
The Effects of Chemotherapeutics on the Ovarian Cancer Microenvironment



Department of Obstetrics and Gynecology/Section of Gynecologic Oncology, University of Chicago, Chicago, IL 60637, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(13), 3136; https://doi.org/10.3390/cancers13133136
Submission received: 29 April 2021
/
Revised: 6 June 2021
/
Accepted: 7 June 2021
/
Published: 23 June 2021
(This article belongs to the Special Issue Cancer-Associated Fibroblasts: Mechanisms of Tumor Progression and Novel Therapeutic Targets)
{kind=link}
{kind=link}
Abstract
:Simple Summary
Cancer cells are the target of most approved therapies. A growing body of evidence suggests that these agents have important roles in modulating the biology of host cells and their interactions with cancer cells, including blood vessels, fibroblasts, immune and fat cells, among others. This review provides an overview of potential roles of commonly used therapeutics in the tumor microenvironment, with a focus on cancer-associated fibroblasts. This includes an emphasis on therapies commonly used for the treatment of high-grade serous ovarian cancers (e.g., platinum, taxanes, PARP inhibitors, and anti-angiogenic agents). In vitro, in vivo, and clinical studies are included, and perspectives offered on how to best interpret the influence of therapeutics on normal cells.
Abstract
High-grade serous ovarian cancer (HGSOC) is characterized by a complex and dynamic tumor microenvironment (TME) composed of cancer-associated fibroblasts (CAFs), immune cells, endothelial cells, and adipocytes. Although most approved therapies target cancer cells, a growing body of evidence suggests that chemotherapeutic agents have an important role in regulating the biology of the diverse cells that compose the TME. Understanding how non-transformed cells respond and adapt to established therapeutics is necessary to completely comprehend their action and develop novel therapeutics that interrupt undesired tumor–stroma interactions. Here, we review the effects of chemotherapeutic agents on normal cellular components of the host-derived TME focusing on CAFs. We concentrate on therapies used in the treatment of HGSOC and synthesize findings from studies focusing on other cancer types and benign tissues. Agents such as platinum derivatives, taxanes, and PARP inhibitors broadly affect the TME and promote or inhibit the pro-tumorigenic roles of CAFs by modifying the bidirectional cross-talk between tumor and stromal cells in the tumor organ. While most chemotherapy research focuses on cancer cells, these studies emphasize the need to consider all cell types within the tumor organ when evaluating chemotherapeutics.
1. Introduction
High-grade serous ovarian cancer (HGSOC) is the most lethal gynecologic malignancy. It is diagnosed at an advanced stage in 75% of women, which substantially contributes to the poor five-year survival of less than 50%. Despite aggressive treatment with cytoreductive surgery and platinum/taxane-based chemotherapy, 70–85% of patients with HGSOC will experience a recurrence [1]. Furthermore, the chemotherapeutic agents used during treatment can elicit serious off-target effects, including fatigue, myelosuppression, and neuropathy [2]. A small but growing body of literature has identified important biological effects of chemotherapeutics on the non-transformed cells of the tumor microenvironment (TME) [3]. Several chemotherapeutic agents can regulate cancer-associated fibroblasts (CAFs) by modifying their biology in ways that either enhance or restrain their pro-tumorigenic behavior. Dissecting these changes will help us better understand how chemotherapy influences non-malignant host cell populations to modulate disease progression, therapeutic response, and side effects of therapy.
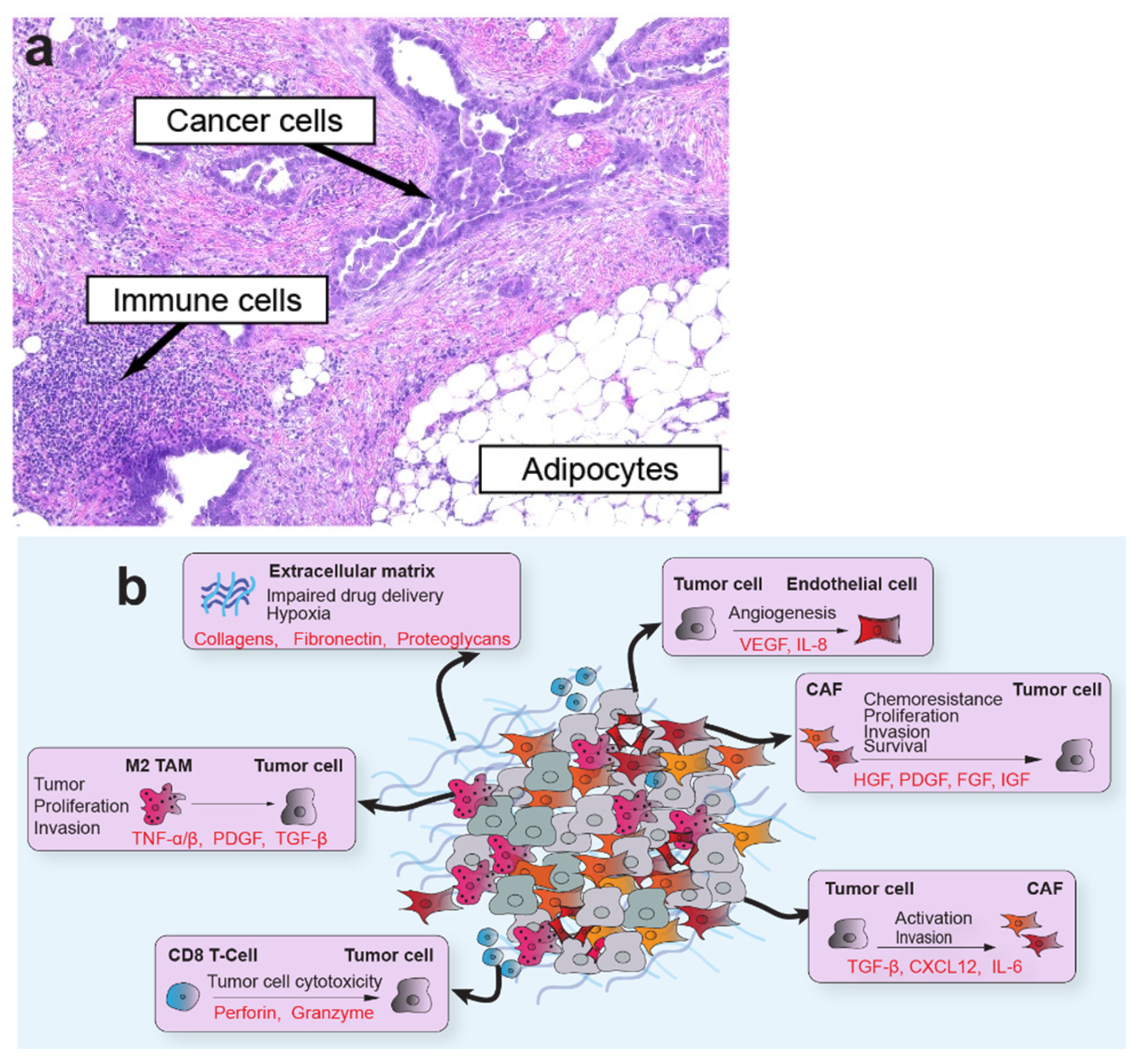
The TME is a multicellular network of tumor cells and host-derived cells, including CAFs, fibroblasts, myofibroblasts, adipocytes, endothelial cells, and immune cells, embedded in a distinct extracellular matrix (ECM; Figure 1). The host cells often comprise the most significant proportion of cells within the tumor organ. As one of the most abundant cellular components of the TME, CAFs are of particular interest in tumor progression [4]. CAFs encompass a highly dynamic populations of activated fibroblasts with pleiotropic functions that shape tumor behavior and engage in a complex network of cross-talk with cellular and non-cellular components of the TME. Their rich secretome supports interactions with multiple cell types in the TME to induce tumor growth, epithelial-to-mesenchymal transition (EMT), angiogenesis, immunosuppression, and ECM remodeling. These elongated cells do not express epithelial, endothelial, or leukocyte markers, and generally maintain stable genomes that lack mutations found in nearby tumor cells [5]. In addition, reactive oxygen species secreted by tumor cells alter CAF metabolism and induce oxidative stress, autophagy, and upregulation of glycolytic enzymes [6]. Although most studies support a pro-tumorigenic role for CAFs in the TME, other studies have found compelling evidence that, in some situations or model systems, CAFs restrain tumor progression [7,8,9]. Several markers have been used to characterize distinct subtypes of CAFs, with smooth muscle actin (αSMA) and fibroblastic activation protein (FAP) among the most widely used. Other markers, such as platelet-derived growth factor receptor beta (PDGFR-β), vimentin, caveolin 1, CD10, GPR77, and tenascin C, are also useful to resolve organ-specific CAF subtypes. Several comprehensive reviews addressing CAF origins, functions, and subtypes can be found elsewhere [4,5,8].
Advanced HGSOC is treated with an aggressive combination of chemotherapy and surgery. Depending on tumor volume at presentation, patients with localized or low-volume metastatic disease undergo surgery followed by six cycles of chemotherapy. Patients with high-volume metastatic disease first receive three cycles of carboplatin and paclitaxel prior to surgery, with three additional cycles after surgery (“neoadjuvant chemotherapy”). A debulking surgery that leaves behind no visible disease confers the most survival benefit to the patient, and the choice of treatment sequence is weighted to maximize the benefit and minimize the morbidity of surgery [10,11]. Unfortunately, recurrence rates are high, being greater than 70%, and long-term survival is infrequent, with only 15% of women with advanced stage cancer surviving 7–10 years [12]. An important determinant of overall survival in HGSOC is persistent susceptibility of the disease to platinum-based agents [13]. Recurrence therapy is chosen based on the interval from the last platinum agent, overall health, acquired toxicities, germline/somatic mutations, and other factors. Second-line or maintenance treatments can involve anti-angiogenics, anti-metabolites, PARP (poly ADP-ribose polymerase) inhibitors, topoisomerase inhibitors, and occasionally radiation therapy [14,15,16,17,18]. The diverse treatment options used in patient care and clinical trials highlight the relevance of understanding their potential impact on the non-transformed components of the TME.
In this review, we assess experimental data that examines how commonly used ovarian cancer chemotherapeutics, including platinum derivatives, taxanes, PARP inhibitors, and anti-angiogenics, alter the function of genetically normal cells in the TME. Although we focus on therapies used in the treatment of ovarian cancer, we incorporate evidence from other histological types of cancer to broadly understand therapy-induced changes in CAF biology and their impact on modifying cancer progression across different tumor types. The challenges in understanding these complex interactions highlight the need for improved model systems that recapitulate the heterogeneity of the TME. Single-cell or compartment-resolved approaches, combined with high-fidelity preclinical models of cancer treatment, will be essential to unravel the biological and clinical relevance of these effects.
2. Alkylating Agents (Cisplatin and Carboplatin)
Alkylating agents, including cisplatin and carboplatin, act by forming DNA adducts and DNA strand cross links which lead to DNA breakage or cross-linking [19]. If a cell cannot repair the lesion, RNA synthesis and DNA replication stall, and the cell undergoes apoptosis. The most common and most efficient primary therapeutic regimen for HGSOC is carboplatin in combination with paclitaxel every three weeks for a total of six cycles [20]. Cisplatin can be used with equivalent efficacy, particularly in patients with myelosuppression that cannot be overcome with carboplatin dose reduction but is associated with a worse overall side effect profile [21]. Platinum compounds cross the fibroblast cell membrane via the CTR1 and CTR2 copper membrane transporters [22,23]. CAFs, in particular, express less CTR1 than adjacent normal fibroblasts and cancer cells, which contributes to their generally chemoresistant phenotype [24].
Platinum agents have been found to alter the CAF secretome, inducing the secretion of protease inhibitors, cytokines, and miRNA-containing exosomes. In primary esophageal squamous cell cancer (ESCC), cisplatin-treated CAFs secreted high levels of plasminogen activator inhibitor-1 (PAI-1), which subsequently promoted cancer cell proliferation and protection from cisplatin-induced apoptosis via inhibition of caspase-3 and activation of AKT and ERK1/2 pathways [25]. Clinical analysis of 49 ESCC patients indicated that those with high expression of PAI-1 in CAFs had significantly worse progression-free survival (PFS). Masuda et al. further found that in vitro inhibition of PAI-1 in lung CAFs increased cancer cell apoptosis and reduced CAF α-SMA expression. Treatment of a co-culture system with PAI-1 inhibitors consequently increased the efficacy of cisplatin killing cancer cells [26].
Multiple studies have found that platinum-based agents can regulate the cytokine profile of CAFs. Lung CAFs treated with cisplatin upregulated IL-11 expression in a time- and dose-dependent manner, promoting pro-survival STAT3 signaling in lung cancer cells [27,28]. In a longitudinal study of ovarian cancer patients from whom tumor tissue was collected before and after chemotherapy, levels of IL-6 were elevated in αSMA+ stromal cells following platinum treatment [29]. In vitro treatment of CAFs from primary human ovarian cancer with cisplatin enhanced their chemoprotective properties in an IL-6-dependent manner. Systematic profiling of cytokines upregulated by HGSOC CAFs following cisplatin treatment found significant elevation of CCL5, IL-8, and MIP [30]. Ovarian cancer cells treated with CCL5 showed decreased apoptosis when exposed to cisplatin, which was consistent with the finding that platinum-resistant patient samples expressed elevated CCL5 levels compared to platinum-sensitive samples. Moreover, CAFs may respond differently than normal fibroblasts to platinum agents, as was observed when oxaliplatin treatment upregulated sDTK, IL-17A, and TGF-β in CAFs but not in normal fibroblasts [31]. In colorectal cancer patient samples, IL-17A level was also found to increase following chemotherapy.
In addition to growth factors and cytokines, CAFs also secrete exosomes containing RNA molecules that may influence the gene expression profile and behavior of cancer and normal cells in the TME [32]. Cisplatin- or paclitaxel-treated gastric CAFs secreted exosomes containing high levels of miR-522, which suppressed ferroptosis in cancer cells and promoted chemo-resistance [33]. Although miR-522 expression in primary CAFs was not elevated, treated CAF exosomes contained nearly a four-fold higher level of miR-522, indicating a preferential enrichment of miR-522 in exosomes. Another miRNA, miR-196a, was upregulated in exosomes from cisplatin-treated head-and-neck cancer (HNC) CAFs, which ultimately enhanced the proliferation and chemo-resistance of HNC cells via targeting CDKN1B and ING5 [34].
Platinum-based interventions may also contribute to transforming normal fibroblasts into CAFs through increasing the expression of CAF-related markers such as FAP and α-SMA, altering the metabolic activity of normal fibroblasts, and inducing some aspects of senescence. Co-culture experiments with bladder cancer suggest that cisplatin accelerates normal fibroblasts’ transition to CAFs and increased the expression of α-SMA and FAP [24]. In immortalized human foreskin fibroblasts, cisplatin and carboplatin treatment increased L-lactate production and glucose consumption [35]. Using glycolytic flux analysis, the authors found that platinum treatment elevated fibroblast glycolysis and reduced oxygen consumption rates, indicating a metabolic switch. A secreted fibroblast factor, the ECM, and metabolism are linked in a report describing a novel role for the ECM protein collagen (COL) 11A1.The CAFs secrete COL11A1 that binds to the cancer cells through a discoidin receptor, which leads to upregulation of fatty acid oxidation, enabling the cancer cells to withstand carboplatin treatment [36].
Because some features of the CAF phenotype are similar to the senescence-associated secretory phenotype (SASP), it may be significant that platinum treatment has also been found to induce senescence and autophagy in CAFs. Treatment of human lung fibroblasts with platinum agents caused a prematurely senescent phenotype, evidenced by elevated p53 expression, loss of the membrane gap junction protein connexin 43 (Cx43), and morphological changes including flattening, filopodia extensions, and cytoplasmic vacuole formation [37]. Similarly, cisplatin treatment of normal oral fibroblasts and foreskin fibroblasts upregulated senescence markers and increased α-SMA expression [38]. An important consideration when interpreting these studies is that carboplatin and cisplatin may exert distinct effects on CAFs due to differences in uptake, bioactivity, and mechanism [39,40,41]. For example, carboplatin, but not cisplatin, was found to augment the glycolytic reserve, upregulate senescence and CAF markers, and promote HIF, SMAD, and STAT signaling in immortalized fibroblasts [35].
As CAFs comprise a highly heterogeneous cell population [42], CAF subpopulations may respond differently to platinum-based therapies. Indeed, Su et al. demonstrated that CD10+GPR77+ CAFs associated with poor prognosis also exhibited docetaxel and cisplatin resistance compared to CD10−GPR77− CAFs [43]. When challenged with cisplatin, CD10+GPR77+ breast cancer CAFs demonstrated significantly lower levels of apoptosis and growth inhibition. Furthermore, not all studies have found that platinum agents have tumor-promoting effects on the TME. In a study focused on lung cancer, cisplatin attenuated the ability of CAFs to promote the adhesion and invasion of cancer cells and reduced cancer cell AKT and NF-κB signaling via an unidentified paracrine mechanism [44]. In vivo experiments demonstrated that while co-implantation of CAFs and lung cancer cells increased tumor volume by over seven-fold, co-implantation with cisplatin-treated CAFs was comparable to implantation of cancer cells alone. Mesothelial cell-derived CAFs in the omental TME decreased cisplatinum sensitivity of HGSOC cells through secretion of fibronectin, which induced the PI3K pathway in the cancer cells [45].
Fully elucidating the mechanistic effects of therapeutic agents on CAFs will require understanding CAF heterogeneity and how therapies might balance cell subpopulations. It is important to note that all these studies indicate that some chemotherapy effects are tumor-promoting.
The effects of alkylating agents on other cellular components of the TME also deserve further attention. Notably, many studies demonstrate alkylating agents cause bone marrow toxicity, myelosuppression, and inhibition of the self-renewal capacity of hematopoietic stem cells [46,47,48]. Cisplatin has multifactorial roles in promoting a tumor-suppressive immune response, including increasing the range of antigen recognition, enhancing macrophage tumoricidal ability, promoting Th1 cytokine secretion, and regulating the recruitment of M1 macrophages, T regulatory cells, and CD8+ T cells [49,50,51,52]. In adipocytes, cisplatin may increase lipolysis while impeding lipogenesis [53]. Platinum treatment of endothelial cells induces features associated with angiogenesis, including dose-dependent decreases in migration and upregulation of ICAM-1, VEGF, and several cytokines such as IL-1 and IL-6 [54,55,56,57]. Independently, CAFs can promote the leakiness of blood vessels through the secretion of microfibrillar-associated protein (MFAP) 5 which binds to integrin receptors present on endothelial cells [58]. All of these effects on other cell types in the TME may also have the potential to regulate the behavior of CAFs.
3. Paclitaxel and Docetaxel
Paclitaxel, a plant alkaloid derived initially from the yew tree, is a cornerstone of upfront and recurrent treatment of HGSOC. Taxanes act during the M phase of the cell cycle by binding to intracellular microtubules to promote their assembly and stabilization, thus disrupting mitosis and leading to cell death [19]. Docetaxel is a therapeutically equivalent choice with a lower risk of peripheral neuropathy [59]. Taxanes can be used in the recurrent setting in both platinum-sensitive and -resistant patients [60].
There is emerging evidence that, in many situations, CAFs cooperate with tumor cells to enhance resistance to anticancer treatments [61]. In response to chemotherapy, CAFs may secrete cytokines, metabolites, and exosomes to potentiate stemness, metabolic reprogramming, and pro-survival signaling in tumor cells to orchestrate chemo-resistance [62]. Co-culture of taxane-treated normal lung fibroblasts with non-small-cell lung carcinoma tumor cells led to increased paclitaxel resistance of cancer cells, suggesting that paclitaxel may promote chemo-resistance via paracrine signaling [63]. Taxane-based chemotherapeutics may also regulate other aspects of the CAF phenotype by transcriptionally attenuating pro-tumorigenic paracrine signaling. Treatment of primary human breast cancer CAFs with docetaxel led to significant decreases in the expression of CXCL2, MMP1, IL-8, FF1, and CXCR7, among other cytokines. Co-culture experiments revealed that docetaxel-treated CAFs promoted the adhesion, invasion, and proliferation of MDA-MB-231 breast cancer cells [64]. A study of primary breast CAFs treated with docetaxel found that upregulation of MMP-1 and collagen IV was important for mediating chemo-resistance through ECM remodeling [65]. Caution should be applied to the interpretation of any in vitro or ex vivo experiments, however, as one study found divergent effects of paclitaxel treatment on primary CAFs compared to intact tissue sections, with increased apoptosis of CAFs observed in the ex vivo model system [66].
Several studies have suggested that taxanes may also regulate the behavior of normal fibroblasts and promote some aspects of the CAF phenotype. In an approach using label-free, quantitative proteomics, treatment of benign foreskin fibroblasts led to profound changes in energy metabolism, autophagy, senescence, myofibroblastic differentiation, and expression of inflammatory markers [67]. This included increased expression of common CAF markers such as α-SMA, fibronectin, and vimentin and upregulation of interleukin 6 (IL-6) and STAT3 signaling. In parallel to the metabolic reprogramming that occurs in CAFs [4,68,69], taxane treatment of dermal fibroblasts increased glycolysis, autophagy, and pro-inflammatory signaling [35,67].
In contrast, studies investigating the effects of taxanes on normal fibroblasts in non-cancer conditions have found evidence of anti-fibrotic effects. In a model of extrahepatic bile duct fibrosis, treatment of human gallbladder myofibroblasts with paclitaxel led to decreased autocrine TGFβ-1 signaling and reduced collagen 1 production associated with fibrosis [70]. In another study examining renal interstitial fibrosis, paclitaxel treatment led to decreased SMAD signaling and suppression of pro-inflammatory cytokine production accompanied by a strong reduction in α-SMA and collagen 1 expression [71]. The mechanistic basis for the differential effects of taxanes on normal fibroblasts and CAFs has not yet been explored.
In addition to effects on fibroblast components of the TME, taxanes may also regulate endothelial cells by exerting intrinsic anti-angiogenic effects. Interestingly, human endothelial cells accumulate higher intracellular levels of paclitaxel than non-endothelial cells, suggesting selectivity and increased susceptibility of endothelial cells to the drug [72]. Multiple studies have found that taxanes can directly compromise endothelial cells by inducing apoptotic cell death [73,74,75,76]. Functionally, paclitaxel attenuates endothelial cell migration, inhibits endothelial tube formation [77], and induces some aspects of the senescent phenotype [78]. Although taxanes may reduce the angiogenic activities of endothelial cells, there is also evidence indicating paclitaxel can increase vascular endothelial growth factor (VEGF) production in cervical cancer tumor cells by regulating hypoxia-inducible factor 1α (HIF-1α) and NF-κB signaling, thereby increasing angiogenesis and promoting chemo-resistance [79].
4. Poly ADP-Ribose Polymerases Inhibitors
Poly ADP-ribose polymerases (PARPs) are enzymes critical for the repair of single-stranded DNA breaks, as well as contributing to the repair of double-stranded breaks and the stabilization of replications forks [80]. In cells with double-stranded DNA repair deficiencies, such as those with BRCA1/2 gene mutations, PARP inhibition leads to synthetic lethality. PARP inhibitors (PARPi) are oral medications that act to trap or inhibit PARP enzymatic action. Maintenance PARPi treatment after upfront chemotherapy has recently proven effective at lowering the risk of disease progression or death by 70% at 41 months in HGSOC patients with a BRCA1/2 mutation. They are now standard in the frontline setting [11,15].
Most studies examining the effects of PARP inhibition on stromal cell populations have focused on normal fibroblasts and found that PARPi affected the regulation of TNFα and TGF-β signaling. Whereas PARP inhibition attenuates the TNFα-induced fibroblast response, its impact on TGF-β mediated effects is less clear. Isolated fibroblast-like synoviocytes treated with TNFα and a PARPi (DPQ or ANI) had reduced expression of inflammatory mediators such as MMP-3, IL8, and MCP-1 [81]. This was associated with diminished TNFα-induced proliferation, JNK phosphorylation, and AP-1 and NF-kB binding. Similar trends were observed in another study which found that pretreating murine fibroblasts with the PARPi INH2BP also suppressed JNK activation and AP-1 DNA binding [82]. In murine fibroblasts, the PARPi 3AB reduced levels of TNF-induced ATP depletion and death [83]. Broadly, TNFα-induced signaling in fibroblasts is attenuated by PARPi.
In human skin fibroblasts, PARP inhibition by 3-AB increased the stimulatory effects of TGF-β, including upregulation of α-SMA transcription, expression, and stress fiber formation compared to TGF-β alone [84]. Additional synergistic effects included upregulation of collagen expression, increased collagen release, and elevated SMAD3 signaling. In vitro, treatment with 3-AB exacerbated fibrosis induced by topoisomerase or bleomycin, resulting in increased dermal thickness and hydroxyproline content. Other studies, however, indicate anti-fibrotic roles for PARP inhibition. Knockdown of PARP1 in cardiac fibroblasts repressed TGF-β1-induced proliferation, migration, and differentiation [85]. In rat models of myocardial infarction (MI), 4-AB alleviated fibrosis and reduced collagen deposition, with an associated decrease in α-SMA expression. In addition, 4-AB increased p62 levels and reduced the LC3-II/LC3-I ratio, suggesting that PARP inhibition may increase autophagy. These divergent phenotypes may be due to differences in cellular context, fibroblast type, or experimental design considerations.
PARPi also demonstrates profound immunomodulatory effects, promoting anti-tumor immune responses by upregulating cytotoxic immune cells such as CD8+ T-cells, B-cells, and NK cells, while decreasing the number of myeloid-derived suppressor cells [86,87]. This anti-tumor response is at least in part due to upregulation of the STING pathway by PARPi [88]. While PARPi show promise in cancer treatment, they are also associated, albeit rarely, with severe side effects such as myeloid leukemia and myelodysplastic syndrome [89]. Importantly, maintenance PARPi treatment can be of extended duration, for two or more years, raising the possibility that long-term, global inhibition of PARP may have distinct influences on the biology of normal cells.
5. Anti-Angiogenic Agents (Bevacizumab)
Bevacizumab, an antibody against VEGF and angiogenesis, is often used to treat recurrent HGSOC as an additional treatment option to target the angiogenic potential of the TME [90]. In upfront therapy for HGSOC, there is some evidence that it benefits poor-prognosis patients with high tumor volume, stage IV, or incompletely resected disease [91]. Bevacizumab is also used in combination with PARPi for maintenance therapy in the upfront setting, with a 19.5-month progression-free survival benefit [92]. A PFS benefit has been seen when used in combination therapy for both platinum-sensitive and -resistant recurrence [93,94]. The lack of a consistent overall survival benefit and the rare risk of serious adverse events, including gastrointestinal perforation, bleeding diathesis, and poor wound healing, has limited this agent’s use. Although anti-VEGF inhibitors are meant to target endothelial cells, fibroblasts also express the receptors VEGFR1 and VEGFR3 [95]. No studies have directly investigated the effects of anti-angiogenic agents on CAFs, but bevacizumab exerts some direct effects on normal fibroblast populations. Bevacizumab-treated rat conjunctival fibroblasts exhibit reduced growth, ECM remodeling, and metabolic activity associated with decreased expression of VEGF, VEGFR1, VEGFR2, TGF-β1, and TGF-β2 [96]. Pharmacologically blocking VEGFR in human lung fibroblasts from patients with idiopathic pulmonary fibrosis suppressed the proliferative effects of secreted factors, including PDGF and bFGF [97]. VEGFR inhibition upregulated pro-MMP-2 activity, downregulated TIMP-2 secretion, and suppressed TGF-β-induced collagen secretion. Another study examining human tendon fibroblasts found that treatment with bevacizumab reduced metabolic activity and viability in a dose-dependent manner [98]. Bevacizumab also decreased the expression of MMP-1, MMP-2, and laminin, suggesting that it may play a role in ECM remodeling.
6. Topoisomerase Inhibitors (Doxorubicin, Ropotecan, and Mitoxantrone)
Topoisomerase inhibitors, including doxorubicin, topotecan, and mitoxantrone, act by inhibiting topoisomerase enzymes, which are responsible for the winding of DNA, leading to DNA strand breaks [99]. Doxorubicin has the additional effect of distorting the DNA double helix and generating free radicals. Liposomal doxorubicin and topotecan are both used for recurrent ovarian cancer with limited efficacy [100]. Although the effects of topoisomerase inhibitors on CAFs have not been extensively investigated, several studies of normal fibroblasts suggest that topoisomerase inhibitors may regulate the TME. Fibroblasts appear to be sensitive to the topoisomerase II inhibitor mitoxantrone, with the treatment of dermal fibroblasts leading to senescence and enhanced glycolysis [101]. Mitoxantrone-treated fibroblasts have been found to induce a cancer stem cell phenotype in MCF7 cells, as assessed using a luciferase reporter system in tumor–stromal co-culture conditions [35]. Mitoxantrone treatment of prostate, ovarian, and breast primary CAFs upregulated WNT16B, thereby mediating acquired resistance, suggesting a role for mitoxantrone in the regulation of Wnt signaling [102]. In a study that directly examined the effects of topoisomerase inhibitors on CAFs in a mouse model of desmoplastic melanoma, a combination of mitoxantrone and the triterpenoid celastrol decreased CAF-mediated collagen production and was associated with a CD8 T-lymphocyte- and dendritic cell-mediated immunogenic response [103]. The effects of topoisomerase inhibitors on other components of the TME have not been systematically investigated.
7. Antimetabolites (Gemcitabine)
Gemcitabine is a pyrimidine analog that primarily acts by inhibiting DNA synthesis through direct incorporation into the DNA backbone. It may also induce activation of mitogen-activated protein kinase (MAPK), triggering apoptosis in response to cellular stress in tumor cells [104]. Gemcitabine is commonly used in the recurrent setting and has a single-agent response rate of about 19%. Common dose-limiting toxicities include neutropenia, nausea, appetite suppression, and a flu-like syndrome [105]. Relatively few studies have examined the effects of gemcitabine on stromal cell populations. CAFs appear to be resistant to gemcitabine treatment when compared to normal fibroblasts or cancer cells [106]. It has been suggested that scavenger molecules from pancreatic CAFs may modify gemcitabine accumulation in tumors by entrapping the active drug and reducing its delivery to cancer cells [107]. In co-culture model systems, pre-treatment of immortalized rat CAFs protected tumor cells from the cytotoxic effects of gemcitabine. Interestingly, inhibition of autophagy in CAFs with chloroquine also reduced cancer cell death in response to gemcitabine [108]. Several studies have also found roles for gemcitabine in the regulation of CAF exosome production. The treatment of pancreatic CAFs with gemcitabine resulted in the increased release of exosomes containing the EMT regulator Snail. Uptake of these exosomes by tumor cells reduced the cytotoxic effects of gemcitabine treatment [109]. Similarly, Fang et al. found that CAF-derived exosomes transferred miRNA-106b to tumor cells and regulated gemcitabine resistance in a TP53INP1-dependent manner [106].
8. Radiotherapy
Radiotherapy involves targeted high-frequency ionizing radiation which ejects electrons from atoms to create ions which then form free radicals, leading to DNA damage. The advent of highly active chemotherapy for HGSOC in the 1990s sidelined radiation therapy in this disease. Its current use is limited to treating isolated recurrences or for symptom palliation [18]. Radiation induces significant changes in the CAF secretome, including upregulation of factors such as IGF-1, bFGF, IL-6, IL-8, GRO, HDGF, and potentially HGF [110,111,112,113]. Furthermore, radiation promotes MMP-3 and possibly MMP-1 expression in CAFs [114,115]. Consistent with the role of CAFs in the regulation of ECM remodeling, it has also been reported that radiotherapy elevates CAF expression of integrins β1, α5, and, most significantly, α2 [114]. Irradiated CAFs produced a stiffer collagen matrix when grown in a 3D culture system [116]. Radiotherapy also increases premature senescence in CAFs [114,117]. Cumulatively, these data suggest that CAFs resist radiation treatment through acquiring a senescent phenotype and that they can simultaneously contribute to cancer cell resistance by secreting pro-tumorigenic factors and remodeling the ECM.
9. Discussion
Studies investigating the effects of anticancer agents on normal cells in the tumor microenvironment have focused on a wide range of molecular and phenotypic features, including metabolic and transcriptional reprogramming and intracellular signaling (Figure 2). These studies have primarily found that CAFs, in response to first-line chemotherapies, secrete multiple cytokines, metabolites, ECM-remodeling enzymes, and exosomes that transform the TME and generally promote chemo-resistance. Platinum derivatives and taxanes appear to promote a precancerous metabolic phenotype in stromal cells, resulting in augmented glycolysis, glucose consumption, lactate production, and activity of several pro-tumorigenic pathways [35,67]. In response to these agents, normal fibroblasts adopt at least some aspects of the CAF phenotype, but it is unclear if these changes are long-lasting or reversible [24,35,67,83]. Other therapies, including PARP inhibitors, anti-angiogenic agents, and topoisomerase inhibitors, have been less studied; nevertheless, some evidence suggests that they have TME-modifying capabilities. Of note, alongside cytotoxic chemotherapeutics, many patients receive 5-HT3 antagonists, steroids, and NK1 antagonists to manage nausea and emesis [118]. The effects of these agents on the TME have not been well investigated, although several stromal cell types express the relevant cognate receptors [119,120,121,122,123].
Additional research using more physiologically relevant models will be necessary if we are to illuminate the complex processes that occur in the treated TME. Organotypic model systems that incorporate both tumor and stromal cell types have proven to be particularly valuable in studies that reveal heterotypic cross-talk, cancer progression, and opportunities for drug discovery [26,63]. For instance, using a complex model system, Gao et al., found that CAFs can form metastatic units with ascitic tumor cells and drive peritoneal metastasis formation, which is common in ovarian cancer [124]. Using such complex models can also identify mechanisms of chemo-resistance and pinpoint drug candidates more likely to be effective in vivo, since malignant cells often exhibit profound differences in sensitivity to therapeutic agents depending on the culture system utilized [125,126,127,128]. Furthermore, while patients receive well-defined cycles of chemotherapy, most studies, both in vitro and in vivo, do not mirror the concentrations and durations of treatment typical of clinical exposures.
In addition, preclinical examinations of chemotherapy-induced changes in the TME have been largely restricted to subcutaneous xenograft models that do not fully recapitulate all components of the TME. Studies using orthotopic or genetically engineered mouse models are needed to more fully understand disease processes. In HGSOC, most tumor cells harbor mutations in TP53, and mouse models have been recently developed with syngeneic murine ovarian cancer cells engineered to express mutant p53 protein via CRISPR/Cas9 [129]. Researchers must carefully consider candidate cell lines for their experiments, since unique and characteristic TMEs arise depending on the HGSOC model employed [130].
CAFs originate from numerous sources and are composed of discrete subpopulations. Therefore, it is difficult to generalize microenvironmental responses to chemotherapy due to heterogeneity within single tissues and between anatomic sites. For example, while cisplatin treatment may downregulate α-SMA in lung fibroblasts, studies in bladder and foreskin fibroblasts have found upregulation of α-SMA or no changes at all [24,35,44]. Furthermore, few studies have examined differences in CAFs between humans and other model organisms [131]. Research in the ovarian cancer TME has identified at least four different CAF subpopulations that express distinct molecular signatures characterized by variable expression of CD29, CD10, FAP, α-SMA, FSP1, PDGFR-β, podoplanin, and caveolin-1. This suggests that the use of single markers such as α-SMA to define CAF identity is imprecise. Embracing multiple CAF markers with complementary functional studies has the potential to elucidate whether therapy-induced changes reflect reprogramming of stromal fibroblasts or the selection of discrete populations. We anticipate that single-cell sequencing will serve to spatially and temporally resolve CAF subpopulations, as well as shed light on the epigenetic and evolutionary dynamics within specific microenvironments [132,133,134]. Advances in mass spectrometry-based proteomics have generated opportunities to characterize the tumor and stromal proteomes in situ [135], quantify the phosphorylation levels of critical signaling pathways [136,137], and identify secreted factors derived from both tumor and stromal cells [138]. In addition, imaging mass cytometry enables the multiplexed, spatial characterization of proteins in both tumor and stromal compartments and has already revealed important aspects of cellular communication and “cellular neighborhoods” in breast cancer [139].
Understanding how chemotherapeutics impact cell types in the TME other than CAFs may also inform opportunities for therapeutic intervention. Patients can experience varying degrees of myelosuppression, mucous membrane reactions, and alopecia, all evidence of unintended cellular targets [19,140]. In addition, taxanes and platinum-based agents can have significant neurotoxicity [141,142]. Because neurons interact with the TME in myriad ways, releasing neurotransmitters, peptides, and growth factors [143,144,145], there are likely to be unexamined roles for chemotherapeutic agents in the regulation of the communication between neurons and cancer cells [146]. Despite a growing literature showing that chemotherapeutics perturb nonmalignant TME cells, in most situations, the mechanistic basis of these effects remains unclear. As well as cross-linking DNA, cisplatin is also known to generate reactive oxygen species, lower GSH and NADH levels, disrupt calcium homeostasis, and activate ERK, JNK, and AKT signaling pathways [147]. PARP inhibitors may disrupt the roles of PARP in DNA damage repair and influence epigenetic remodeling. Several of these therapy-induced effects on fibroblasts are therefore unsurprising, whereas others require further elucidation. For example, the general upregulation of α-SMA may be due to cisplatin-induced changes in SMAD3 signaling, PAI-1 autocrine signaling, or other mechanisms [148]. To understand these aspects of treatment, future studies should investigate the transcriptional and translational changes as well as the epitranscriptomic and epigenetic remodeling that occurs in response to anticancer agents. How chemotherapeutic agents may remodel the pre-metastatic niche has not yet been explored.
Beyond their likely prognostic and diagnostic value [149], CAFs have garnered interest as targets of cancer therapy [150]. This could be of particular relevance to OvCa, since there are few targetable mutations. Rather than aiming to ablate whole CAF populations, it may be possible to reprogram CAFs towards an anti-tumorigenic phenotype [151]. Indeed, emerging research provides a rationale for targeting the TME with all-trans retinoic acid to induce CAF quiescence [152] or using CXCL12 receptor inhibitors to disrupt CAF-mediated immune evasion, as well as targeting many other CAF behaviors [153]. Fully realizing the clinical efficacy of these emerging chemotherapies will require a robust understanding of how common treatments influence the tumor microenvironment.
10. Conclusions
In summary, chemotherapy and radiation does not only affect the cancer cells, but has a profound imapct on the TME. Understanding the effect of various treatments on cancer cells is necessary to completely comprehend their action and develop novel therapeutics that interrupt undesired tumor–stroma interactions.
Author Contributions
Conceptualization, M.A.E.; writing—original draft preparation, M.A.E., C.O., M.J., and J.X.; writing—review and editing, M.A.E., C.O., J.X., M.J., and E.L.; funding acquisition, E.L. and M.A.E. All authors have read and agreed to the published version of the manuscript.
Funding
E.L. was supported by the National Cancer Institute (NCI) grants R01CA211916 and R01CA169604. M.A.E. was supported by the Ovarian Cancer Research Alliance (OCRA) Liz Tilberis Early Career Award 650339.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
E.L. receives funding from Arsenal Bio and Abbvie to support preclinical research unrelated to the manuscript. All other authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Foley, O.W.; Rauh-Hain, J.A.; Del Carmen, M.G. Recurrent epithelial ovarian cancer: An update on treatment. Oncology 2013, 27, 288. [Google Scholar]
- Kayl, A.E.; Meyers, C.A. Side-effects of chemotherapy and quality of life in ovarian and breast cancer patients. Curr. Opin. Obstet. Gynecol. 2006, 18, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; De Nardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Santi, A.; Kugeratski, F.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Gerling, M.; Büller, N.V.J.A.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergström, Å.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.B.; et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef]
- Vergote, I.; Coens, C.; Nankivell, M.; Kristensen, G.B.; Parmar, M.K.B.; Ehlen, T.; Jayson, G.C.; Johnson, N.; Swart, A.M.; Verheijen, R.; et al. Neoadjuvant chemotherapy versus debulking surgery in advanced tubo-ovarian cancers: Pooled analysis of individual patient data from the EORTC 55971 and CHORUS trials. Lancet Oncol. 2018, 19, 1680–1687. [Google Scholar] [CrossRef] [Green Version]
- Kurnit, K.C.; Fleming, G.F.; Lengyel, E. Updates and New Options in Advanced Epithelial Ovarian Cancer Treatment. Obstet. Gynecol. 2021, 137, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Javellana, M.; Hoppenot, C.; Lengyel, E. The road to long-term survival: Surgical approach and longitudinal treatments of long-term survivors of advanced-stage serous ovarian cancer. Gynecol. Oncol. 2019, 152, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Luvero, D.; Milani, A.; Ledermann, J.A. Treatment options in recurrent ovarian cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2014, 6, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- González-Martín, A.; Sánchez-Lorenzo, L. Immunotherapy with checkpoint inhibitors in patients with ovarian cancer: Still promising? Cancer 2019, 125, 4616–4622. [Google Scholar] [CrossRef] [Green Version]
- Doo, D.W.; Norian, L.A.; Arend, R.C. Checkpoint inhibitors in ovarian cancer: A review of preclinical data. Gynecol. Oncol. Rep. 2019, 29, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Fields, E.C.; McGuire, W.P.; Lin, L.; Temkin, S.M. Radiation Treatment in Women with Ovarian Cancer: Past, Present, and Future. Front. Oncol. 2017, 7, 177. [Google Scholar] [CrossRef] [Green Version]
- Chi, D.; Berchuck, A.; Dizon, D.; Yasher, C. Principles and Practice of Gynecologic Oncology, 7th ed.; Wolters Kluwer: Baltimore, MD, USA, 2017. [Google Scholar]
- NCCN. Ovarian Cancer Including Fallopian Tube Cancer and Primary Peritoneal Cancer. Version 2. 2013. Available online: https://www2.tri-kobe.org/nccn/guideline/archive/gynecological2012/english/ovarian.pdf (accessed on 3 May 2021).
- Neijt, J.P.; Engelholm, S.A.; Tuxen, M.K.; Sørensen, P.G.; Hansen, M.; Sessa, C.; De Swart, C.A.M.; Hirsch, F.R.; Lund, B.; Van Houwelingen, H.C. Exploratory Phase III Study of Paclitaxel and Cisplatin Versus Paclitaxel and Carboplatin in Advanced Ovarian Cancer. J. Clin. Oncol. 2000, 18, 3084–3092. [Google Scholar] [CrossRef]
- Holzer, A.K.; Manorek, G.H.; Howell, S.B. Contribution of the Major Copper Influx Transporter CTR1 to the Cellular Accumulation of Cisplatin, Carboplatin, and Oxaliplatin. Mol. Pharmacol. 2006, 70, 1390–1394. [Google Scholar] [CrossRef]
- Blair, B.G.; Larson, C.A.; Safaei, R.; Howell, S.B. Copper Transporter 2 Regulates the Cellular Accumulation and Cytotoxicity of Cisplatin and Carboplatin. Clin. Cancer Res. 2009, 15, 4312–4321. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375. [Google Scholar] [CrossRef]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Nakashima, T.; Namba, M.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Iwamoto, H.; Fujitaka, K.; Miyata, Y.; et al. Inhibition of PAI-1 limits chemotherapy resistance in lung cancer through suppressing myofibroblast characteristics of cancer-associated fibroblasts. J. Cell. Mol. Med. 2019, 23, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef]
- Yan, H.; Guo, B.-Y.; Zhang, S. Cancer-associated fibroblasts attenuate Cisplatin-induced apoptosis in ovarian cancer cells by promoting STAT3 signaling. Biochem. Biophys. Res. Commun. 2016, 470, 947–954. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Z.-Y.; Jin, P.; Yang, X.; Li, X.; Wei, X.; Wang, Y.; Long, S.; Zhang, T.; Chen, G.; et al. Metformin Suppresses Tumor Progression by Inactivating Stromal Fibroblasts in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 1291–1302. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Sun, C.; Li, N.; Shan, W.; Lu, H.; Guo, L.; Guo, E.; Xia, M.; Weng, D.; Meng, L.; et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int. J. Oncol. 2016, 48, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, M.; Yin, T.; Shi, H.; Wen, Y.; Zhang, B.; Chen, M.; Xu, G.; Ren, K.; Wei, Y. Targeting of cancer-associated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol. Med. Rep. 2016, 13, 2476–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of Exosomes in Crosstalk Between Cancer-Associated Fibroblasts and Cancer Cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Guo, H.; Wang, X.; Zhu, X.; Yan, M.; Wang, X.; Xu, Q.; Shi, J.; Lu, E.; Chen, W.; et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019, 20, 1–21. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Sotgia, F.; Lisanti, M. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget 2015, 6, 10728–10745. [Google Scholar] [CrossRef] [Green Version]
- Nallanthighal, S.; Rada, M.; Heiserman, J.P.; Cha, J.; Sage, J.; Zhou, B.; Yang, W.; Hu, Y.; Korgaonkar, C.; Hanos, C.T.; et al. Inhibition of collagen XI alpha 1-induced fatty acid oxidation triggers apoptotic cell death in cisplatin-resistant ovarian cancer. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Lin, Z.X.; Zhang, Z.Q. Cisplatin-induced premature senescence with concomitant reduction of gap junctions in human fibroblasts. Cell Res. 2004, 14, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellone, M.; Hanley, C.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Burger, H.; Zoumaro-Djayoon, A.; Boersma, A.; Helleman, J.; Berns, P.; Mathijssen, A.; Loos, W.; Wiemer, E. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2). Br. J. Pharmacol. 2010, 159, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perde-Schrepler, M.; Fischer-Fodor, E.; Virag, P.; Brie, I.; Cenariu, M.; Pop, C.; Valcan, A.; Gurzau, E.; Maniu, A. The expression of copper transporters associated with the ototoxicity induced by platinum-based chemotherapeutic agents. Heart Res. 2020, 388, 107893. [Google Scholar] [CrossRef]
- Qin, Z.; Ren, G.; Yuan, J.; Chen, H.; Lu, Y.; Li, N.; Zhang, Y.; Chen, X.; Zhao, D. Systemic Evaluation on the Pharmacokinetics of Platinum-Based Anticancer Drugs from Animal to Cell Level: Based on Total Platinum and Intact Drugs. Front. Pharmacol. 2020, 10, 1485. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Li, X.; Song, Q.; Guo, X.; Wang, L.; Zhang, Q.; Cao, L.; Ren, Y.; Wu, X.; Meng, Z.; Xu, K. The Metastasis Potential Promoting Capacity of Cancer-Associated Fibroblasts Was Attenuated by Cisplatin via Modulating KRT8. OncoTargets Ther. 2020, 13, 2711–2723. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, M.; Kajiyama, H.; Yokoi, A.; Sugiyama, M.; Koya, Y.; Yamakita, Y.; Liu, W.; Nakamura, K.; Moriyama, Y.; Yasui, H.; et al. Ovarian cancer-associated mesothelial cells induce acquired platinum-resistance in peritoneal metastasis via the FN1/Akt signaling pathway. Int. J. Cancer 2020, 146, 2268–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tricot, G.; Jagannath, S.; Vesole, D.; Nelson, J.; Tindle, S.; Miller, L.; Cheson, B.; Crowley, J.; Barlogie, B. Peripheral blood stem cell transplants for multiple myeloma: Identification of favorable variables for rapid engraftment in 225 patients. Blood 1995, 85, 588–596. [Google Scholar] [CrossRef] [Green Version]
- Botnick, L.; Hannon, E.C.; Vigneulle, R.; Hellman, S. Differential effects of cytotoxic agents on hematopoietic progenitors. Cancer Res. 1981, 41, 2338–2342. [Google Scholar]
- Probin, V.; Zhou, D. Cancer Therapy-Induced Residual Bone Marrow Injury: Mechanisms of Induction and Implication for Therapy. Curr. Cancer Ther. Rev. 2006, 2, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Krisl, J.C.; Doan, V.P. Chemotherapy and Transplantation: The Role of Immunosuppression in Malignancy and a Review of Antineoplastic Agents in Solid Organ Transplant Recipients. Arab. Archaeol. Epigr. 2017, 17, 1974–1991. [Google Scholar] [CrossRef] [PubMed]
- Raudenska, M.; Balvan, J.; Fojtu, M.; Gumulec, J.; Masarik, M. Unexpected therapeutic effects of cisplatin. Metallomics 2019, 11, 1182–1199. [Google Scholar] [CrossRef]
- Suresh, A.; Sodhi, A. Production of interleukin-1 and tumor necrosis factor by bone marrow-derived macrophages: Effect of cisplatin and lipopolysaccharide. Immunol. Lett. 1991, 30, 93–100. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Punt, C.J.; Hato, S.V.; Eleveld-Trancikova, D.; Jansen, B.J.; Nierkens, S.; Schreibelt, G.; De Boer, A.; Van Herpen, C.M.; Kaanders, J.H.; et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J. Clin. Investig. 2011, 121, 3100–3108. [Google Scholar] [CrossRef]
- Garcia, J.M.; Scherer, T.; Chen, J.-A.; Guillory, B.; Nassif, A.; Papusha, V.; Smiechowska, J.; Asnicar, M.; Buettner, C.; Smith, R.G. Inhibition of Cisplatin-Induced Lipid Catabolism and Weight Loss by Ghrelin in Male Mice. Endocrinology 2013, 154, 3118–3129. [Google Scholar] [CrossRef] [Green Version]
- Montiel, M.; Urso, L.; De La Blanca, E.P.; Marsigliante, S.; Jiménez, E. Cisplatin Reduces Endothelial Cell Migration Via Regulation of Type 2-Matrix Metalloproteinase Activity. Cell. Physiol. Biochem. 2009, 23, 441–448. [Google Scholar] [CrossRef]
- Yu, M.; Han, J.; Cui, P.; Dai, M.; Li, H.; Zhang, J.; Xiu, R. Cisplatin up-regulates ICAM-1 expression in endothelial cell via a NF-κB dependent pathway. Cancer Sci. 2008, 99, 391–397. [Google Scholar] [CrossRef]
- Wild, R.; Dings, R.P.; Subramanian, I.; Ramakrishnan, S. Carboplatin selectively induces the VEGF stress response in endothelial cells: Potentiation of antitumor activity by combination treatment with antibody to VEGF. Int. J. Cancer 2004, 110, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, S.; Shinozaki, R.; Fukue, K.; Kougo, T. Release of Cytokines from Human Umbilical Vein Endothelial Cells Treated with Platinum Compoundsin vitro. Jpn. J. Cancer Res. 1998, 89, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Yeung, T.-L.; Yip, K.-P.; Wong, K.-K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2017, 128, 589–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasey, P.A.; Jayson, G.C.; Gordon, A.; Gabra, H.; Coleman, R.; Atkinson, R.; Parkin, D.; Paul, J.; Hay, A.; Kaye, S.B.; et al. Phase III Randomized Trial of Docetaxel-Carboplatin Versus Paclitaxel-Carboplatin as First-line Chemotherapy for Ovarian Carcinoma. J. Natl. Cancer Inst. 2004, 96, 1682–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markman, M.; Hall, J.; Spitz, D.; Weiner, S.; Carson, L.; Van Le, L.; Baker, M. Phase II Trial of Weekly Single-Agent Paclitaxel in Platinum/Paclitaxel-Refractory Ovarian Cancer. J. Clin. Oncol. 2002, 20, 2365–2369. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.-S.; Hsu, C.-C.; Pai, V.C.; Liao, W.-Y.; Huang, S.-S.; Tan, K.-T.; Yen, C.-J.; Hsu, S.-C.; Chen, W.-Y.; Shan, Y.-S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef] [PubMed]
- Bartling, B.; Hofmann, H.-S.; Silber, R.-E.; Simm, A. Differential impact of fibroblasts on the efficient cell death of lung cancer cells induced by paclitaxel and cisplatin. Cancer Biol. Ther. 2008, 7, 1250–1261. [Google Scholar] [CrossRef] [Green Version]
- Rong, G.; Kang, H.; Wang, Y.; Hai, T.; Sun, H. Candidate markers that associate with chemotherapy resistance in breast cancer through the study on taxotere-induced damage to tumor microenvironment and gene expression profiling of carcino-ma-associated fibroblasts (CAFs). PLoS ONE 2013, 8, e70960. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Wang, B.; Li, K.; Sun, H.; Hai, T.; Zhang, Y.; Kang, H. Upregulating MMP-1 in carcinoma-associated fibroblasts reduces the efficacy of Taxotere on breast cancer synergized by Collagen IV. Oncol. Lett. 2018, 16, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, M.; Van Der Kuip, H.; Haubeiß, S.; Fritz, P.; Schroth, W.; Friedel, G.; Simon, W.E.; Mürdter, T.E.; Aulitzky, W. Highly variable response to cytotoxic chemotherapy in carcinoma-associated fibroblasts (CAFs) from lung and breast. BMC Cancer 2008, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pagès, M.; Smith, D.L.; Győrffy, B.; Sotgia, F.; Lisanti, M.P. Proteomic identification of prognostic tumour biomarkers, using chemotherapy-induced cancer-associated fibroblasts. Aging 2015, 7, 816–838. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Bera, S. CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumor Biol. 2016, 37, 8503–8514. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-S.; Savard, C.E.; Choi, J.-W.; Kuver, R.; Lee, S.P. Paclitaxel Interrupts TGF-β1 Signaling Between Gallbladder Epithelial Cells and Myofibroblasts. J. Surg. Res. 2007, 141, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Zhang, L.; Yang, R.; Xu, X.; Chen, J.; Wang, S.; He, Z.; Dong, Z.; Yang, J.; Zhao, Y.; et al. Paclitaxel attenuates renal interstitial fibroblast activation and interstitial fibrosis by inhibiting STAT3 signaling. Drug Des. Dev. Ther. 2015, 9, 2139–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchan, J.R.; Jayaram, D.R.; Supko, J.G.; He, X.; Bubley, G.J.; Sukhatme, V.P. Increased endothelial uptake of paclitaxel as a potential mechanism for its antiangiogenic effects: Potentiation by Cox-2 inhibition. Int. J. Cancer 2004, 113, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.-I.; Yamamoto, A.; You, F.; Yamashita, K.; Ikegame, Y.; Tawada, M.; Yoshimori, T.; Shimizu, S.; Nakashima, S. The Stent-Eluting Drugs Sirolimus and Paclitaxel Suppress Healing of the Endothelium by Induction of Autophagy. Am. J. Pathol. 2009, 175, 2226–2234. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.-H.; Chan, S.-H.; Chu, P.-M.; Tsai, K.-L. Docetaxel Facilitates Endothelial Dysfunction through Oxidative Stress via Modulation of Protein Kinase C Beta: The Protective Effects of Sotrastaurin. Toxicol. Sci. 2015, 145, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.S.; Yoo, J.-M.; Lim, Y.; Tudev, M.; Yoo, H.-S.; Hong, J.T.; Yun, Y.-P. Inhibitory effects of docetaxel on platelet-derived growth factor (PDGF)-BB-induced proliferation of vascular smooth muscle cells through blocking PDGF-receptor? Phosphorylation. J. Pharmacol. Sci. 2011, 116, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, E.; Carré, M.; Pourroy, B.; Camoin, L.; Rebaï, O.; Briand, C.; Braguer, D. Antiangiogenic activity of paclitaxel is associated with its cytostatic effect, mediated by the initiation but not completion of a mitochondrial apoptotic signaling pathway. Mol. Cancer Ther. 2004, 3, 1301–1310. [Google Scholar]
- Michailidou, M.; Brown, H.K.; Lefley, D.V.; Evans, A.; Cross, S.S.; Coleman, R.E.; Brown, N.J.; Holen, I. Microvascular Endothelial Cell Responses in vitro and in vivo: Modulation by Zoledronic Acid and Paclitaxel? J. Vasc. Res. 2010, 47, 481–493. [Google Scholar] [CrossRef]
- Ota, H.; Eto, M.; Ako, J.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Sirolimus and Everolimus Induce Endothelial Cellular Senescence Via Sirtuin 1 Down-Regulation. J. Am. Coll. Cardiol. 2009, 53, 2298–2305. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Oh, J.M.; Jin, D.H.; Yang, K.-H.; Moon, E.-Y. Paclitaxel Induces Vascular Endothelial Growth Factor Expression through Reactive Oxygen Species Production. Pharmacology 2008, 81, 317–324. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Nussenzweig, A.R.C.A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- García, S.; Bodaño, A.; Pablos, J.L.; Gómez-Reino, J.J.; Conde, C. Poly (ADP-ribose) polymerase inhibition reduces tumor necrosis factor-induced inflammatory response in rheumatoid synovial fibroblasts. Ann. Rheum. Dis. 2007, 67, 631–637. [Google Scholar] [CrossRef]
- Andreone, T.L.; O’Connor, M.; Denenberg, A.; Hake, P.W.; Zingarelli, B. Poly (ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J. Immunol. 2003, 170, 2113–2120. [Google Scholar] [CrossRef] [Green Version]
- Los, M.J.; Mozoluk, M.; Ferrari, D.; Stepczynska, A.; Stroh, C.; Renz, A.; Herceg, Z.; Wang, Z.-Q.; Schulze-Osthoff, K. Activation and Caspase-mediated Inhibition of PARP: A Molecular Switch between Fibroblast Necrosis and Apoptosis in Death Receptor Signaling. Mol. Biol. Cell 2002, 13, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pötter, S.; Chen, C.-W.; Liang, R.; Gelse, K.; Ludolph, I.E.; Horch, R.; Distler, O.; Schett, G.; Distler, J.H.W.; et al. Poly (ADP-ribose) polymerase-1 regulates fibroblast activation in systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 744–751. [Google Scholar] [CrossRef]
- Wang, H.; Yang, X.; Yang, Q.; Gong, L.; Xu, H.; Wu, Z. PARP-1 inhibition attenuates cardiac fibrosis induced by myocardial infarction through regulating autophagy. Biochem. Biophys. Res. Commun. 2018, 503, 1625–1632. [Google Scholar] [CrossRef]
- Césaire, M.; Thariat, J.; Candéias, S.M.; Stefan, D.; Saintigny, Y.; Chevalier, F. Combining PARP inhibition, radiation, and immunotherapy: A possible strategy to improve the treatment of cancer? Int. J. Mol. Sci. 2018, 19, 3793. [Google Scholar] [CrossRef] [Green Version]
- Evans, T.; Matulonis, U. PARP inhibitors in ovarian cancer: Evidence, experience and clinical potential. Ther. Adv. Med. Oncol. 2017, 9, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972.e5–2980.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricks, T.K.; Chiu, H.-J.; Ison, G.; Kim, G.; McKee, A.E.; Kluetz, P.; Pazdur, R. Successes and Challenges of PARP Inhibitors in Cancer Therapy. Front. Oncol. 2015, 5, 222. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, G.; Patruno, R.; Ruggieri, E.; Montemurro, S.; Valerio, P.; Ribatti, D. Vascular Endothelial Growth Factor (VEGF) as a Target of Bevacizumab in Cancer: From the Biology to the Clinic. Curr. Med. Chem. 2006, 13, 1845–1857. [Google Scholar] [CrossRef]
- Tewari, K.S.; Burger, R.A.; Enserro, D.; Norquist, B.M.; Swisher, E.M.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Huang, H.; Homesley, H.D.; et al. Final Overall Survival of a Randomized Trial of Bevacizumab for Primary Treatment of Ovarian Cancer. J. Clin. Oncol. 2019, 37, 2317–2328. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab Combined with Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer: The AURELIA Open-Label Randomized Phase III Trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A Randomized, Double-Blind, Placebo-Controlled Phase III Trial of Chemotherapy with or Without Bevacizumab in Patients with Platinum-Sensitive Recurrent Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [Green Version]
- Lalla, R.V.; Boisoneau, D.S.; Spiro, J.D.; Kreutzer, D.L. Expression of Vascular Endothelial Growth Factor Receptors on Tumor Cells in Head and Neck Squamous Cell Carcinoma. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Xiang, H.; Yang, G.; Ma, J.; Zhao, J. Direct Effects of Bevacizumab on Rat Conjunctival Fibroblast. Cell Biophys. 2015, 73, 45–50. [Google Scholar] [CrossRef]
- Hostettler, K.K.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef]
- Park, Y.M.; Kim, C.D.; Lee, J.S. Effect of Bevacizumab on Human Tenon’s Fibroblasts Cultured from Primary and Recurrent Pterygium. Korean J. Physiol. Pharmacol. 2015, 19, 357–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.N.; Fleagle, J.T.; Guthrie, D.; Parkin, D.E.; Gore, M.E.; Lacave, A.J. Recurrent Epithelial Ovarian Carcinoma: A Randomized Phase III Study of Pegylated Liposomal Doxorubicin Versus Topotecan. J. Clin. Oncol. 2001, 19, 3312–3322. [Google Scholar] [CrossRef]
- Seifrtova, M.; Havelek, R.; Soukup, T.; Filipova, A.; Mokry, J.; Rezacova, M. Mitoxantrone ability to induce premature se-nescence in human dental pulp stem cells and human dermal fibroblasts. J. Physiol. Pharmacol. 2013, 64, 255–266. [Google Scholar]
- Johnson, L.M.; Price, U.K.; Figg, W.D. Treatment-induced secretion of WNT16B promotes tumor growth and acquired resistance to chemotherapy: Implications for potential use of inhibitors in cancer treatment. Cancer Biol. Ther. 2013, 14, 90–91. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Chen, F.; Hou, L.; Shen, L.; Zhang, X.; Wang, D.; Huang, L. Nanocarrier-Mediated Chemo-Immunotherapy Arrested Cancer Progression and Induced Tumor Dormancy in Desmoplastic Melanoma. ACS Nano 2018, 12, 7812–7825. [Google Scholar] [CrossRef]
- Giovannetti, E.; Peters, G.J. Molecular Targets of Gemcitabine Action: Rationale for Development of Novel Drugs and Drug Combinations. Curr. Pharm. Des. 2012, 18, 2811–2829. [Google Scholar] [CrossRef]
- Thigpen, T. The Role of Gemcitabine in First-Line Treatment of Advanced Ovarian Carcinoma. Semin. Oncol. 2006, 33, 26–32. [Google Scholar] [CrossRef]
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543. [Google Scholar] [CrossRef]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.E.; Bapiro, T.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2017, 67, 497–507. [Google Scholar] [CrossRef]
- Zhang, X.; Schönrogge, M.; Eichberg, J.; Wendt, E.H.U.; Kumstel, S.; Stenzel, J.; Lindner, T.; Jaster, R.; Krause, B.J.; Vollmar, B.; et al. Blocking Autophagy in Cancer-Associated Fibroblasts Supports Chemotherapy of Pancreatic Cancer Cells. Front. Oncol. 2018, 8, 590. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Tommelein, J.; De Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2017, 78, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, C.-H.; Wang, X.-T.; Ma, W.; Wang, N.-N.; Nesa, E.U.; Wang, J.-B.; Wang, C.; Jia, Y.-B.; Wang, K.; Tian, H.; et al. Irradiated fibroblasts promote epithelial–mesenchymal transition and HDGF expression of esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2015, 458, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Liska, D.; Xiang, S.; Karagkounis, G.; Signs, S.; Kalady, M.; Huang, E. Cancer Associated Fibroblasts Mediate Resistance to Radiotherapy in Rectal Cancer Cells. Gastroenterology 2017, 152, S1234. [Google Scholar] [CrossRef]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.-W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor–stromal interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef] [Green Version]
- Hellevik, T.; Pettersen, I.; Berg, V.; Winberg, J.O.; Moe, B.T.; Bartnes, K.; Paulssen, R.H.; Busund, L.-T.; Bremnes, R.; Chalmers, A.; et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat. Oncol. 2012, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kletsas, D. Human lung fibroblasts prematurely senescent after exposure to ionizing radiation enhance the growth of malignant lung epithelial cells in vitro and in vivo. Int. J. Oncol. 2011, 39, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Qayyum, M.A.; Insana, M.F. Stromal responses to fractionated radiotherapy. Int. J. Radiat. Biol. 2012, 88, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zubiaurre, I.; Fenton, C.G.; Taman, H.; Pettersen, I.; Hellevik, T.; Paulssen, R.H. Tumorigenic Responses of Can-cer-Associated Stromal Fibroblasts after Ablative Radiotherapy: A Transcriptome-Profiling Study. J. Cancer Ther. 2013, 4, 208–250. [Google Scholar] [CrossRef] [Green Version]
- Moradian, S.; Howell, D. Prevention and management of chemotherapy-induced nausea and vomiting. Int. J. Palliat. Nurs. 2015, 21, 216–224. [Google Scholar] [CrossRef]
- Rosso, M.; Muñoz, M.; Berger, M. The Role of Neurokinin-1 Receptor in the Microenvironment of Inflammation and Cancer. Sci. World J. 2012, 2012, 381434. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Xia, H.; Ni, D.; Hu, Y.; Liu, J.; Qin, Y.; Zhou, Q.; Yi, Q.; Xie, Y. High-Dose Dexamethasone Manipulates the Tumor Microenvironment and Internal Metabolic Pathways in Anti-Tumor Progression. Int. J. Mol. Sci. 2020, 21, 1846. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.D.; Panagi, M.; Wang, C.; Khan, T.T.; Martin, M.R.; Voutouri, C.; Toh, K.; Papageorgis, P.; Mpekris, F.; Polydorou, C.; et al. Dexamethasone Increases Cisplatin-Loaded Nanocarrier Delivery and Efficacy in Metastatic Breast Cancer by Normalizing the Tumor Microenvironment. ACS Nano 2019, 13, 6396–6408. [Google Scholar] [CrossRef]
- Stratz, C.; Bhatia, H.S.; Akundi, R.S.; Nührenberg, T.; Trenk, D.; Muñoz, E.; Fiebich, B.L. The anti-inflammatory effects of the 5-HT3 receptor antagonist tropisetron are mediated by the inhibition of p38 MAPK activation in primary human monocytes. Int. Immunopharmacol. 2012, 13, 398–402. [Google Scholar] [CrossRef]
- Stratz, C.; Anakwue, J.; Bhatia, H.S.; Pitz, S.; Fiebich, B.L. Anti-inflammatory effects of 5-HT3 receptor antagonists in interleukin-1beta stimulated primary human chondrocytes. Int. Immunopharmacol. 2014, 22, 160–166. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastasis of ovarian cancer. J. Exp. Med. 2019, 216, 688–703. [Google Scholar] [CrossRef] [Green Version]
- Howes, A.L.; Richardson, R.D.; Finlay, D.; Vuori, K. 3-Dimensional Culture Systems for Anti-Cancer Compound Profiling and High-Throughput Screening Reveal Increases in EGFR Inhibitor-Mediated Cytotoxicity Compared to Monolayer Culture Systems. PLoS ONE 2014, 9, e108283. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Lal-Nag, M.; White, E.A.; Shen, M.; Chiang, C.-Y.; Mitra, A.K.; Zhang, Y.; Curtis, M.W.; Schryver, E.M.; Bettis, S.; et al. Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Horman, S.R.; To, J.; Orth, A.P. An HTS-Compatible 3D Colony Formation Assay to Identify Tumor-Specific Chemotherapeutics. J. Biomol. Screen. 2013, 18, 1298–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krausz, E.; De Hoogt, R.; Gustin, E.; Cornelissen, F.; Grand-Perret, T.; Janssen, L.; Vloemans, N.; Wuyts, D.; Frans, S.; Axel, A.; et al. Translation of a Tumor Microenvironment Mimicking 3D Tumor Growth Co-culture Assay Platform to High-Content Screening. J. Biomol. Screen. 2013, 18, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniati, E.; Berlato, C.; Gopinathan, G.; Heath, O.; Kotantaki, P.; Lakhani, A.; McDermott, J.; Pegrum, C.; Delaine-Smith, R.M.; Pearce, O.M.; et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020, 30, 525–540.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roswall, P.; Pietras, K. Of mice and men: A comparative study of cancer-associated fibroblasts in mammary carcinoma. Upsala J. Med. Sci. 2012, 117, 196–201. [Google Scholar] [CrossRef]
- González-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020, 6, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Eckert, M.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; Delgado, B.D.L.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef] [Green Version]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141.e9–155.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, S.; Bartolomé, R.A.; Mendes, M.; Barderas, R.; Fernandez-Aceñero, M.J.; Peláez-García, A.; Peña, C.; Lopez-Lucendo, M.; Villar-Vázquez, R.; De Herreros, A.G.; et al. Proteome Profiling of Cancer-Associated Fibroblasts Identifies Novel Proinflammatory Signatures and Prognostic Markers for Colorectal Cancer. Clin. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, H.R.; Jackson, H.W.; Zanotelli, V.R.T.; Danenberg, E.; Fischer, J.R.; Bardwell, H.; Provenzano, E.; CRUK IMAXT Grand Challenge Team; Rueda, O.M.; Chin, S.-F.; et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat. Cancer 2020, 1, 163–175. [Google Scholar] [CrossRef]
- Annunziata, C.; Chu, C.; Rubin, S. Chemotherapy for Gynecologic Cancers: Society of Gynecologic Oncology Handbook, 3rd ed.; Society of Gynecologic Oncology: Chicago, IL, USA, 2017. [Google Scholar]
- McWhinney, S.R.; Goldberg, R.M.; McLeod, H.L. Platinum neurotoxicity pharmacogenetics. Mol. Cancer Ther. 2009, 8, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [Green Version]
- Pundavela, J.; Roselli, S.; Faulkner, S.; Attia, J.; Scott, R.J.; Thorne, R.F.; Forbes, J.F.; Bradshaw, R.A.; Walker, M.M.; Jobling, P.; et al. Nerve fibers infiltrate the tumor microenvironment and are associated with nerve growth factor production and lymph node invasion in breast cancer. Mol. Oncol. 2015, 9, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.W.; Nagaraja, A.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 2015, 15, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Arese, M.; Bussolino, F.; Pergolizzi, M.; Bizzozero, L.; Pascal, D. Tumor progression: The neuronal input. Ann. Transl. Med. 2018, 6, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanat, O.; Ertas, H.; Caner, B. Platinum-induced neurotoxicity: A review of possible mechanisms. World J. Clin. Oncol. 2017, 8, 329–335. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Wu, Z.; Phan, S. Smad3 Mediates Transforming Growth Factor-β–Induced α-Smooth Muscle Actin Expression. Am. J. Respir. Cell Mol. Biol. 2003, 29, 397–404. [Google Scholar] [CrossRef]
- Paulsson, J.; Micke, P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin. Cancer Biol. 2014, 25, 61–68. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.; Wu, S.Z.; Roden, D.; Chan, C.-L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic Acid–Induced Pancreatic Stellate Cell Quiescence Reduces Paracrine Wnt–β-Catenin Signaling to Slow Tumor Progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Cellular heterogeneity and bidirectional communication in the TME. (a) Representative image of a high-grade serous ovarian cancer metastasis to the omentum exemplifies the diverse ecosystem of tumors that includes cancer cells, immune cells, CAFs, adipocytes, endothelial cells, and other cells embedded in an ECM. CAFs and endothelial cells are present throughout the TME. (1:100) (b) Cross-talk between cell types in the ECM involves bidirectional signaling between tumor and stromal cell types that enforce a pro-tumorigenic microenvironment.
Figure 1.
Cellular heterogeneity and bidirectional communication in the TME. (a) Representative image of a high-grade serous ovarian cancer metastasis to the omentum exemplifies the diverse ecosystem of tumors that includes cancer cells, immune cells, CAFs, adipocytes, endothelial cells, and other cells embedded in an ECM. CAFs and endothelial cells are present throughout the TME. (1:100) (b) Cross-talk between cell types in the ECM involves bidirectional signaling between tumor and stromal cell types that enforce a pro-tumorigenic microenvironment.
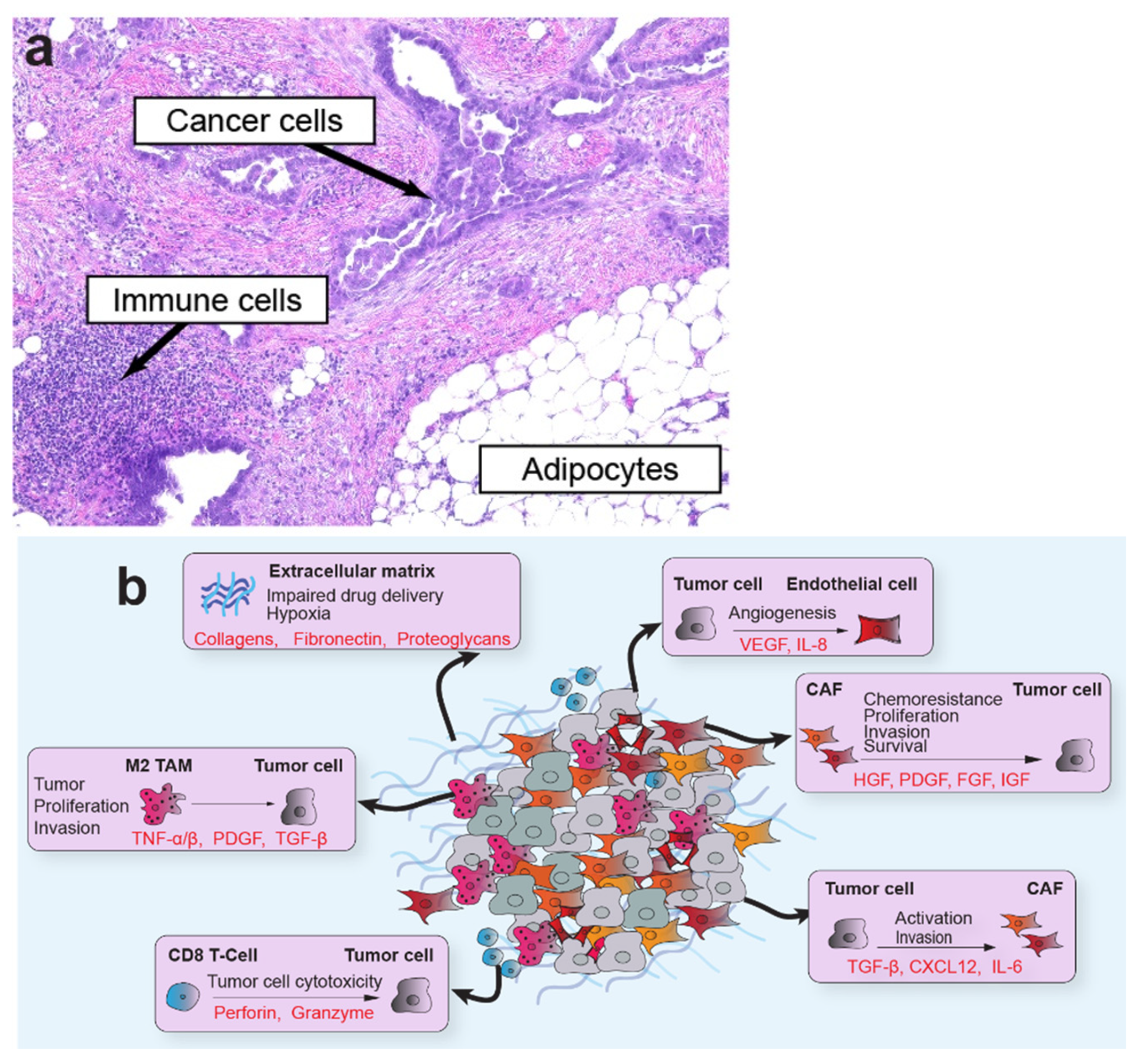
Figure 2.
Effects of therapeutic agents on cancer associated fibroblasts (CAF)s. The diverse therapeutics used in the treatment of ovarian cancer, including platinum agents, taxanes, Poly ADP-ribose polymerases (PARP) inhibitors, tomoisomerase inhibitors, and anti-angiogenics, may have (a) pro-tumorigenic or (b) anti-tumorigenic effects on CAFs. * Experimental or clinical observations in normal fibroblasts or fibroblasts from disease states other than cancer.
Figure 2.
Effects of therapeutic agents on cancer associated fibroblasts (CAF)s. The diverse therapeutics used in the treatment of ovarian cancer, including platinum agents, taxanes, Poly ADP-ribose polymerases (PARP) inhibitors, tomoisomerase inhibitors, and anti-angiogenics, may have (a) pro-tumorigenic or (b) anti-tumorigenic effects on CAFs. * Experimental or clinical observations in normal fibroblasts or fibroblasts from disease states other than cancer.
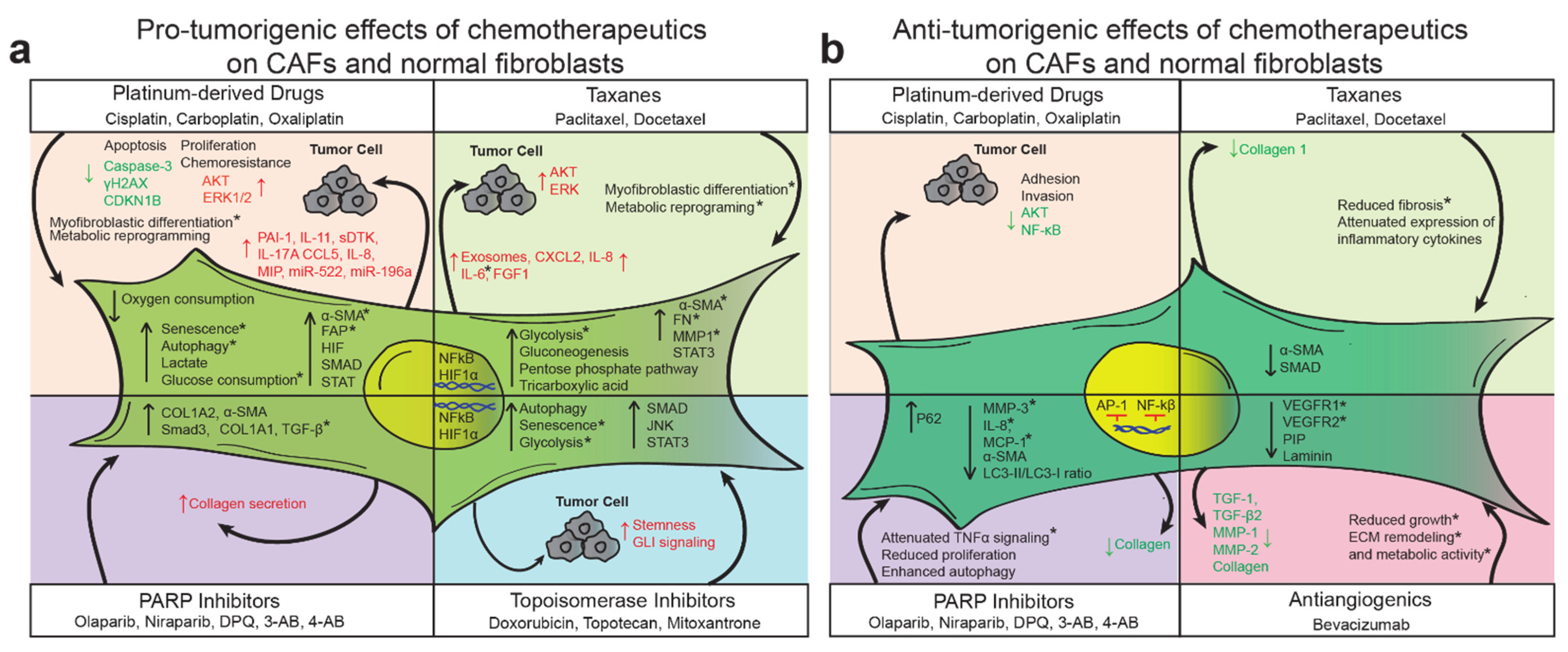
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Eckert, M.A.; Orozco, C.; Xiao, J.; Javellana, M.; Lengyel, E. The Effects of Chemotherapeutics on the Ovarian Cancer Microenvironment. Cancers 2021, 13, 3136. https://doi.org/10.3390/cancers13133136
AMA Style
Eckert MA, Orozco C, Xiao J, Javellana M, Lengyel E. The Effects of Chemotherapeutics on the Ovarian Cancer Microenvironment. Cancers. 2021; 13(13):3136. https://doi.org/10.3390/cancers13133136
Chicago/Turabian StyleEckert, Mark A., Carlos Orozco, Jason Xiao, Melissa Javellana, and Ernst Lengyel. 2021. "The Effects of Chemotherapeutics on the Ovarian Cancer Microenvironment" Cancers 13, no. 13: 3136. https://doi.org/10.3390/cancers13133136
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.