
SurvCNN: A Discrete Time-to-Event Cancer Survival Estimation Framework Using Image Representations of Omics Data



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets and Study Design
2.2. Data Retrieval and Preprocessing
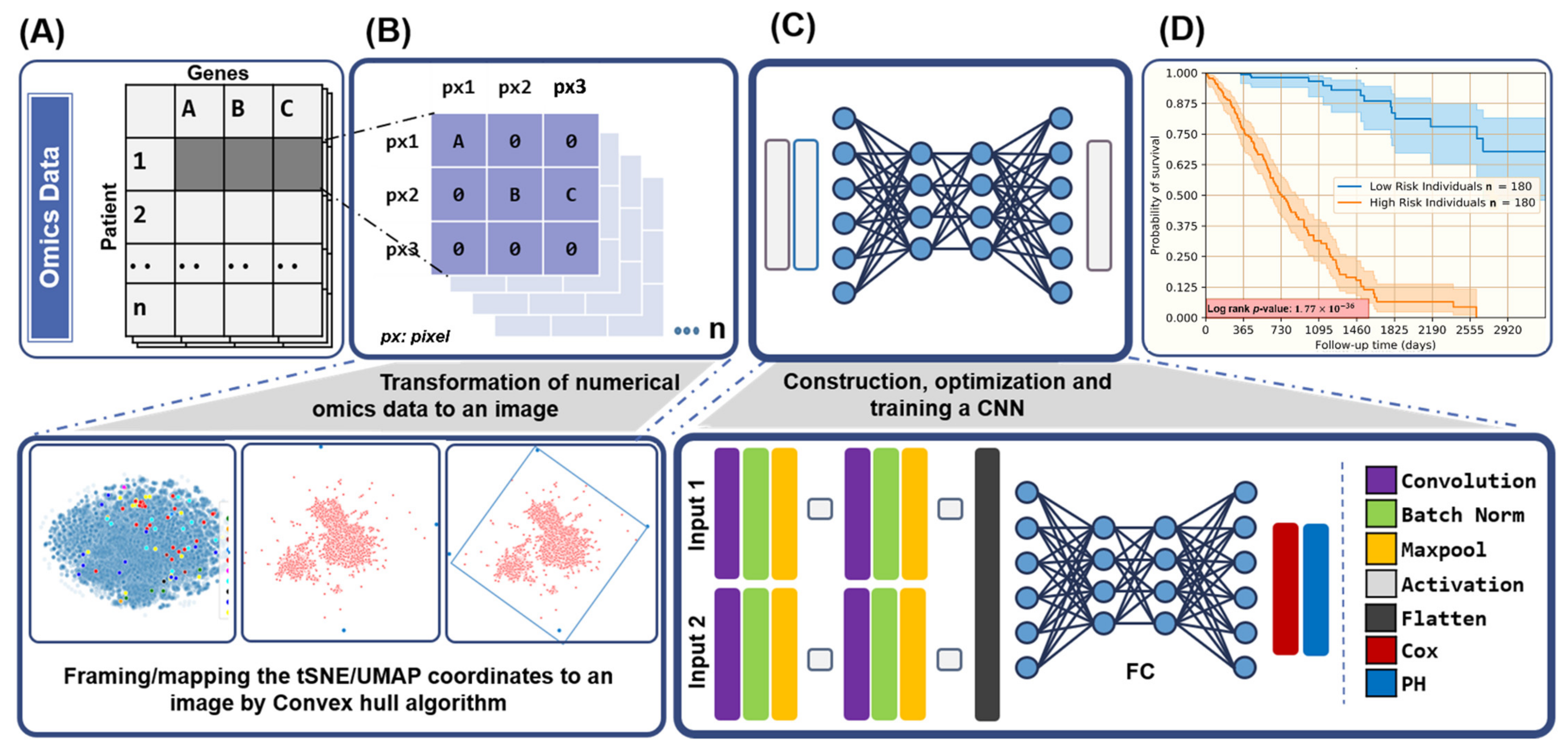
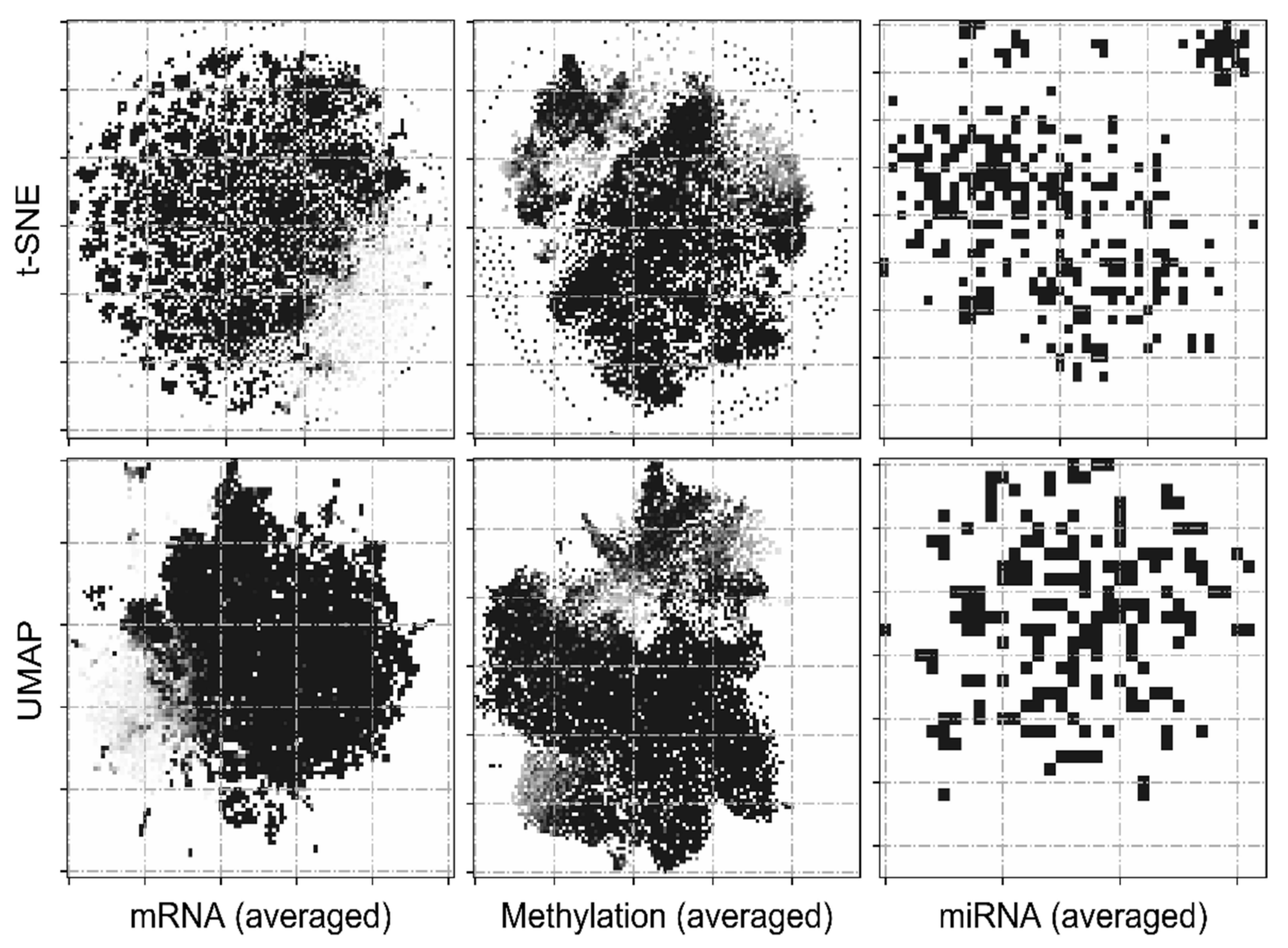
2.3. Feature Transformation
2.4. CNN Designs, Architecture, and Evaluation
2.5. Survival Analysis
2.6. Loss Function
2.7. Hazard Probability
2.8. Dichotomizing Patient Groups Using Kaplan Meier Estimates
2.9. Performance Metrics
2.10. Functional Enrichment and Gene Ontology Analysis for Identified Biomarkers
3. Results
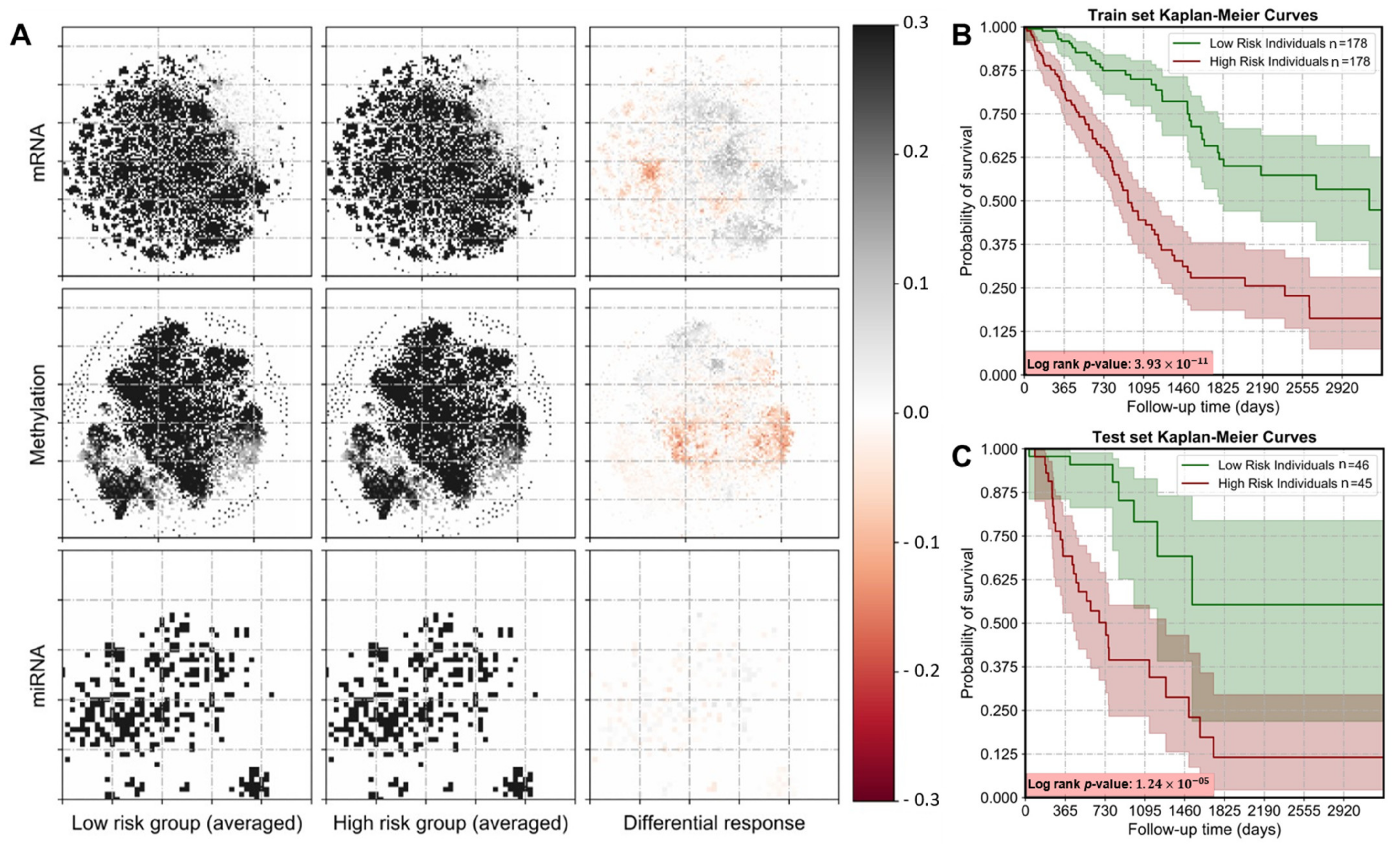
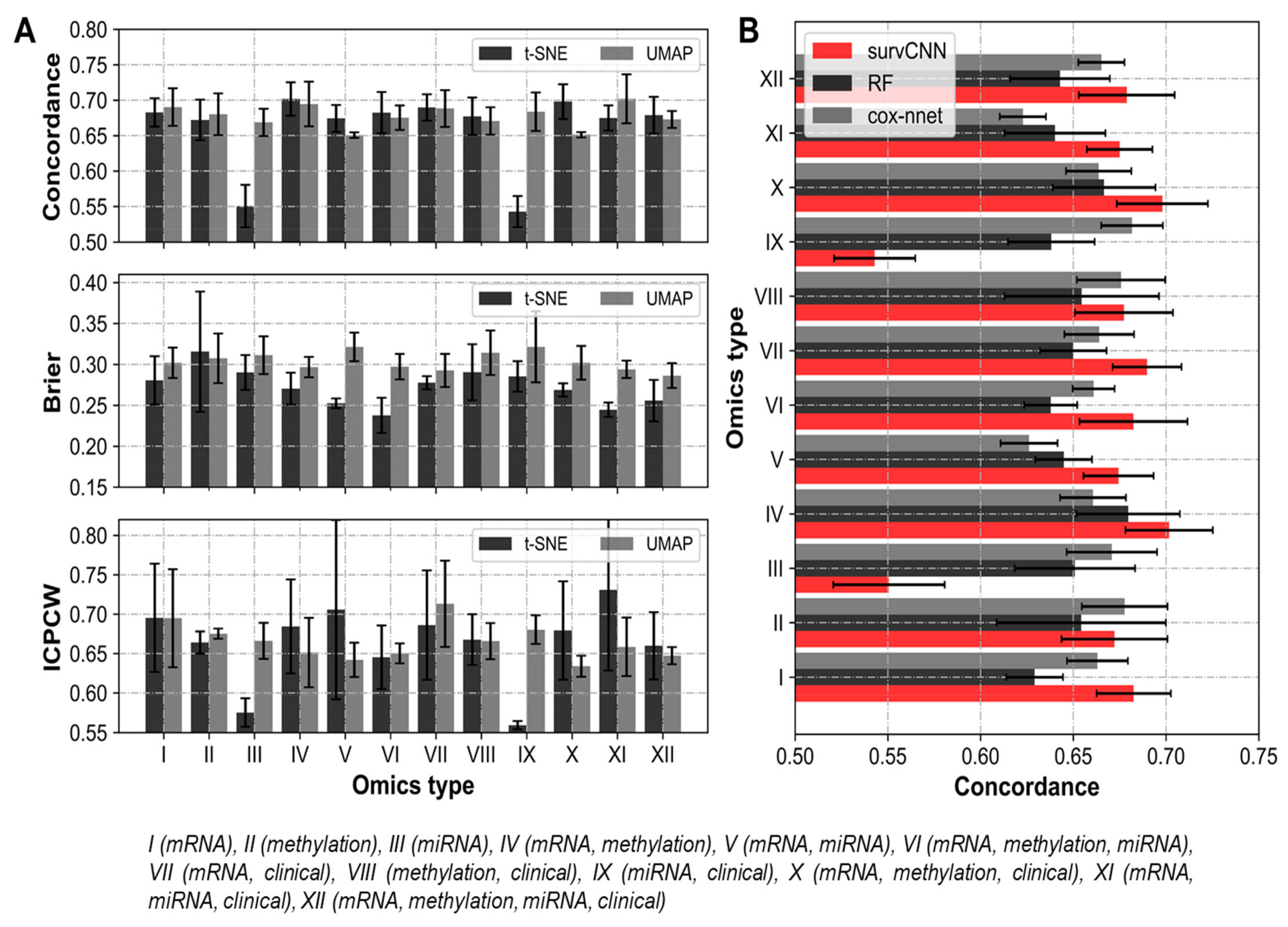
3.1. Image Representations of Omics-Data Have Predictive Ability
3.2. Identification of Prognostic Subtypes in Lung Adenocarcinoma
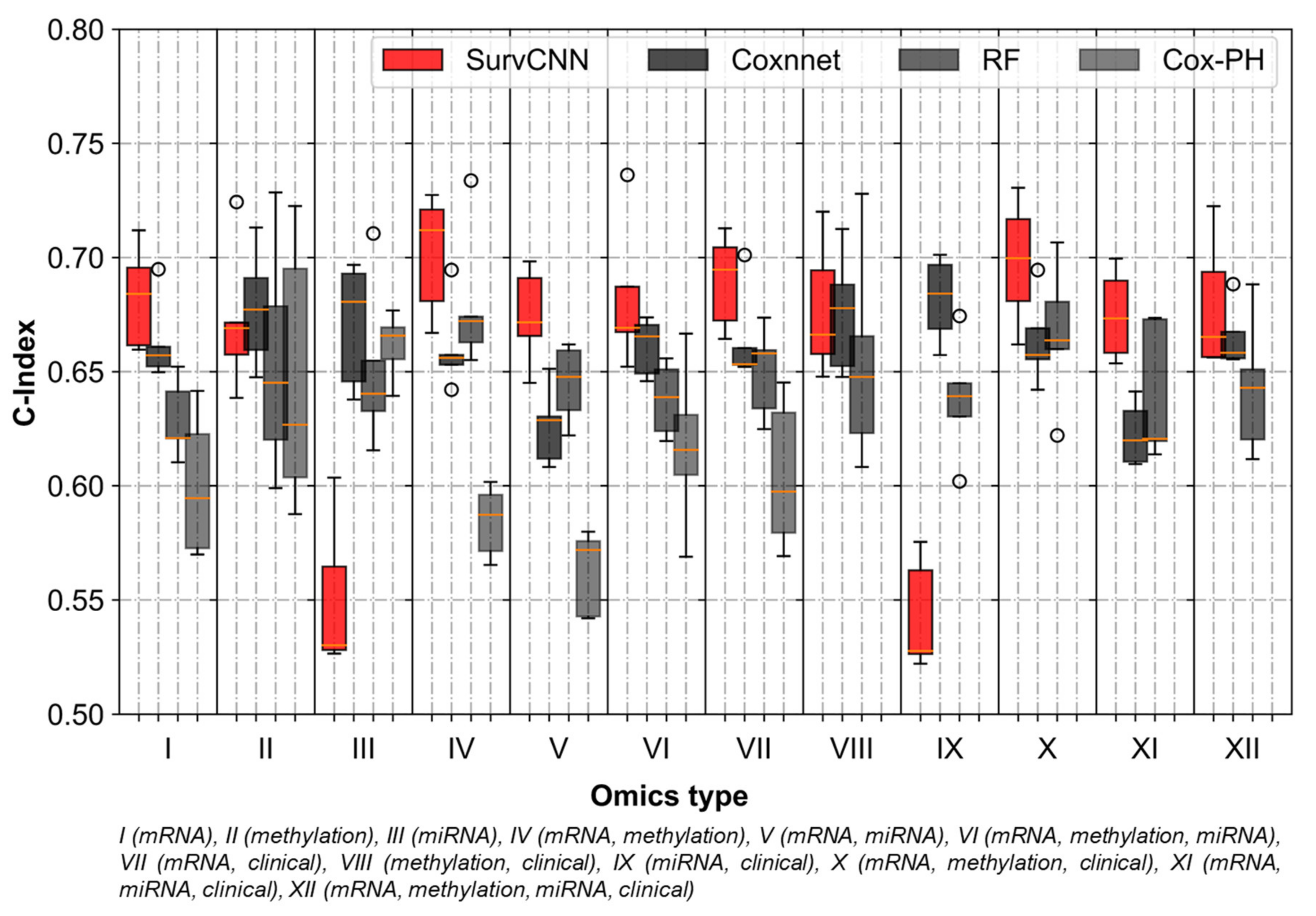
3.3. Integrating Omics-Types Affects the Performance of Prognostic Models
3.4. Transformation Based Multi-Omics Integration Outperforms Alternative Approaches
3.5. SurvCNN Identifies LUAD Associated Pathways, GO Terms, and Cell Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- O’Donnell, S.T.; Ross, R.P.; Stanton, C. The progress of multi-omics technologies: Determining function in lactic acid bacteria using a systems level approach. Front. Microbiol. 2020, 10, 3084. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, H.; Patjoshi, S.; Yeghiazaryan, K.; Kehrer, C.; Kuhn, W.; Golubnitschaja, O. Premenopausal breast cancer: Potential clinical utility of a multi-omics based machine learning approach for patient stratification. EPMA J. 2018, 9, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Luo, H.; Zhou, H.; Li, G.; Bu, D.; Yang, X.; Zhao, X.; Zhang, H.; Liu, S.; Zhong, Y.; et al. Identification of prognostic biomarkers in hepatitis B virus-related hepatocellular carcinoma and stratification by integrative multi-omics analysis. J. Hepatol. 2014, 61, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Bair, E.; Tibshirani, R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004, 2, e108. [Google Scholar] [CrossRef]
- Cheng, W.-Y.; Yang, T.-H.O.; Anastassiou, D. Development of a prognostic model for breast cancer survival in an open challenge environment. Sci. Transl. Med. 2013, 5, 181ra50. [Google Scholar] [CrossRef] [PubMed]
- Royston, P.; Altman, D.G. External validation of a Cox prognostic model: Principles and methods. BMC Med. Res. Methodol. 2013, 13, 33. [Google Scholar] [CrossRef]
- Yeh, R.W.; Secemsky, E.A.; Kereiakes, D.J.; Normand, S.-L.T.; Gershlick, A.H.; Cohen, D.J.; Spertus, J.A.; Steg, P.G.; Cutlip, D.E.; Rinaldi, M.J.; et al. Development and validation of a prediction rule for benefit and harm of dual antiplatelet therapy beyond 1 year after percutaneous coronary intervention. JAMA 2016, 315, 1735–1749. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.; Thompson, I.R.; et al. Genomics of DRUG SENSITIVITY IN CANCER (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization classification of lung tumors. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Hackshaw, A.K.; Law, M.R.; Wald, N.J. The accumulated evidence on lung cancer and environmental tobacco smoke. BMJ 1997, 315, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Wu, H.; Wang, M.D. Integrating multi-omics data by learning modality invariant representations for improved prediction of overall survival of cancer. Methods 2021, 189, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Wang, L.-N.; Ling, X.; Dong, J. An overview on data representation learning: From traditional feature learning to recent deep learning. J. Financ. Data Sci. 2016, 2, 265–278. [Google Scholar] [CrossRef]
- Beale, D.J.; Karpe, A.V.; Ahmed, W. Beyond metabolomics: A review of multi-omics-based approaches. In Microbial Metabolomics; Springer: Cham, Switzerland, 2016; pp. 289–312. [Google Scholar]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef]
- Legendre, P.; Legendre, L.F. Numerical Ecology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 24. [Google Scholar]
- Jolliffe, I. Principal component analysis. In International Encyclopedia of Statistical Science; Lovric, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1094–1096. [Google Scholar]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, C.-C.; Li, W.; Shen, H.; Laird, P.W.; Zhou, X.J. Discovery of multi-dimensional modules by integrative analysis of cancer genomic data. Nucleic Acids Res. 2012, 40, 9379–9391. [Google Scholar] [CrossRef]
- Louhimo, R.; Hautaniemi, S. CNAmet: An R package for integrating copy number, methylation and expression data. Bioinformatics 2011, 27, 887–888. [Google Scholar] [CrossRef]
- Mankoo, P.K.; Shen, R.; Schultz, N.; Levine, D.A.; Sander, C. Time to recurrence and survival in serous ovarian tumors predicted from integrated genomic profiles. PLoS ONE 2011, 6, e24709. [Google Scholar] [CrossRef]
- Djebbari, A.; Quackenbush, J. Seeded Bayesian Networks: Constructing genetic networks from microarray data. BMC Syst. Biol. 2008, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Jung, Y.-S.; Sungur, E.A.; Han, K.-H.; Park, C.; Sohn, I. A copula method for modeling directional dependence of genes. BMC Bioinform. 2008, 9, 225. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Haffner, P.; Bottou, L.; Bengio, Y. Object Recognition with Gradient-Based Learning. In Shape, Contour and Grouping in Computer Vision; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 1999; Volume 1681, pp. 319–345. [Google Scholar] [CrossRef]
- Cox, D.R. Regression models and life-tables. J. R. Stat. Soc. Ser. B 1972, 34, 187–220. [Google Scholar] [CrossRef]
- Chin, S.F.; Teschendorff, A.; Marioni, J.C.; Wang, Y.; Barbosa-Morais, N.L.; Thorne, N.P.; Costa, J.L.; Pinder, S.; Van De Wiel, M.; Green, A.R.; et al. High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007, 8, R215. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Narasimhan, B.; Chu, G. Impute: Imputation for microarray data. 2016. R package version 1.48.0.
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426 2018. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Sharma, A.; Vans, E.; Shigemizu, D.; Boroevich, K.A.; Tsunoda, T. DeepInsight: A methodology to transform a non-image data to an image for convolution neural network architecture. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. In Proceedings of the 32nd International Conference on International Conference on Machine Learning, Lille, France, 7–9 July 2015; pp. 448–456. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Caruana, R.; Lawrence, S.; Giles, L. Overfitting in neural nets: Backpropagation, conjugate gradient, and early stopping. In Proceedings of the 13th International Conference on Neural Information Processing Systems, Denver, CO, USA, 27–30 November 2000; MIT Press: Cambridge, MA, USA, 2000; pp. 381–387. [Google Scholar]
- Zhu, X.; Yao, J.; Huang, J. Deep convolutional neural network for survival analysis with pathological images. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzen, China, 15–18 December 2016; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2016; Volume 2016, pp. 544–547. [Google Scholar]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-nnet: An artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef]
- Gensheimer, M.F.; Narasimhan, B. A scalable discrete-time survival model for neural networks. PeerJ 2019, 7, e6257. [Google Scholar] [CrossRef] [PubMed]
- Bewick, V.; Cheek, L.; Ball, J. Statistics review 12: Survival analysis. Crit. Care 2004, 8, 389–394. [Google Scholar] [CrossRef]
- Gerds, T.A.; Kattan, M.; Schumacher, M.; Yu, C. Estimating a time-dependent concordance index for survival prediction models with covariate dependent censoring. Stat. Med. 2013, 32, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Graf, E.; Schmoor, C.; Sauerbrei, W.; Schumacher, M. Assessment and comparison of prognostic classification schemes for survival data. Stat. Med. 1999, 18, 2529–2545. [Google Scholar] [CrossRef]
- Wei, R.; De Vivo, I.; Huang, S.; Zhu, X.; Risch, H.; Moore, J.H.; Yu, H.; Garmire, L.X. Meta-dimensional data integration identifies critical pathways for susceptibility, tumorigenesis and progression of endometrial cancer. Oncotarget 2016, 7, 55249–55263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dong, G.; Mao, L.; Huang, B.; Gamalo-Siebers, M.; Wang, J.; Yu, G.; Hoaglin, D.C. The inverse-probability-of-censoring weighting (IPCW) adjusted win ratio statistic: An unbiased estimator in the presence of independent censoring. J. Biopharm. Stat. 2020, 30, 882–899. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Mogensen, U.B.; Ishwaran, H.; Gerds, T.A. Evaluating random forests for survival analysis using prediction error curves. J. Stat. Softw. 2012, 50, 1–23. [Google Scholar] [CrossRef]
- Heffernan, T.P.; Simpson, D.A.; Frank, A.R.; Heinloth, A.N.; Paules, R.S.; Cordeiro-Stone, M.; Kaufmann, W.K. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 2002, 22, 8552–8561. [Google Scholar] [CrossRef] [PubMed]
- Ishimi, Y. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J. Biol. Chem. 1997, 272, 24508–24513. [Google Scholar] [CrossRef]
- Mossi, R.; Jónsson, Z.O.; Allen, B.L.; Hardin, S.H.; Hübscher, U. Replication factor C interacts with the C-terminal side of proliferating cell nuclear antigen. J. Biol. Chem. 1997, 272, 1769–1776. [Google Scholar] [CrossRef]
- Wang, Q.; Su, L.; Liu, N.; Zhang, L.; Xu, W.; Fang, H. Cyclin dependent kinase 1 inhibitors: A review of recent progress. Curr. Med. Chem. 2011, 18, 2025–2043. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, H.; Lyu, Z.; Su, J.; He, J.; Pei, Y.; Cheng, X.; Zhou, N.; Lu, X.; Zhou, S.; Zhao, Y. Effect of lung squamous cell carcinoma tumor microenvironment on the CD105+endothelial cell proteome. J. Proteome Res. 2014, 13, 4717–4729. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I | II | III | IV | V | VI | VII | VIII | IX | X | XI | XII | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mRNA | + | + | + | + | + | + | + | + | ||||
| meth | + | + | + | + | + | + | ||||||
| miRNA | + | + | + | + | + | + | ||||||
| Clinical | + | + | + | + | + | + |
| Omics Type * | Total Cases | Living | Deceased | Features Before Processing | Features After Processing | Age (yrs.) | Survival (yrs.) | ||
|---|---|---|---|---|---|---|---|---|---|
| Median | Range | Median | Range | ||||||
| I | 515 | 328 | 187 | 20,172 | 123 × 123 | 66.0 | 38–88 | 1.803 | 0–2 |
| II | 458 | 293 | 165 | 17,052 | 123 × 123 | 1.786 | |||
| III | 450 | 286 | 164 | 477 | 42 × 42 | 1.789 | |||
| IV | 454 | 290 | 164 | 37,224 | (123 × 123) × 2 | 1.785 | |||
| V | 446 | 283 | 163 | 20,649 | (123 × 123) × 2 | 1.788 | |||
| VI | 446 | 283 | 163 | 37,701 | (123 × 123) + (42 × 42) | 1.788 | |||
| VII | 515 | 328 | 187 | 20,192 | (123 × 123) + 20 | 1.802 | |||
| VIII | 458 | 293 | 165 | 17,072 | (123 × 123) + 20 | 1.786 | |||
| IX | 450 | 286 | 164 | 497 | (42 × 42) + 20 | 1.789 | |||
| X | 454 | 290 | 164 | 37,244 | (123 × 123) × 2 + 20 | 1.784 | |||
| XI | 446 | 283 | 163 | 20,669 | (123 × 123) + (42 × 42) + 20 | 1.787 | |||
| XII | 446 | 283 | 163 | 37,721 | (123 × 123) × 2 + (42 × 42) + 20 | 1.787 | |||
| Omics Type * (Set 1) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II | III | IV | V | VI | VII | VIII | IX | X | XI | XII | ||
| Omics type * (Set 2) | I | 1.56 | 18.23 | −5.21 | 2.32 | 0.021 | −3.73 | 1.18 | 32.72 | −4.50 | 4.50 | 0.80 |
| II | - | 20.99 | −3.64 | −0.28 | −3.78 | −2.25 | −1.00 | 17.19 | −4.09 | −0.36 | −1.20 | |
| III | - | - | −15.78 | −14.89 | −38.86 | −15.70 | −36.67 | 1.49 | −19.84 | −16.03 | −49.08 | |
| IV | - | - | - | 6.50 | 2.15 | 4.29 | 3.90 | 23.57 | 1.40 | 6.33 | 3.13 | |
| V | - | - | - | - | −1.05 | −5.56 | −0.56 | 30.39 | −7.21 | −0.23 | −0.73 | |
| VI | - | - | - | - | - | 0.87 | 1.34 | 22.37 | −2.31 | 0.97 | 0.91 | |
| VII | - | - | - | - | - | - | 2.14 | 28.09 | −2.42 | 8.36 | 1.72 | |
| VIII | - | - | - | - | - | - | - | 40.89 | −5.17 | 0.46 | −0.84 | |
| IX | - | - | - | - | - | - | - | - | −30.35 | −35.70 | −43.84 | |
| X | - | - | - | - | - | - | - | - | - | 5.88 | 3.55 | |
| XI | - | - | - | - | - | - | - | - | - | - | −0.74 | |
| Omics Type * | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | VIII | IX | X | XI | XII | |
| RF | 13.52 | 1.78 | −22.69 | 2.29 | 11.36 | 4.84 | 28.40 | 2.71 | −15.36 | 8.46 | 5.66 | 9.16 |
| Cox-PH | 18.81 | 1.63 | −10.23 | 23.75 | 25.03 | 12.15 | 13.61 | - | - | - | - | - |
| Cox-nnet | 3.96 | −1.10 | −12.68 | 5.23 | 11.59 | 2.02 | 3.64 | 0.44 | −26.87 | 6.61 | 19.10 | 1.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalakoti, Y.; Yadav, S.; Sundar, D. SurvCNN: A Discrete Time-to-Event Cancer Survival Estimation Framework Using Image Representations of Omics Data. Cancers 2021, 13, 3106. https://doi.org/10.3390/cancers13133106
Kalakoti Y, Yadav S, Sundar D. SurvCNN: A Discrete Time-to-Event Cancer Survival Estimation Framework Using Image Representations of Omics Data. Cancers. 2021; 13(13):3106. https://doi.org/10.3390/cancers13133106
Chicago/Turabian StyleKalakoti, Yogesh, Shashank Yadav, and Durai Sundar. 2021. "SurvCNN: A Discrete Time-to-Event Cancer Survival Estimation Framework Using Image Representations of Omics Data" Cancers 13, no. 13: 3106. https://doi.org/10.3390/cancers13133106
APA StyleKalakoti, Y., Yadav, S., & Sundar, D. (2021). SurvCNN: A Discrete Time-to-Event Cancer Survival Estimation Framework Using Image Representations of Omics Data. Cancers, 13(13), 3106. https://doi.org/10.3390/cancers13133106