
Hampering Stromal Cells in the Tumor Microenvironment as a Therapeutic Strategy to Destem Cancer Stem Cells


Abstract
:Simple Summary
Abstract
1. Introduction of the Relationship between CSCs and TME Components
2. The Interplay between Stromal Cells and CSCs within the TME
2.1. Endothelial Cells
2.2. Cancer Associated Fibroblasts
2.3. Adipocytes
3. The Interplay between Immune Cells and CSCs within the TME
3.1. Tumor Associated Macrophages
3.2. Natural Killer Cells
3.3. Myeloid-Derived Suppressor Cells
3.4. Tumor-Associated Neutrophils
3.5. Regulatory T Cells
4. Interplay of Various Cellular Factors within the TME in the Regulation of Cancer Stemness and Immune Evasion
5. Conclusions
6. Future Perspectives on CSC-Targeted Therapies
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Bhattacharya, R.; Ye, X.; Fan, F.; Boulbes, D.R.; Xia, L.; Ellis, L.M. Endothelial cells activate the cancer stem cell-associated NANOGP8 pathway in colorectal cancer cells in a paracrine fashion. Mol. Oncol. 2017, 11, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Castillejo, C.; Sanchez-Sanchez, F.; Andreu-Agullo, C.; Ferron, S.R.; Aroca-Aguilar, J.D.; Sanchez, P.; Mira, H.; Escribano, J.; Farinas, I. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat. Neurosci. 2006, 9, 331–339. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef]
- Fessler, E.; Borovski, T.; Medema, J.P. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol. Cancer 2015, 14, 157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Dong, Z.; Lauxen, I.S.; Filho, M.S.; Nor, J.E. Endothelial cell-secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype. Cancer Res. 2014, 74, 2869–2881. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Pan, J.; Yu, L.; Liu, H.; Shu, X.; Sun, L.; Lou, J.; Yang, Z.; Ran, Y. Tumor endothelial cells promote metastasis and cancer stem cell-like phenotype through elevated Epiregulin in esophageal cancer. Am. J. Cancer Res. 2016, 6, 2277. [Google Scholar]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Zeh, H.J., 3rd; et al. Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers 2020, 12, 2652. [Google Scholar] [CrossRef]
- Lewis, S.; Jones, D. The Role of Cancer Associated Fibroblasts in Maintaining Cancer Stem Cells. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Huang, T.X.; Guan, X.Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 2019, 9, 1889–1904. [Google Scholar]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Li, M.; Cao, L.M.; Gu, Q.H.; Deng, P.B.; Tan, Y.; Hu, C.P. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. Int. J. Med. 2019, 112, 581–590. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Kadel, D.; Zhang, Y.; Sun, H.-R.; Zhao, Y.; Dong, Q.Z.; Qin, L.X. Current perspectives of cancer-associated fibroblast in therapeutic resistance: Potential mechanism and future strategy. Cell Biol. Toxicol. 2019, 35, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct interactions with cancer-associated fibroblasts lead to enhanced pancreatic cancer stem cell function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Louisa Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.M.; Attieh, Y.; Barbazan, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef]
- Kinugasa, Y.; Matsui, T.; Takakura, N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem Cells 2014, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Yashiro, M.; Nishii, T.; Matsuoka, J.; Fuyuhiro, Y.; Morisaki, T.; Fukuoka, T.; Shimizu, K.; Shimizu, T.; Miwa, A.; et al. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-β signaling. Int. J. Cancer 2014, 134, 1785–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, E.Y.; Lo, J.; Cheng, B.Y.; Ma, M.K.; Lee, J.M.; Ng, J.K.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Barone, I.; Vircillo, V.; Panza, S.; Malivindi, R.; Gelsomino, L.; Pellegrino, M.; Rago, V.; Mauro, L.; Lanzino, M.; et al. Activated FXR Inhibits Leptin Signaling and Counteracts Tumor-promoting Activities of Cancer-Associated Fibroblasts in Breast Malignancy. Sci. Rep. 2016, 6, 21782. [Google Scholar] [CrossRef]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.L.; Dive, C.; Renehan, A.G. Biological mechanisms linking obesity and cancer risk: New perspectives. Annu. Rev. Med. 2010, 61, 301–316. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Caers, J.; Deleu, S.; Belaid, Z.; De Raeve, H.; Van Valckenborgh, E.; De Bruyne, E.; Defresne, M.P.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 2007, 21, 1580–1584. [Google Scholar] [CrossRef] [Green Version]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J. Methodol. 2016, 6, 43. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [Green Version]
- He, J.Y.; Wei, X.H.; Li, S.J.; Liu, Y.; Hu, H.L.; Li, Z.Z.; Kuang, X.H.; Wang, L.; Shi, X.; Yuan, S.T.; et al. Adipocyte-derived IL-6 and leptin promote breast Cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun. Signal 2018, 16, 100. [Google Scholar] [CrossRef] [Green Version]
- Cardenas, C.; Montagna, M.K.; Pitruzzello, M.; Lima, E.; Mor, G.; Alvero, A.B. Adipocyte microenvironment promotes Bclxl expression and confers chemoresistance in ovarian cancer cells. Apoptosis 2017, 22, 558–569. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, Y.; Yan, X.; Yan, F.; Sun, Y.; Zeng, J.; Waigel, S.; Yin, Y.; Fraig, M.M.; Egilmez, N.K. Circulating adipose fatty acid binding protein is a new link underlying obesity-associated breast/mammary tumor development. Cell Metab. 2018, 28, 689–705.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Mo, Y.; Li, M.T.; Zou, S.W.; Cheng, Z.L.; Sun, Y.P.; Xiong, Y.; Guan, K.L.; Lei, Q.Y. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J. Clin. Investig. 2014, 124, 5453–5465. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Abarzua-Catalan, L.; Trigo, C.; Delpiano, A.; Sanhueza, C.; García, K.; Ibañez, C.; Hormazábal, K.; Diaz, D.; Brañes, J. Leptin stimulates migration and invasion and maintains cancer stem-like properties in ovarian cancer cells: An explanation for poor outcomes in obese women. Oncotarget 2015, 6, 21100. [Google Scholar] [CrossRef] [Green Version]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef] [Green Version]
- Quatromoni, J.G.; Eruslanov, E. Tumor-associated macrophages: Function, phenotype, and link to prognosis in human lung cancer. Am. J. Transl. Res. 2012, 4, 376–389. [Google Scholar]
- Jung, K.Y.; Cho, S.W.; Kim, Y.A.; Kim, D.; Oh, B.C.; Park, D.J.; Park, Y.J. Cancers with Higher Density of Tumor-Associated Macrophages Were Associated with Poor Survival Rates. J. Pathol. Transl. Med. 2015, 49, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Ge, Z.; Ding, S. The Crosstalk between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Lee, H.W.; Choi, H.J.; Ha, S.J.; Lee, K.T.; Kwon, Y.G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim. Biophys. Acta. Rev. Cancer 2013, 1835, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizutani, K.; Sud, S.; McGregor, N.A.; Martinovski, G.; Rice, B.T.; Craig, M.J.; Varsos, Z.S.; Roca, H.; Pienta, K.J. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia (New York N.Y.) 2009, 11, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Tsang, J.Y.S.; Li, D.; Niu, N.; Ho, D.H.H.; Lau, K.F.; Lui, V.C.H.; Lamb, J.R.; Chen, Y.; Tam, P.K.H. Inhibition of TGF-β signaling in combination with TLR7 ligation re-programs a tumoricidal phenotype in tumor-associated macrophages. Cancer Lett. 2013, 331, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J. Targeting TGF-β Signaling for Therapeutic Gain. Cold Spring Harb. Perspect. Biol. 2017, 9, a022301. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ding, Z.Y.; Li, S.; Liu, S.; Xiao, C.; Li, Z.; Zhang, B.X.; Chen, X.P.; Yang, X. Targeting transforming growth factor-β signaling for enhanced cancer chemotherapy. Theranostics 2021, 11, 1345–1363. [Google Scholar] [CrossRef]
- Sarode, P.; Zheng, X.; Giotopoulou, G.A.; Weigert, A.; Kuenne, C.; Günther, S.; Friedrich, A.; Gattenlöhner, S.; Stiewe, T.; Brüne, B.; et al. Reprogramming of tumor-associated macrophages by targeting β-catenin/FOSL2/ARID5A signaling: A potential treatment of lung cancer. Sci. Adv. 2020, 6, eaaz6105. [Google Scholar] [CrossRef]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2017, 8, 8633–8647. [Google Scholar] [CrossRef] [Green Version]
- Higashino, N.; Koma, Y.I.; Hosono, M.; Takase, N.; Okamoto, M.; Kodaira, H.; Nishio, M.; Shigeoka, M.; Kakeji, Y.; Yokozaki, H. Fibroblast activation protein-positive fibroblasts promote tumor progression through secretion of CCL2 and interleukin-6 in esophageal squamous cell carcinoma. Lab. Investig. 2019, 99, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFNγ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Shan, S.; Yang, Z.; Gu, X.; Wang, Y.; Wang, C.; Ren, T. IL-33 blockade suppresses tumor growth of human lung cancer through direct and indirect pathways in a preclinical model. Oncotarget 2017, 8, 68571–68582. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zeng, Y.; Zhang, M.; Ma, H.; Xu, B.; Jiang, H.; Wang, J.; Li, G. CD56brightCD16− natural killer cells are shifted toward an IFN-γ-promoting phenotype with reduced regulatory capacity in osteoarthritis. Hum. Immunol. 2019, 80, 871–877. [Google Scholar] [CrossRef]
- Bassani, B.; Baci, D.; Gallazzi, M.; Poggi, A.; Bruno, A.; Mortara, L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities into Potent Anti-Tumor Effects. Cancers 2019, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Stabile, H.; Nisti, P.; Morrone, S.; Pagliara, D.; Bertaina, A.; Locatelli, F.; Santoni, A.; Gismondi, A. Multifunctional human CD56 low CD16 low natural killer cells are the prominent subset in bone marrow of both healthy pediatric donors and leukemic patients. Haematologica 2015, 100, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef]
- Bruno, A.; Bassani, B.; D’Urso, D.G.; Pitaku, I.; Cassinotti, E.; Pelosi, G.; Boni, L.; Dominioni, L.; Noonan, D.M.; Mortara, L.; et al. Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J. 2018, 32, 5365–5377. [Google Scholar] [CrossRef] [Green Version]
- Keskin, D.B.; Allan, D.S.; Rybalov, B.; Andzelm, M.M.; Stern, J.N.; Kopcow, H.D.; Koopman, L.A.; Strominger, J.L. TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3378–3383. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.; Ferlazzo, G.; Albini, A.; Noonan, D.M. A think tank of TINK/TANKs: Tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J. Natl. Cancer Inst. 2014, 106, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Herszényi, L.; Hritz, I.; Lakatos, G.; Varga, M.Z.; Tulassay, Z. The behavior of matrix metalloproteinases and their inhibitors in colorectal cancer. Int. J. Mol. Sci. 2012, 13, 13240–13263. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.F.; Yip, C.W.; Wong, N.C.; Fong, D.Y.; Ng, L.W.; Wan, A.M.; Wong, C.K.; Cheung, T.T.; Ng, I.O.; Poon, R.T.; et al. Granulin-epithelin precursor renders hepatocellular carcinoma cells resistant to natural killer cytotoxity. Cancer Immunol. Res. 2014, 2, 1209–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK Cells Recognize and Kill Human Glioblastoma Cells with Stem Cell-Like Properties. J. Immunol. 2009, 182, 3530. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, S.; Rasool, S.; Maccalli, C. The Cross Talk between Cancer Stem Cells/Cancer Initiating Cells and Tumor Microenvironment: The Missing Piece of the Puzzle for the Efficient Targeting of these Cells with Immunotherapy. Cancer Microenviron. 2019, 12, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Targeting Cancer Stem Cells with Natural Killer Cell Immunotherapy. Expert Opin. Biol. Ther. 2017, 17, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Tallerico, R.; Garofalo, C.; Carbone, E. A New Biological Feature of Natural Killer Cells: The Recognition of Solid Tumor-Derived Cancer Stem Cells. Front. Immunol. 2016, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Chen, M.; Smith, R.C.; Hagino, T.; Perez-Cunningham, J.; Sckisel, G.D.; Urayama, S.; et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J. Immunol. 2015, 195, 4010–4019. [Google Scholar] [CrossRef] [Green Version]
- Tallerico, R.; Todaro, M.; Di Franco, S.; Maccalli, C.; Garofalo, C.; Sottile, R.; Palmieri, C.; Tirinato, L.; Pangigadde, P.N.; La Rocca, R.; et al. Human NK cells selective targeting of colon cancer-initiating cells: A role for natural cytotoxicity receptors and MHC class I molecules. J. Immunol. 2013, 190, 2381–2390. [Google Scholar] [CrossRef] [Green Version]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA Interference Targeting Transforming Growth Factor-β Enhances NKG2D-Mediated Antiglioma Immune Response, Inhibits Glioma Cell Migration and Invasiveness, and Abrogates Tumorigenicity In vivo. Cancer Res. 2004, 64, 7596. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.; Manzini, C.; Vitale, M.; Balsamo, M.; Ognio, E.; Boitano, M.; Queirolo, P.; Moretta, L.; Mingari, M.C. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 2009, 21, 793–801. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Komura, N.; Mabuchi, S.; Shimura, K.; Yokoi, E.; Kozasa, K.; Kuroda, H.; Takahashi, R.; Sasano, T.; Kawano, M.; Matsumoto, Y.; et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol. Immunother. 2020, 69, 2477–2499. [Google Scholar] [CrossRef]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef] [Green Version]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Cao, B.; Liang, X.; Lu, S.; Luo, H.; Wang, Z.; Wang, S.; Jiang, J.; Lang, J.; Zhu, G. Microenvironmental oxygen pressure orchestrates an anti-and pro-tumoral γδ T cell equilibrium via tumor-derived exosomes. Oncogene 2019, 38, 2830–2843. [Google Scholar] [CrossRef]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Ai, L.; Mu, S.; Sun, C.; Fan, F.; Yan, H.; Qin, Y.; Cui, G.; Wang, Y.; Guo, T.; Mei, H.; et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol. Cancer 2019, 18, 88. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, E.; Mabuchi, S.; Komura, N.; Shimura, K.; Kuroda, H.; Kozasa, K.; Takahashi, R.; Sasano, T.; Kawano, M.; Matsumoto, Y.; et al. The role of myeloid-derived suppressor cells in endometrial cancer displaying systemic inflammatory response: Clinical and preclinical investigations. Oncoimmunology 2019, 8, e1662708. [Google Scholar] [CrossRef]
- Granot, Z.; Jablonska, J. Distinct functions of neutrophil in cancer and its regulation. Mediat. Inflamm. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef] [Green Version]
- Bekes, E.M.; Schweighofer, B.; Kupriyanova, T.A.; Zajac, E.; Ardi, V.C.; Quigley, J.P.; Deryugina, E.I. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am. J. Pathol. 2011, 179, 1455–1470. [Google Scholar] [CrossRef]
- Yu, P.; Huang, Y.; Han, Y.; Lin, L.; Sun, W.; Rabson, A.; Wang, Y.; Shi, Y. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils. Oncogene 2017, 36, 482–490. [Google Scholar] [CrossRef]
- Jablonska, J.; Wu, C.F.; Andzinski, L.; Leschner, S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Huh, S.J.; Liang, S.; Sharma, A.; Dong, C.; Robertson, G.P. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 2010, 70, 6071–6082. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, J.; Yang, L.; Li, J.; Wu, W.; Huang, M.; Lin, L.; Su, S. Tumor-Contacted Neutrophils Promote Metastasis by a CD90-TIMP-1 Juxtacrine-Paracrine Loop. Clin. Cancer Res. 2019, 25, 1957–1969. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, J.K.; Jeon, H.Y.; Ham, S.W.; Kim, H. CD133 Regulates IL-1β signaling and neutrophil recruitment in glioblastoma. Mol. Cells 2017, 40, 515. [Google Scholar]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, D.; Tsun, A.; Li, B. FOXP3+ regulatory T cells and their functional regulation. Cell. Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Napoletano, C.; Bellati, F.; Ruscito, I.; Pernice, M.; Zizzari, I.G.; Caponnetto, S.; Tomao, F.; Frigerio, L.; Liberati, M.; Rughetti, A.; et al. Immunological and Clinical Impact of Cancer Stem Cells in Vulvar Cancer: Role of CD133/CD24/ABCG2-Expressing Cells. Anticancer Res. 2016, 36, 5109–5116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.K.; Yang, B.; Liu, H.; Hu, Y.; Yang, J.L.; Wu, L.L.; Zhou, Z.H.; Jiao, S.C. Regulatory T cells increase in breast cancer and in stage IV breast cancer. Cancer Immunol. Immunother. 2012, 61, 911–916. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, X.; Qi, P.; Ye, Y.; Shen, W.; Leng, L.; Wang, L.; Li, X.; Luo, X.; Chen, Y.; et al. Sox2 Communicates with Tregs Through CCL1 to Promote the Stemness Property of Breast Cancer Cells. Stem Cells 2017, 35, 2351–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhang, C.; Wang, B.; Zhang, H.; Qin, G.; Li, C.; Cao, L.; Gao, Q.; Ping, Y.; Zhang, K.; et al. Regulatory T cells promote glioma cell stemness through TGF-beta-NF-kappaB-IL6-STAT3 signaling. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Hong, J.; Yun, C.O. Regulatory T Cells Induce Metastasis by Increasing Tgf-beta and Enhancing the Epithelial-Mesenchymal Transition. Cells 2019, 8, 1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wang, B.; Guan, C.; Wu, B.; Cai, C.; Wang, M.; Zhang, B.; Liu, T.; Yang, P. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J. Leukoc. Biol. 2011, 89, 85–91. [Google Scholar] [CrossRef]
- Xu, Y.; Wan, Y.; Liu, J.; Wang, Y.; Li, S.; Xing, H.; Tang, K.; Tian, Z.; Rao, Q.; Wang, M. Regulatory T cells promote the stemness of acute myeloid leukemia cells through IL10 cytokine related signaling pathway. Blood 2017, 130, 2727. [Google Scholar]
- Beck, B.; Driessens, G.; Goossens, S.; Youssef, K.K.; Kuchnio, A.; Caauwe, A.; Sotiropoulou, P.A.; Loges, S.; Lapouge, G.; Candi, A.; et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 2011, 478, 399–403. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.-P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T reg cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Ji, J.; Eggert, T.; Budhu, A.; Forgues, M.; Ji, J.; Eggert, T.; Budhu, A.; Forgues, M.; Takai, A.; Dang, H.; et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 2015, 62, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Wei, Y.; Han, D.; Li, Y.; Shi, S.; Jiao, D.; Wu, J.; Zhang, Q.; Shi, C.; Yang, L.; et al. Interaction with CD68 and regulation of GAS6 expression by endosialin in fibroblasts drives recruitment and polarization of marophages in hepatocellular carcinoma. Cancer Res. 2020, 80, 3892–3905. [Google Scholar]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.S.; Yeung, T.L.; Yip, K.P.; Wong, K.K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2018, 128, 589–606. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; He, J.; Pan, Q.Z.; Yang, J.; Zhao, J.; Zhang, Y.J.; Huang, Y.; Tang, Y.; Wang, Q.; He, J.; et al. Cancer-associated fibroblast-mediated cellular crosstalk supports hepatocellular carcinoma progression. Hepatology 2021, 73, 1717–1735. [Google Scholar] [CrossRef]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-associated fibroblasts affect intratumoral CD8+ and FoxP3+ T cells via IL6 in the tumor microenvironment. Clin. Cancer Res. 2018, 24, 4820–4833. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-meidated feeforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef]
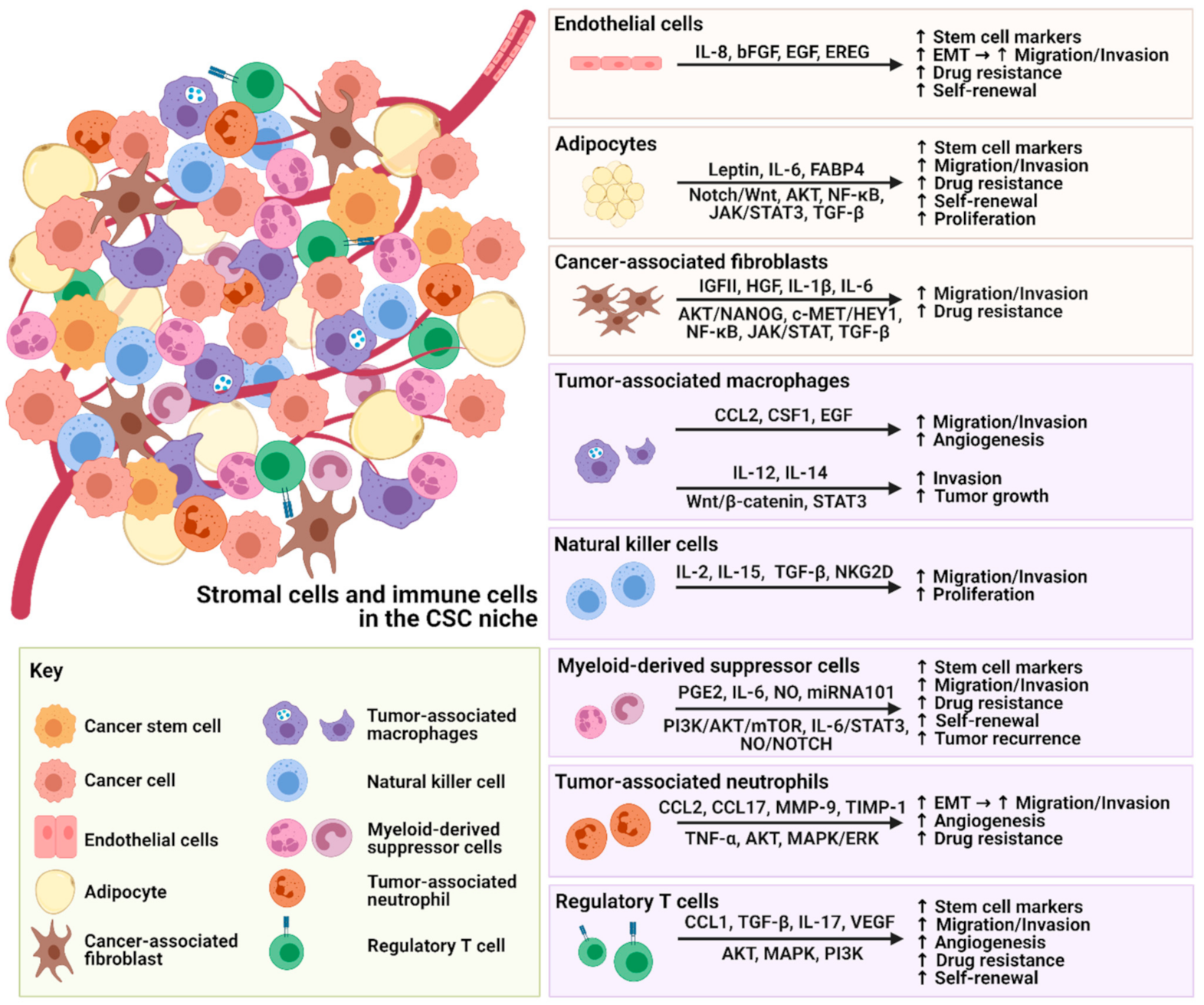

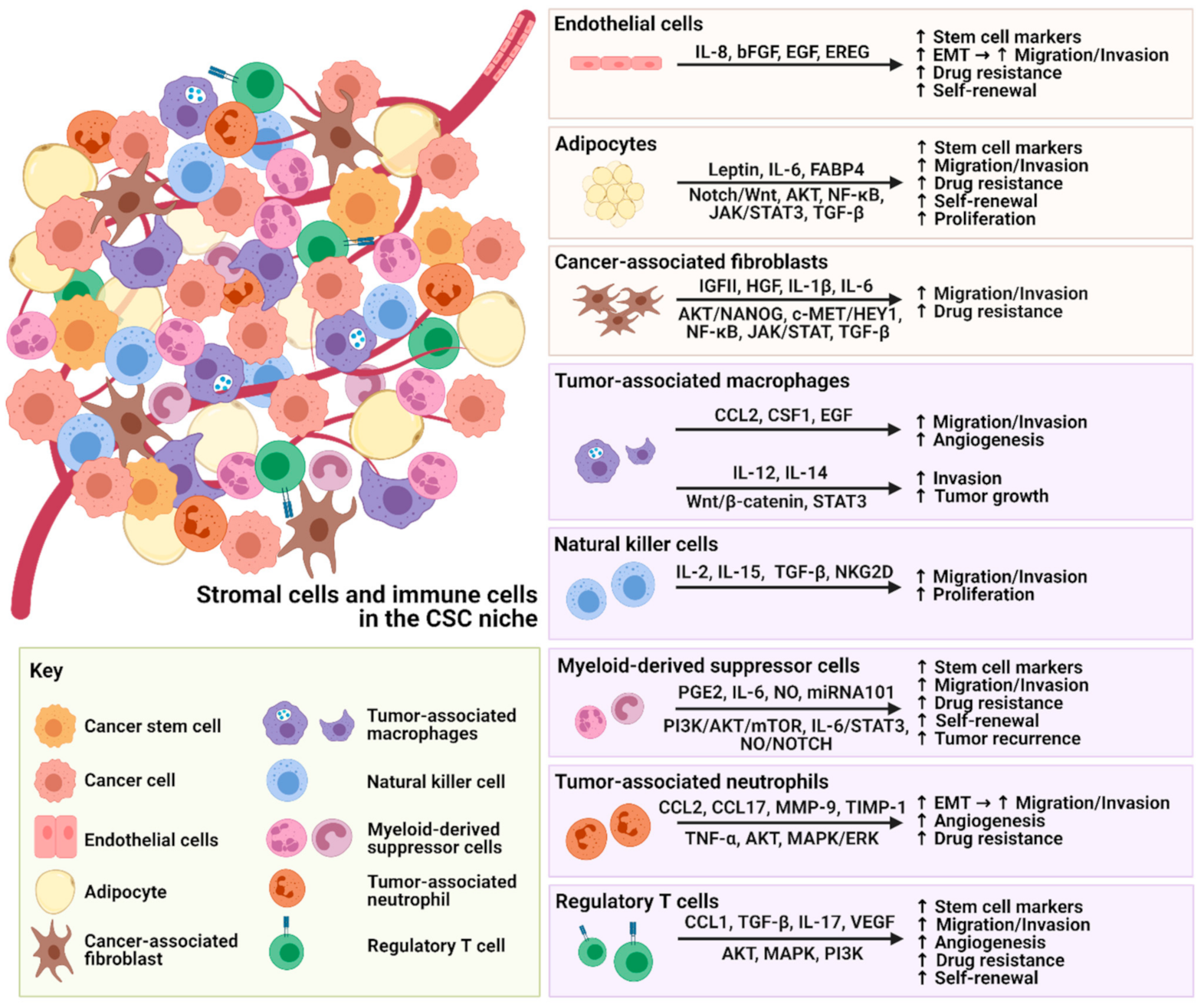{kind=link}
| Type of Targeted Cell | Cancer Type | Novel Therapeutic Strategies | Mode of Action | Effects on Cancer Stemness | References |
|---|---|---|---|---|---|
| Targeted Stromal Cells | - | ||||
| Endothelial cells | Glioblastoma | IL-8 neutralizing antibody | Blockade of endothelial cell-secreted IL-8 | Reduced spheroid size and tumor growth in vivo | [13] |
| Adipocytes | Breast/Mammary cancer | BMS309403 | Inhibiting FABP4 functions | Suppressed tumor growth and tumor volume with decreased IL-6 level and tumor ALDH1 activity | [48] |
| Ovarian cancer | Anti-OB-R blocking peptide | Blockade of leptin receptor | Decreased leptin-induced cell migration and invasion abilities | [51] | |
| CAFs | Breast Cancer | GW4064 | Agonist of FXR | Reduced progression and motility of tumors | [37,38] |
| Pirfenidone (PFD) and doxorubicin | Inhibiting collagen production and tumor growth | Reduced components of ECM, inhibited tumor growth and lung metastasis | [39] | ||
| Targeted Immune Cells | - | ||||
| TAMs | Pancreatic carcinoma | CD40 Agonists | Activating and inducing macrophages | Degraded ECM and improved tumor infiltration of immune cells | [72] |
| Non-small cell lung cancer (NSCLC) | IL-33 neutralizing antibody | Blockade of IL-33 | Inhibited M2-like macrophages polarization via inhibition of IL-10 and VEGF as well as reduced accumulation of Treg cells | [73] | |
| NK cells | Colon cancer | Chondrocytes | Expressing a high level of IL-12 | Increased the infiltrations of both T cells and NK cells, reduced cancer cells and tumor angiogenesis | [91] |
| MDSCs | Breast cancer | Combination of anti-IL-6 antibody and iNOS inhibitor | Targeting MDSC-derived IL-6 and nitric oxide | Reduced spheroid formation stimulated by MDSCs | [97] |
| Endometrial cancer | Doxorubicin and Gr-1 neutralizing antibody or celecoxib | Blockade of MDSCs or inhibiting MDSC functions | Reduced ALHD+ expression and sensitized tumor cells to chemotherapy | [105] | |
| TANs | Breast cancer | Zileuton | Inhibiting neutrophil Alox5 | Reduced lung metastasis | [115] |
| Treg cells | Glioma | Tocilizumab | Blockade of IL-6 receptor | Inhibited tumor growth and CD133 expression induced by Treg cells | [122] |
| Acute myeloid leukemia (AML) | IL-10R neutralizing antibody | Neutralizing IL-10 receptor functions | Reduced side population, sphere formation ability, and expression of OCT4 and NANOG | [125] | |
| Ovarian cancer | CD25 neutralizing antibody | Inhibiting CD25+ Treg cells | Reduced angiogenesis and inhibited tumor growth | [127] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, K.P.S.; Leung, R.W.H.; Lee, T.K.W. Hampering Stromal Cells in the Tumor Microenvironment as a Therapeutic Strategy to Destem Cancer Stem Cells. Cancers 2021, 13, 3191. https://doi.org/10.3390/cancers13133191
Chung KPS, Leung RWH, Lee TKW. Hampering Stromal Cells in the Tumor Microenvironment as a Therapeutic Strategy to Destem Cancer Stem Cells. Cancers. 2021; 13(13):3191. https://doi.org/10.3390/cancers13133191
Chicago/Turabian StyleChung, Katherine Po Sin, Rainbow Wing Hei Leung, and Terence Kin Wah Lee. 2021. "Hampering Stromal Cells in the Tumor Microenvironment as a Therapeutic Strategy to Destem Cancer Stem Cells" Cancers 13, no. 13: 3191. https://doi.org/10.3390/cancers13133191
APA StyleChung, K. P. S., Leung, R. W. H., & Lee, T. K. W. (2021). Hampering Stromal Cells in the Tumor Microenvironment as a Therapeutic Strategy to Destem Cancer Stem Cells. Cancers, 13(13), 3191. https://doi.org/10.3390/cancers13133191