900425. [Google Scholar] [CrossRef]
- Choi, Y.P.; Lee, J.H.; Gao, M.-Q.; Gil Kim, B.; Kang, S.; Kim, S.H.; Cho, N.H. Cancer-associated fibroblast promote transmigration through endothelial brain cells in three-dimensionalin vitromodels. Int. J. Cancer 2014, 135, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Casar, J.M.; Alvarez, E.; Junquera, S.; Marin, L.; Bongera, M.; Vazquez, J.; Vizoso, F.J.; González, L.O.; González, L. Comparative analysis and clinical value of the expression of metalloproteases and their inhibitors by intratumor stromal fibroblasts and those at the invasive front of breast carcinomas. Breast Cancer Res. Treat. 2009, 116, 39–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Mosher, R.; Seo, S.; Beebe, D.; Friedl, A. Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility. Am. J. Pathol. 2011, 178, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Ahirwar, D.K.; Nasser, M.W.; Ouseph, M.M.; Elbaz, M.; Cuitiño, M.C.; Kladney, R.D.; Varikuti, S.; Kaul, K.; Satoskar, A.R.; Ramaswamy, B.; et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene 2018, 37, 4428–4442. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. MCR 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Gui, Y.; Aguilar-Mahecha, A.; Krzemien, U.; Hosein, A.; Buchanan, M.; LaFleur, J.; Pollak, M.N.; Ferrario, C.; Basik, M. Metastatic breast carcinoma–Associated fibroblasts have enhanced protumorigenic properties related to increased IGF2 Expression. Clin. Cancer Res. 2019, 25, 7229–7242. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Peterse, J.L.; van ’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nat. Cell Biol. 2011, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, T.; Zhang, P.; Sun, Y.; Wang, Y.; Tong, J.; Dai, H.; Hua, Z. High throughput sequencing identifies breast cancer-secreted exosomal LncRNAs initiating pulmonary pre-metastatic niche formation. Gene 2019, 710, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.; Allan, A.L. Molecular mechanisms of breast cancer metastasis to the lung: Clinical and experimental perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef] [Green Version]
- Qi, F.; He, T.; Jia, L.; Song, N.; Guo, L.; Ma, X.; Wang, C.; Xuhui, M.; Fu, Y.; Lin, J.; et al. The miR-30 family inhibits pulmonary vascular hyperpermeability in the premetastatic phase by direct targeting of Skp2. Clin. Cancer Res. 2015, 21, 3071–3080. [Google Scholar] [CrossRef] [Green Version]
- Canesin, G.; Cuevas, E.P.; Santos, V.; López-Menéndez, C.; Moreno-Bueno, G.; Huang, Y.; Csiszar, K.; Portillo, F.; Peinado, H.; Lyden, D.; et al. Lysyl oxidase-like 2 (LOXL2) and E47 EMT factor: Novel partners in E-cadherin repression and early metastasis colonization. Oncogene 2014, 34, 951–964. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.-F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Xiong, S.; Mao, Y.; Chen, M.; Ma, X.; Zhou, X.; Ma, Z.; Liu, F.; Huang, Z.; Luo, Q.; et al. Periostin promotes immunosuppressive premetastatic niche formation to facilitate breast tumour metastasis. J. Pathol. 2016, 239, 484–495. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.D.P.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.; Sahai, E.; et al. Mesenchymal cancer cell-stroma crosstalk promotes niche activation, epithelial reversion, and metastatic colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, C.; Zha, H.; Long, H.; Wang, X.; Yang, F.; Gao, J.; Hu, C.; Zhou, L.; Guo, B.; Zhu, B. C3a-C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J. Exp. Clin. Cancer Res. 2020, 39, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Han, Y.; Kang, Y. Bone marrow niches in the regulation of bone metastasis. Br. J. Cancer 2021, 124, 1912–1920. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.C.; Kothari, A.N.; Kuo, P.C.; Mi, Z. Cancer stemness in bone marrow micrometastases of human breast cancer. Surgery 2018, 163, 330–335. [Google Scholar] [CrossRef]
- Sasaki, S.; Baba, T.; Nishimura, T.; Hayakawa, Y.; Hashimoto, S.-I.; Gotoh, N.; Mukaida, N. Essential roles of the interaction between cancer cell-derived chemokine, CCL4, and intra-bone CCR5-expressing fibroblasts in breast cancer bone metastasis. Cancer Lett. 2016, 378, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Mi, Z.; Bhattacharya, S.D.; Kim, V.M.; Guo, H.; Talbot, L.J.; Kuo, P.C. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 2011, 32, 477–487. [Google Scholar] [CrossRef]
- Hauge, A.; Rofstad, E.K. Antifibrotic therapy to normalize the tumor microenvironment. J. Transl. Med. 2020, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Singha, N.C.; Nekoroski, T.; Zhao, C.; Symons, R.; Jiang, P.; Frost, G.I.; Huang, Z.; Shepard, H.M.; Goglia, A.G.; Delsite, R.; et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol. Cancer Ther. 2015, 14, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabets, M.; Gifford, A.M.; Scheel, C.; Nilsson, B.; Reinhardt, F.; Bray, M.-A.; Carpenter, A.; Jirström, K.; Magnusson, K.; Ebert, B.L.; et al. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J. Clin. Investig. 2011, 121, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Qassab, Y.; Grassilli, S.; Brugnoli, F.; Vezzali, F.; Capitani, S.; Bertagnolo, V. Protective role of all-trans retinoic acid (ATRA) against hypoxia-induced malignant potential of non-invasive breast tumor derived cells. BMC Cancer 2018, 18, 1194. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liao, S.; Diop-Frimpong, B.; Chen, W.; Goel, S.; Naxerova, K.; Ancukiewicz, M.; Boucher, Y.; Jain, R.K.; Xu, L. TGF- blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2012, 109, 16618–16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Schiemann, W.P. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-β signaling and metastasis. Breast Cancer Res. 2009, 11, R68. [Google Scholar] [CrossRef] [Green Version]
- Van der Spek, Y.M.; Kroep, J.R.; Tollenaar, R.A.E.M.; Mesker, W.E. Chemotherapy resistance and stromal targets in breast cancer treatment: A review. Mol. Biol. Rep. 2020, 47, 8169–8177. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Panza, S.; Augimeri, G.; Giordano, F.; Malivindi, R.; Gelsomino, L.; Marsico, S.; Győrffy, B.; Bonofiglio, D.; Andò, S.; et al. Phosphodiesterase 5 (PDE5) is Highly Expressed in Cancer-Associated Fibroblasts and Enhances Breast Tumor Progression. Cancers 2019, 11, 1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantziarka, P.; Sukhatme, V.; Crispino, S.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)—Selective PDE5 inhibitors as. Ecancermedicalscience 2018, 12, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Nogueira, P.; Fuster, G.; Gutierrez-Uzquiza, Á.; Gascón, P.; Carbó, N.; Bragado, P. Cancer-Associated Fibroblasts in Breast Cancer Treatment Response and Metastasis. Cancers 2021, 13, 3146. https://doi.org/10.3390/cancers13133146
Fernández-Nogueira P, Fuster G, Gutierrez-Uzquiza Á, Gascón P, Carbó N, Bragado P. Cancer-Associated Fibroblasts in Breast Cancer Treatment Response and Metastasis. Cancers. 2021; 13(13):3146. https://doi.org/10.3390/cancers13133146
Chicago/Turabian StyleFernández-Nogueira, Patricia, Gemma Fuster, Álvaro Gutierrez-Uzquiza, Pere Gascón, Neus Carbó, and Paloma Bragado. 2021. "Cancer-Associated Fibroblasts in Breast Cancer Treatment Response and Metastasis" Cancers 13, no. 13: 3146. https://doi.org/10.3390/cancers13133146
APA StyleFernández-Nogueira, P., Fuster, G., Gutierrez-Uzquiza, Á., Gascón, P., Carbó, N., & Bragado, P. (2021). Cancer-Associated Fibroblasts in Breast Cancer Treatment Response and Metastasis. Cancers, 13(13), 3146. https://doi.org/10.3390/cancers13133146