
FOXM1: A Multifunctional Oncoprotein and Emerging Therapeutic Target in Ovarian Cancer



Abstract
Simple Summary
Abstract
1. Introduction
1.1. FOX Proteins and FOXM1 Discovery
1.2. FOXM1 Structure and Transcriptional Activity
1.3. FOXM1 Function and Regulation
1.4. Transgenic Mouse Models Reveal Functions of FOXM1
2. Ovarian Cancer
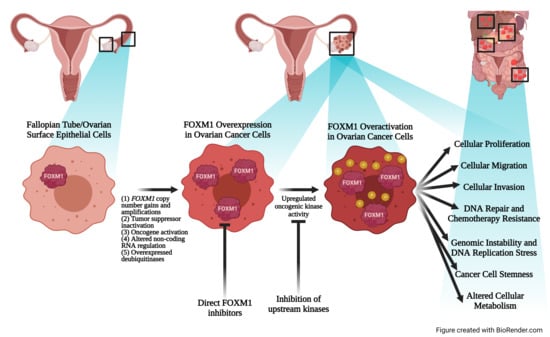
3. FOXM1 Is Overexpressed and Activated in Ovarian Cancer
3.1. The FOXM1 Gene Experiences Copy Number Gains and Amplifications
3.2. Inactivation of Upstream Tumor Suppressor Promotes FOXM1 Gene Expression
3.3. Upstream Oncogenes Promote FOXM1 Expression
3.4. FOXM1 Is Post-Transcriptionally Regulated by Non-Coding RNAs (ncRNA)
3.5. FOXM1 Is Stabilized and Activated by Post-Translational Mechanisms
4. FOXM1 Oncogenic Functions
4.1. FOXM1 Expression Is Associated with Tumor Progression and Poor Prognosis in Ovarian Cancer
4.2. FOXM1 Promotes Cellular Proliferation, Migration, and Invasion
4.3. FOXM1 Promotes DNA Repair and Chemotherapy Resistance
4.4. FOXM1 Promotes Cancer Cell Stemness
4.5. FOXM1 Promotes Genomic Instability and DNA Replication Stress
4.6. FOXM1 Is Linked to Altered Cellular Metabolism
4.7. FOXM1 Isoform Expression and Function in Cancer
5. Clinical Translation
5.1. FOXM1 Has Potential as a Prognostic Biomarker in Ovarian Cancer
5.2. In Vivo Studies of FOXM1 in Ovarian Cancer Are Limited
5.3. Therapeutic Targeting of FOXM1 in Ovarian Cancer
5.4. Inhibitors of Upstream Signaling Pathways
5.5. Direct FOXM1 Inhibitors
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Weigel, D.; Jürgens, G.; Küttner, F.; Seifert, E.; Jäckle, H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 1989, 57, 645–658. [Google Scholar] [CrossRef]
- Kaestner, K.H.; Lee, K.H.; Schlondorff, J.; Hiemisch, H.; Monaghan, A.P.; Schutz, G. Six members of the mouse forkhead gene family are developmentally regulated. Proc. Natl. Acad. Sci. USA 1993, 90, 7628–7631. [Google Scholar] [CrossRef]
- Clevidence, D.E.; Overdier, D.G.; Tao, W.; Qian, X.; Pani, L.; Lai, E.; Costa, R.H. Identification of nine tissue-specific transcription factors of the hepatocyte nuclear factor 3/forkhead DNA-binding-domain family. Proc. Natl. Acad. Sci. USA 1993, 90, 3948–3952. [Google Scholar] [CrossRef]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nat. Cell Biol. 1993, 364, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Kaestner, K.H.; Knochel, W.; E Martinez, D. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000, 14, 142–146. [Google Scholar] [PubMed]
- Jackson, B.C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse forkhead box (FOX) gene families. Hum. Genom. 2010, 4, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Myatt, S.S.; Lam, E.W.-F. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Friedman, J.R.; Kaestner, K.H. The Foxa family of transcription factors in development and metabolism. Cell. Mol. Life Sci. CMLS 2006, 63, 2317–2328. [Google Scholar] [CrossRef]
- Seo, S.; Fujita, H.; Nakano, A.; Kang, M.; Duarte, A.; Kume, T. The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev. Biol. 2006, 294, 458–470. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J.; Zhang, X.; Sejas, D.P.; Qiu, Y.; Williams, D.A.; Pang, Q. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Fleskens, V.; van Boxtel, R. Forkhead Box P family members at the crossroad between tolerance and immunity: A balancing act. Int. Rev. Immunol. 2014, 33, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Roose, J.; Clevers, H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997, 25, 1715–1719. [Google Scholar] [CrossRef]
- Yao, K.M.; Sha, M.; Lu, Z.; Wong, G.G. Molecular analysis of a novel winged helix protein, WIN. Expression pattern, DNA binding property, and alternative splicing within the DNA binding domain. J. Biol. Chem. 1997, 272, 19827–19836. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Kelly, T.F.; Samadani, U.; Lim, L.; Rubio, S.; Overdier, D.G.; A Roebuck, K.; Costa, R.H. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol. Cell. Biol. 1997, 17, 1626–1641. [Google Scholar] [CrossRef]
- Korver, W.; Roose, J.; Heinen, K.; Weghuis, D.O.; de Bruijn, D.; van Kessel, A.G.; Clevers, H. The HumanTRIDENT/HFH-11/FKHL16Gene: Structure, Localization, and Promoter Characterization. Genomics 1997, 46, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, J.M.; Rao, P.N.; Gerace, L. Cloning of cDNAs for M-phase phosphoproteins recognized by the MPM2 monoclonal antibody and determination of the phosphorylated epitope. Proc. Natl. Acad. Sci. USA 1994, 91, 714–718. [Google Scholar] [CrossRef]
- Ramirez, M.I.; Rishi, A.K.; Cao, Y.X.; Williams, M.C. TGT3, Thyroid Transcription Factor I, and Sp1 Elements Regulate Transcriptional Activity of the 1.3-Kilobase Pair Promoter ofT1α, a Lung Alveolar Type I Cell Gene. J. Biol. Chem. 1997, 272, 26285–26294. [Google Scholar] [CrossRef]
- Korver, W.; Roose, J.; Wilson, A.; Clevers, H. The Winged-Helix Transcription Factor Trident is Expressed in Actively Dividing Lymphocytes. Immunobiology 1997, 198, 157–161. [Google Scholar] [CrossRef]
- Ahn, J.-I.; Lee, K.-H.; Shin, D.-M.; Shim, J.-W.; Kim, C.-M.; Kim, H.; Lee, S.-H.; Lee, Y.-S. Temporal expression changes during differentiation of neural stem cells derived from mouse embryonic stem cell. J. Cell. Biochem. 2004, 93, 563–578. [Google Scholar] [CrossRef]
- Krupczak-Hollis, K.; Wang, X.; Kalinichenko, V.V.; Gusarova, G.A.; Wang, I.C.; Dennewitz, M.B.; Yoder, H.M.; Kiyokawa, H.; Kaestner, K.H.; Costa, R.H. The mouse Forkhead Box m1 transcription factor is essential for hepatoblast mitosis and development of intrahepatic bile ducts and vessels during liver morphogenesis. Dev. Biol. 2004, 276, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-M.; Ramakrishna, S.; Gusarova, G.A.; Yoder, H.M.; Costa, R.H.; Kalinichenko, V.V. The Forkhead Box M1 Transcription Factor Is Essential for Embryonic Development of Pulmonary Vasculature. J. Biol. Chem. 2005, 280, 22278–22286. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Kim, I.-M.; Petrovic, V.; Malin, D.; Wang, I.-C.; Kalin, T.V.; Meliton, L.; Zhao, Y.-Y.; Ackerson, T.; Qin, Y.; et al. Myocardium defects and ventricular hypoplasia in mice homozygous null for theForkhead Box m1 transcription factor. Dev. Dyn. 2007, 236, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, V.V.; Lim, L.; Shin, B.; Costa, R.H. Differential expression of forkhead box transcription factors following butylated hydroxytoluene lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L695–L704. [Google Scholar] [CrossRef] [PubMed]
- Zu, G.; Guo, J.; Zhou, T.; Che, N.; Liu, B.; Wang, D.; Zhang, X. The transcription factor FoxM1 activates Nurr1 to promote intestinal regeneration after ischemia/reperfusion injury. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Chang-Panesso, M.; Kadyrov, F.F.; Lalli, M.; Wu, H.; Ikeda, S.; Kefaloyianni, E.; Abdelmageed, M.M.; Herrlich, A.; Kobayashi, A.; Humphreys, B.D. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J. Clin. Investig. 2019, 129, 5501–5517. [Google Scholar] [CrossRef]
- Chen, Z.; Li, L.; Xu, S.; Liu, Z.; Zhou, C.; Li, Z.; Liu, Y.; Wu, W.; Huang, Y.; Kuang, M.; et al. A Cdh1-FoxM1-Apc axis controls muscle development and regeneration. Cell Death Dis. 2020, 11, 180. [Google Scholar] [CrossRef]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Stahl, M.; Medema, R.H. FoxM1: At the crossroads of ageing and cancer. Biochim. Biophys. Acta (BBA)-Bioenerg. 2007, 1775, 92–102. [Google Scholar] [CrossRef]
- Liao, G.-B.; Li, X.-Z.; Zeng, S.; Liu, C.; Yang, S.-M.; Yang, L.; Hu, C.-J.; Bai, J.-Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Zhang, X.; Zhang, L.; Du, Y.; Zheng, H.; Zhang, P.; Sun, Y.; Wang, Y.; Chen, J.; Ding, P.; Wang, N.; et al. A novel FOXM1 isoform, FOXM1D, promotes epithelial–mesenchymal transition and metastasis through ROCKs activation in colorectal cancer. Oncogene 2016, 36, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. FOXM1 in Cancer: Interactions and Vulnerabilities. Cancer Res. 2017, 77, 3135–3139. [Google Scholar] [CrossRef]
- Kong, X.; Li, L.; Li, Z.; Le, X.; Huang, C.; Jia, Z.; Cui, J.; Huang, S.; Wang, L.; Xie, K. Dysregulated Expression of FOXM1 Isoforms Drives Progression of Pancreatic Cancer. Cancer Res. 2013, 73, 3987–3996. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, P.; Zhang, W.; Zhang, X.; Chen, J.; Ding, P.; Li, L.; Lv, X.; Li, L.; Hu, W. PI3K activation is enhanced by FOXM1D binding to p110 and p85 subunits. Signal Transduct. Target. Ther. 2020, 5, 1–3. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Huang, S.; Chen, J.; Ding, P.; Wang, Q.; Li, L.; Lv, X.; Li, L.; Zhang, P.; et al. FOXM1D potentiates PKM2-mediated tumor glycolysis and angiogenesis. Mol. Oncol. 2021, 15, 1466–1485. [Google Scholar] [CrossRef]
- Barrett, C.L.; DeBoever, C.; Jepsen, K.; Saenz, C.C.; Carson, D.A.; Frazer, K.A. Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proc. Natl. Acad. Sci. USA 2015, 112, E3050–E3057. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Wang, Z.; Costa, R.H.; Tyner, A.; Lau, L.F.; Raychaudhuri, P. An N-terminal inhibitory domain modulates activity of FoxM1 during cell cycle. Oncogene 2007, 27, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef]
- Littler, D.R.; Fernández, M.A.; Stein, A.; Hibbert, R.G.; Heidebrecht, T.; Aloy, P.; Medema, R.; Perrakis, A. Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res. 2010, 38, 4527–4538. [Google Scholar] [CrossRef]
- A Sanders, D.; Ross-Innes, C.S.; Beraldi, D.; Carroll, J.S.; Balasubramanian, S. Genome-wide mapping of FOXM1 binding reveals co-binding with estrogen receptor alpha in breast cancer cells. Genome Biol. 2013, 14, R6. [Google Scholar] [CrossRef]
- Chen, X.; Müller, G.A.; Quaas, M.; Fischer, M.; Han, N.; Stutchbury, B.; Sharrocks, A.D.; Engeland, K. The Forkhead Transcription Factor FOXM1 Controls Cell Cycle-Dependent Gene Expression through an Atypical Chromatin Binding Mechanism. Mol. Cell. Biol. 2012, 33, 227–236. [Google Scholar] [CrossRef]
- Iness, A.; Litovchick, L. MuvB: A Key to Cell Cycle Control in Ovarian Cancer. Front. Oncol. 2018, 8, 223. [Google Scholar] [CrossRef]
- Sanders, D.A.; Gormally, M.V.; Marsico, G.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. FOXM1 binds directly to non-consensus sequences in the human genome. Genome Biol. 2015, 16, 1–23. [Google Scholar] [CrossRef]
- Kang, K.; Choi, Y.; Kim, H.H.; Yoo, K.H.; Yu, S. Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines. Int. J. Mol. Sci. 2020, 21, 6141. [Google Scholar] [CrossRef]
- Mullen, D.J.; Yan, C.; Kang, D.S.; Zhou, B.; Borok, Z.; Marconett, C.N.; Farnham, P.J.; Offringa, I.A.; Rhie, S.K. TENET 2.0: Identification of key transcriptional regulators and enhancers in lung adenocarcinoma. PLoS Genet. 2020, 16, e1009023. [Google Scholar] [CrossRef]
- Ye, H.; Holterman, A.X.; Yoo, K.W.; Franks, R.R.; Costa, R.H. Premature Expression of the Winged Helix Transcription Factor HFH-11B in Regenerating Mouse Liver Accelerates Hepatocyte Entry into S Phase. Mol. Cell. Biol. 1999, 19, 8570–8580. [Google Scholar] [CrossRef]
- Wang, X.; Arceci, A.; Bird, K.; Mills, C.A.; Choudhury, R.; Kernan, J.L.; Zhou, C.; Bae-Jump, V.; Bowers, A.; Emanuele, M.J. VprBP/DCAF1 Regulates the Degradation and Nonproteolytic Activation of the Cell Cycle Transcription Factor FoxM1. Mol. Cell. Biol. 2017, 37, e00609-16. [Google Scholar] [CrossRef]
- Jeffery, J.M.; Kalimutho, M.; Johansson, P.; Cardenas, D.G.; Kumar, R.; Khanna, K.K. FBXO31 protects against genomic instability by capping FOXM1 levels at the G2/M transition. Oncogene 2016, 36, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Lüscher-Firzlaff, J.M.; Lilischkis, R.; Lüscher, B. Regulation of the transcription factor FOXM1c by Cyclin E/CDK2. FEBS Lett. 2006, 580, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Dominguez-Brauer, C.; Wang, Z.; Asara, J.M.; Costa, R.H.; Tyner, A.L.; Lau, L.F.; Raychaudhuri, P. A Conserved Phosphorylation Site within the Forkhead Domain of FoxM1B Is Required for Its Activation by Cyclin-CDK1. J. Biol. Chem. 2009, 284, 30695–30707. [Google Scholar] [CrossRef]
- Major, M.L.; Lepe, R.; Costa, R.H. Forkhead Box M1B Transcriptional Activity Requires Binding of Cdk-Cyclin Complexes for Phosphorylation-Dependent Recruitment of p300/CBP Coactivators. Mol. Cell. Biol. 2004, 24, 2649–2661. [Google Scholar] [CrossRef]
- Fernández, M.A.; Halim, V.A.; Aprelia, M.; Mohammed, S.; Medema, R.H. Protein Phosphatase 2A (B55α) Prevents Premature Activation of Forkhead Transcription Factor FoxM1 by Antagonizing Cyclin A/Cyclin-dependent Kinase-mediated Phosphorylation. J. Biol. Chem. 2011, 286, 33029–33036. [Google Scholar] [CrossRef]
- Laoukili, J.; Alvarez, M.; Meijer, L.A.T.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.R.; Medema, R.H. Activation of FoxM1 during G 2 Requires Cyclin A/Cdk-Dependent Relief of Autorepression by the FoxM1 N-Terminal Domain. Mol. Cell. Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Raychaudhuri, P.; Costa, R.H. Chk2 Mediates Stabilization of the FoxM1 Transcription Factor to Stimulate Expression of DNA Repair Genes. Mol. Cell. Biol. 2007, 27, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.Y.M.; Tong, T.H.K.; Cheung, A.M.S.; Tsang, A.C.C.; Leung, W.Y.; Yao, K.-M. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J. Cell Sci. 2005, 118, 795–806. [Google Scholar] [CrossRef]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; Van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Costa, R.H.; Lau, L.F.; Tyner, A.L.; Raychaudhuri, P. Anaphase-Promoting Complex/Cyclosome-Cdh1-Mediated Proteolysis of the Forkhead Box M1 Transcription Factor Is Critical for Regulated Entry into S Phase. Mol. Cell. Biol. 2008, 28, 5162–5171. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Brison, C.M.; Nerli, S.; E Arsenault, H.; McShan, A.C.; Chen, E.; Lee, H.-W.; A Benanti, J.; Sgourakis, N.G.; Rubin, S.M. An order-to-disorder structural switch activates the FoxM1 transcription factor. eLife 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.C.; Chen, Y.-J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Cost, R.H. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Mol. Cell. Biol. 2005, 25, 10875. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Wonsey, D.R.; Follettie, M.T. Loss of the Forkhead Transcription Factor FoxM1 Causes Centrosome Amplification and Mitotic Catastrophe. Cancer Res. 2005, 65, 5181–5189. [Google Scholar] [CrossRef]
- Schimmel, J.; Eifler, K.; Sigurðsson, J.O.; Cuijpers, S.A.; Hendriks, I.A.; Vries, M.V.-D.; Kelstrup, C.D.; Francavilla, C.; Medema, R.; Olsen, J.V.; et al. Uncovering SUMOylation Dynamics during Cell-Cycle Progression Reveals FoxM1 as a Key Mitotic SUMO Target Protein. Mol. Cell 2014, 53, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-M.; Liu, R.; Wang, L.; Nascimento, L.; Brennan, V.C.; Yang, W.-H. SUMOylation of FOXM1B Alters Its Transcriptional Activity on Regulation of MiR-200 Family and JNK1 in MCF7 Human Breast Cancer Cells. Int. J. Mol. Sci. 2014, 15, 10233–10251. [Google Scholar] [CrossRef]
- Myatt, S.S.; Kongsema, M.; Man, C.W.-Y.; Kelly, D.J.; Gomes, A.R.; Khongkow, P.; Karunarathna, U.; Zona, S.; Langer, J.; Dunsby, C.; et al. SUMOylation inhibits FOXM1 activity and delays mitotic transition. Oncogene 2014, 33, 4316–4329. [Google Scholar] [CrossRef]
- Jaiswal, N.; John, R.; Chand, V.; Nag, A. Oncogenic Human Papillomavirus 16E7 modulates SUMOylation of FoxM1b. Int. J. Biochem. Cell Biol. 2015, 58, 28–36. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, C.; Wu, J.; Elsayed, Z.; Fu, Z. Polo-like Kinase 1-mediated Phosphorylation of Forkhead Box Protein M1b Antagonizes Its SUMOylation and Facilitates Its Mitotic Function. J. Biol. Chem. 2015, 290, 3708–3719. [Google Scholar] [CrossRef]
- Lv, C.; Zhao, G.; Sun, X.; Wang, P.; Xie, N.; Luo, J.; Tong, T. Acetylation of FOXM1 is essential for its transactivation and tumor growth stimulation. Oncotarget 2016, 7, 60366–60382. [Google Scholar] [CrossRef]
- Cohn, O.; Feldman, M.; Weil, L.; Kublanovsky, M.; Levy, D. Chromatin associated SETD3 negatively regulates VEGF expression. Sci. Rep. 2016, 6, 37115. [Google Scholar] [CrossRef]
- Korver, W.; Schilham, M.W.; Moerer, P.; Hoff, M.J.V.D.; Dam, K.; Lamers, W.H.; Medema, R.; Clevers, H. Uncoupling of S phase and mitosis in cardiomyocytes and hepatocytes lacking the winged-helix transcription factor Trident. Curr. Biol. 1998, 8, 1327–1330. [Google Scholar] [CrossRef]
- Kalin, T.V.; Ustiyan, V.; Kalinichenko, V.V. Multiple faces of FoxM1 transcription factor: Lessons from transgenic mouse models. Cell Cycle (Georgetown Tex) 2011, 10, 396–405. [Google Scholar] [CrossRef]
- Ustiyan, V.; Wang, I.-C.; Ren, X.; Zhang, Y.; Snyder, J.; Xu, Y.; Wert, S.E.; Lessard, J.L.; Kalin, T.V.; Kalinichenko, V.V. Forkhead box M1 transcriptional factor is required for smooth muscle cells during embryonic development of blood vessels and esophagus. Dev. Biol. 2009, 336, 266–279. [Google Scholar] [CrossRef]
- Kalin, T.V.; Wang, I.-C.; Meliton, L.; Zhang, Y.; Wert, S.E.; Ren, X.; Snyder, J.; Bell, S.M.; Graf, L.; Whitsett, J.A.; et al. Forkhead Box m1 transcription factor is required for perinatal lung function. Proc. Natl. Acad. Sci. USA 2008, 105, 19330–19335. [Google Scholar] [CrossRef]
- Wang, I.-C.; Zhang, Y.; Snyder, J.; Sutherland, M.J.; Burhans, M.S.; Shannon, J.M.; Park, H.J.; Whitsett, J.A.; Kalinichenko, V.V. Increased expression of FoxM1 transcription factor in respiratory epithelium inhibits lung sacculation and causes Clara cell hyperplasia. Dev. Biol. 2010, 347, 301–314. [Google Scholar] [CrossRef]
- Zhang, H.; Ackermann, A.M.; Gusarova, G.A.; Lowe, D.; Feng, X.; Kopsombut, U.G.; Costa, R.H.; Gannon, M. The FoxM1 Transcription Factor Is Required to Maintain Pancreatic β-Cell Mass. Mol. Endocrinol. 2006, 20, 1853–1866. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Gao, X.-P.; Zhao, Y.D.; Mirza, M.K.; Frey, R.S.; Kalinichenko, V.V.; Wang, I.C.; Costa, R.H.; Malik, A.B. Endothelial cell–restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J. Clin. Investig. 2006, 116, 2333–2343. [Google Scholar] [CrossRef]
- Mirza, M.K.; Sun, Y.; Zhao, Y.D.; Potula, H.-H.; Frey, R.S.; Vogel, S.M.; Malik, A.B.; Zhao, Y.-Y. FoxM1 regulates re-annealing of endothelial adherens junctions through transcriptional control of β-catenin expression. J. Exp. Med. 2010, 207, 1675–1685. [Google Scholar] [CrossRef]
- Wang, X.; Kiyokawa, H.; Dennewitz, M.B.; Costa, R.H. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 16881–16886. [Google Scholar] [CrossRef]
- Wang, X.; Hung, N.; Costa, R.H. Earlier expression of the transcription factor HFH-11B diminishes induction of p21CIP1/WAF1 levels and accelerates mouse hepatocyte entry into S-phase following carbon tetrachloride liver injury. Hepatology 2001, 33, 1404–1414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Quail, E.; Hung, N.-J.; Tan, Y.; Ye, H.; Costa, R.H. Increased levels of forkhead box M1B transcription factor in transgenic mouse hepatocytes prevent age-related proliferation defects in regenerating liver. Proc. Natl. Acad. Sci. USA 2001, 98, 11468–11473. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, V.V.; Gusarova, G.A.; Tan, Y.; Wang, I.C.; Major, M.L.; Wang, X.; Yoder, H.M.; Costa, R.H. Ubiquitous expression of the forkhead box M1B transgene accelerates proliferation of distinct pulmonary cell types following lung injury. J. Biol. Chem. 2003, 278, 37888–37894. [Google Scholar] [CrossRef]
- Ackermann, M.A.; Costa, R.H.; Gannon, M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes 2008, 57, 3069–3077. [Google Scholar] [CrossRef]
- Wang, I.-C.; Meliton, L.; Tretiakova, M.; Costa, R.H.; Kalinichenko, V.V.; Kalin, T.V. Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene 2008, 27, 4137–4149. [Google Scholar] [CrossRef][Green Version]
- Yoshida, Y.; Wang, I.; Yoder, H.M.; Davidson, N.O.; Costa, R.H. The Forkhead Box M1 Transcription Factor Contributes to the Development and Growth of Mouse Colorectal Cancer. Gastroenterology 2007, 132, 1420–1431. [Google Scholar] [CrossRef]
- Kalin, T.V.; Wang, I.-C.; Ackerson, T.J.; Major, M.L.; Detrisac, C.J.; Kalinichenko, V.V.; Lyubimov, A.; Costa, R.H. Increased Levels of the FoxM1 Transcription Factor Accelerate Development and Progression of Prostate Carcinomas in both TRAMP and LADY Transgenic Mice. Cancer Res. 2006, 66, 1712–1720. [Google Scholar] [CrossRef]
- Milewski, D.; Balli, D.; Ustiyan, V.; Le, T.; Dienemann, H.; Warth, A.; Breuhahn, K.; Whitsett, J.A.; Kalinichenko, V.V.; Kalin, T.V. FOXM1 activates AGR2 and causes progression of lung adenomas into invasive mucinous adenocarcinomas. PLoS Genet. 2017, 13, e1007097. [Google Scholar] [CrossRef] [PubMed]
- A Kalinina, O.; A Kalinin, S.; Polack, E.W.; Mikaelian, I.; Panda, S.; Costa, R.H.; Adami, G.R. Sustained hepatic expression of FoxM1B in transgenic mice has minimal effects on hepatocellular carcinoma development but increases cell proliferation rates in preneoplastic and early neoplastic lesions. Oncogene 2003, 22, 6266–6276. [Google Scholar] [CrossRef]
- Kim, I.-M.; Ackerson, T.; Ramakrishna, S.; Tretiakova, M.; Wang, I.-C.; Kalin, T.V.; Major, M.L.; Gusarova, G.A.; Yoder, H.M.; Costa, R.H.; et al. The Forkhead Box m1 Transcription Factor Stimulates the Proliferation of Tumor Cells during Development of Lung Cancer. Cancer Res. 2006, 66, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-C.; Meliton, L.; Ren, X.; Zhang, Y.; Balli, D.; Snyder, J.; Whitsett, J.A.; Kalinichenko, V.V.; Kalin, T.V. Deletion of Forkhead Box M1 Transcription Factor from Respiratory Epithelial Cells Inhibits Pulmonary Tumorigenesis. PLoS ONE 2009, 4, e6609. [Google Scholar] [CrossRef] [PubMed]
- Gusarova, G.A.; Wang, I.C.; Major, M.L.; Kalinichenko, V.V.; Ackerson, T.; Petrovic, V.; Costa, R.H. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J. Clin. Investig. 2007, 117, 99–111. [Google Scholar] [CrossRef]
- Kalinichenko, V.V.; Major, M.L.; Wang, X.; Petrovic, V.; Kuechle, J.; Yoder, H.M.; Dennewitz, M.B.; Shin, B.; Datta, A.; Raychaudhuri, P.; et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004, 18, 830–850. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.; Park, E.Y.; Klinkebiel, D.L.; Pack, S.D.; Shin, Y.-H.; Abdullaev, Z.; Emerson, R.E.; Coffey, D.M.; Kwon, S.Y.; Creighton, C.J.; et al. In vivo modeling of metastatic human high-grade serous ovarian cancer in mice. PLoS Genet. 2020, 16, e1008808. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 70, 7–30. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Suh-Burgmann, E.J.; Alavi, M. Detection of early stage ovarian cancer in a large community cohort. Cancer Med. 2019, 8, 7133–7140. [Google Scholar] [CrossRef]
- Wright, A.A.; Bohlke, K.; Armstrong, D.K.; Bookman, M.A.; Cliby, W.A.; Coleman, R.L.; Dizon, D.S.; Kash, J.J.; Meyer, L.A.; Moore, K.N.; et al. Neoadjuvant Chemotherapy for Newly Diagnosed, Advanced Ovarian Cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 3460–3473. [Google Scholar] [CrossRef]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.-M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 75, 739–741. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian Cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- Baldwin, L.A.; Huang, B.; Miller, R.W.; Tucker, T.; Goodrich, S.T.; Podzielinski, I.; DeSimone, C.P.; Ueland, F.R.; Van Nagell, J.R.; Seamon, L.G. Ten-Year Relative Survival for Epithelial Ovarian Cancer. Obstet. Gynecol. 2012, 120, 612–618. [Google Scholar] [CrossRef]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.-S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995–2009: Analysis of individual data for 25 676 887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef]
- Kurman, R. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 2013, 24, x16–x21. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Soong, T.R.; Howitt, B.E.; Horowitz, N.; Nucci, M.R.; Crum, C.P. The fallopian tube, “precursor escape” and narrowing the knowledge gap to the origins of high-grade serous carcinoma. Gynecol. Oncol. 2019, 152, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Drapkin, R. It’s Totally Tubular… Riding the New Wave of Ovarian Cancer Research. Cancer Res. 2016, 76, 10–17. [Google Scholar] [CrossRef]
- Klinkebiel, D.; Zhang, W.; Akers, S.N.; Odunsi, K.; Karpf, A.R. DNA Methylome Analyses Implicate Fallopian Tube Epithelia as the Origin for High-Grade Serous Ovarian Cancer. Mol. Cancer Res. 2016, 14, 787–794. [Google Scholar] [CrossRef]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial Carcinoma of the Fimbria and Pelvic Serous Carcinoma: Evidence for a Causal Relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007, 211, 26–35. [Google Scholar] [CrossRef]
- Levanon, K.; Crum, C.; Drapkin, R. New Insights into the Pathogenesis of Serous Ovarian Cancer and Its Clinical Impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef]
- Przybycin, C.G.; Kurman, R.J.; Ronnett, B.M.; Shih, I.-M.; Vang, R. Are All Pelvic (Nonuterine) Serous Carcinomas of Tubal Origin? Am. J. Surg. Pathol. 2010, 34, 1407–1416. [Google Scholar] [CrossRef]
- Piek, J.M.J.; Van Diest, P.J.; Zweemer, R.P.; Jansen, J.W.; Poort-Keesom, R.J.J.; Menko, F.H.; Gille, J.J.P.; Jongsma, A.P.M.; Pals, G.; Kenemans, P. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 2001, 195, 451–456. [Google Scholar] [CrossRef]
- Leeper, K.; Garcia, R.; Swisher, E.; Goff, B.; Greer, B.; Paley, P. Pathologic Findings in Prophylactic Oophorectomy Specimens in High-Risk Women. Gynecol. Oncol. 2002, 87, 52–56. [Google Scholar] [CrossRef]
- Medeiros, F.; Muto, M.G.; Lee, Y.; Elvin, A.J.; Callahan, M.J.; Feltmate, C.; E Garber, J.; Cramer, D.W.; Crum, C.P. The Tubal Fimbria Is a Preferred Site for Early Adenocarcinoma in Women with Familial Ovarian Cancer Syndrome. Am. J. Surg. Pathol. 2006, 30, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Drapkin, R. Ovarian Cancer Pathogenesis: A Model in Evolution. J. Oncol. 2009, 2010, 1–13. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; The Australian Ovarian Cancer Study Group; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Ottesen, A.M.; Skakkebæk, N.E.; Lundsteen, C.; Leffers, H.; Larsen, J.; Rajpert-De, M.E. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosomes Cancer 2003, 38, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Jung, E.-M.; Cho, J.; Lee, J.W.; Hwang, K.-T.; Yang, S.-J.; Kang, J.J.; Bae, J.-Y.; Jeon, Y.K.; Park, I.-A.; et al. DNA copy number alterations and expression of relevant genes in triple-negative breast cancer. Genes Chromosomes Cancer 2008, 47, 490–499. [Google Scholar] [CrossRef]
- Green, M.R.; Aya, B.C.; Gandhi, M.K.; Lea, R.A.; Wellwood, J.; Wood, P.; Marlton, P.; Griffiths, L.R. Integrative genomic profiling reveals conserved genetic mechanisms for tumorigenesis in common entities of non-Hodgkin’s lymphoma. Genes Chromosomes Cancer 2011, 50, 313–326. [Google Scholar] [CrossRef]
- Yu, J.; Deshmukh, H.; Payton, J.E.; Dunham, C.; Scheithauer, B.W.; Tihan, T.; Prayson, R.A.; Guha, A.; Bridge, J.A.; Ferner, R.E.; et al. Array-Based Comparative Genomic Hybridization Identifies CDK4 and FOXM1 Alterations as Independent Predictors of Survival in Malignant Peripheral Nerve Sheath Tumor. Clin. Cancer Res. 2011, 17, 1924–1934. [Google Scholar] [CrossRef]
- Liu, Y.; Melin, B.S.; Rajaraman, P.; Wang, Z.; Linet, M.; Shete, S.; Amos, C.I.; Lau, C.C.; Scheurer, M.; The Gliogene Consortium; et al. Insight in glioma susceptibility through an analysis of 6p22.3, 12p13.33-12.1, 17q22-23.2 and 18q23 SNP genotypes in familial and non-familial glioma. Qual. Life Res. 2012, 131, 1507–1517. [Google Scholar] [CrossRef][Green Version]
- Shi, J.; Chatterjee, N.; Rotunno, M.; Wang, Y.; Pesatori, A.C.; Consonni, D.; Li, P.; Wheeler, W.; Broderick, P.; Henrion, M.; et al. Inherited Variation at Chromosome 12p13.33, Including RAD52, Influences the Risk of Squamous Cell Lung Carcinoma. Cancer Discov. 2012, 2, 131–139. [Google Scholar] [CrossRef]
- Delahaye-Sourdeix, M.; Oliver, J.; Timofeeva, M.N.; Gaborieau, V.; Johansson, M.; Chabrier, A.; Wozniak, M.B.; Brenner, D.R.; Vallée, M.P.; Anantharaman, D.; et al. The 12p13.33/RAD52 Locus and Genetic Susceptibility to Squamous Cell Cancers of Upper Aerodigestive Tract. PLoS ONE 2015, 10, e0117639. [Google Scholar] [CrossRef]
- Singh, N.; Sahu, D.K.; Goel, M.; Kant, R.; Gupta, D.K. Retrospective analysis of FFPE based Wilms’ Tumor samples through copy number and somatic mutation related Molecular Inversion Probe Based Array. Gene 2015, 565, 295–308. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Follia, L.; Ferrero, G.; Mandili, G.; Beccuti, M.; Giordano, D.; Spadi, R.; Satolli, M.A.; Evangelista, A.; Katayama, H.; Hong, W.; et al. Integrative Analysis of Novel Metabolic Subtypes in Pancreatic Cancer Fosters New Prognostic Biomarkers. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Barger, C.J.; Zhang, W.; Hillman, J.; Stablewski, A.B.; Higgins, M.J.; Vanderhyden, B.C.; Odunsi, K.; Karpf, A.R. Genetic determinants of FOXM1 overexpression in epithelial ovarian cancer and functional contribution to cell cycle progression. Oncotarget 2015, 6, 27613–27627. [Google Scholar] [CrossRef]
- Paracchini, L.; Beltrame, L.; Grassi, T.; Inglesi, A.; Fruscio, R.; Landoni, F.; Ippolito, D.; Marchette, M.D.; Paderno, M.; Adorni, M.; et al. Genome-wide copy number alterations in circulating tumor DNA as a novel biomarker in high grade serous ovarian cancer patients. Clin. Cancer Res. 2020, 27, 2549–2559. [Google Scholar] [CrossRef]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih, I.-M.; Kurman, R.J. Molecular Alterations of TP53 are a Defining Feature of Ovarian High-Grade Serous Carcinoma: A Rereview of Cases Lacking TP53 Mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef]
- Brachova, P.; Thiel, K.W.; Leslie, K.K. The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 19257–19275. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties onTP53mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Pandit, B.; Halasi, M.; Gartel, A.L. p53 negatively regulates expression of FoxM1. Cell Cycle (Georgetown Tex) 2009, 8, 3425–3427. [Google Scholar] [CrossRef]
- Barsotti, A.M.; Prives, C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene 2009, 28, 4295–4305. [Google Scholar] [CrossRef]
- Elgaaen, B.V.; Olstad, O.K.; Sandvik, L.; Ødegaard, E.; Sauer, T.; Staff, A.C.; Gautvik, K.M. ZNF385B and VEGFA Are Strongly Differentially Expressed in Serous Ovarian Carcinomas and Correlate with Survival. PLoS ONE 2012, 7, e46317. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Lantvit, D.D.; Chae, D.H.; Burdette, J.E. Cadherin-6 type 2, K-cadherin (CDH6) is regulated by mutant p53 in the fallopian tube but is not expressed in the ovarian surface. Oncotarget 2016, 7, 69871–69882. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, L.; Minn, K.; Madan, R.; Godwin, A.K.; Shridhar, V.; Chien, J. Targeting of mutant p53-induced FoxM1 with thiostrepton induces cytotoxicity and enhances carboplatin sensitivity in cancer cells. Oncotarget 2014, 5, 11365–11380. [Google Scholar] [CrossRef]
- Rodgers, L.H.; Hainmhire, E.Ó.; Young, A.N.; Burdette, J.E. Loss of PAX8 in high-grade serous ovarian cancer reduces cell survival despite unique modes of action in the fallopian tube and ovarian surface epithelium. Oncotarget 2016, 7, 32785–32795. [Google Scholar] [CrossRef]
- Ghannam-Shahbari, D.; Jacob, E.; Kakun, R.R.; Wasserman, T.; Korsensky, L.; Sternfeld, O.; Kagan, J.; Bublik, D.R.; Aviel-Ronen, S.; Levanon, K.; et al. PAX8 activates a p53-p21-dependent pro-proliferative effect in high grade serous ovarian carcinoma. Oncogene 2018, 37, 2213–2224. [Google Scholar] [CrossRef]
- Tanaka, N.; Zhao, M.; Tang, L.; Patel, A.A.; Xi, Q.; Van, H.; Takahashi, H.; Osman, A.A.; Zhang, J.; Wang, J.; et al. Gain-of-function mutant p53 promotes the oncogenic potential of head and neck squamous cell carcinoma cells by targeting the transcription factors FOXO3a and FOXM1. Oncogene 2018, 37, 1279–1292. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Endsley, M.P.; Moyle-Heyrman, G.; Karthikeyan, S.; Lantvit, D.D.; Davis, D.A.; Wei, J.-J.; Burdette, J.E. Spontaneous Transformation of Murine Oviductal Epithelial Cells: A Model System to Investigate the Onset of Fallopian-Derived Tumors. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef]
- Millour, J.; De Olano, N.; Horimoto, Y.; Monteiro, L.J.; Langer, J.K.; Aligue, R.; Hajji, N.; Lam, E.W.-F. ATM and p53 Regulate FOXM1 Expression via E2F in Breast Cancer Epirubicin Treatment and Resistance. Mol. Cancer Ther. 2011, 10, 1046–1058. [Google Scholar] [CrossRef]
- de Olano, N.; Koo, C.-Y.; Monteiro, L.J.; Pinto, P.H.; Gomes, A.R.; Aligue, R.; Lam, E.W.-F. The p38 MAPK–MK2 Axis Regulates E2F1 and FOXM1 Expression after Epirubicin Treatment. Mol. Cancer Res. 2012, 10, 1189–1202. [Google Scholar] [CrossRef]
- Bollu, L.R.; Shepherd, J.; Zhao, D.; Ma, Y.; Tahaney, W.; Speers, C.; Mazumdar, A.; Mills, G.B.; Brown, P.H. Mutant P53 induces MELK expression by release of wild-type P53-dependent suppression of FOXM1. NPJ Breast Cancer 2020, 6, 1–12. [Google Scholar] [CrossRef]
- Molinuevo, R.; Freije, A.; De Pedro, I.; Stoll, S.W.; Elder, J.T.; Gandarillas, A. FOXM1 allows human keratinocytes to bypass the oncogene-induced differentiation checkpoint in response to gain of MYC or loss of p53. Oncogene 2016, 36, 956–965. [Google Scholar] [CrossRef]
- McGovern, U.B.; Francis, R.E.; Peck, B.; Guest, S.K.; Wang, J.; Myatt, S.S.; Krol, J.; Kwok, J.M.-M.; Polychronis, A.; Coombes, R.C.; et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol. Cancer Ther. 2009, 8, 582–591. [Google Scholar] [CrossRef]
- Delpuech, O.; Griffiths, B.; East, P.; Essafi, A.; Lam, E.W.-F.; Burgering, B.; Downward, J.; Schulze, A. Induction of Mxi1-SRα by FOXO3a Contributes to Repression of Myc-Dependent Gene Expression. Mol. Cell. Biol. 2007, 27, 4917–4930. [Google Scholar] [CrossRef]
- Lam, E.W.-F.; Brosens, J.; Gomes, A.R.; Koo, C.-Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef]
- Zou, Y.; Tsai, W.-B.; Cheng, C.-J.; Hsu, C.; Chung, Y.M.; Li, P.-C.; Lin, S.-H.; Hu, M.C.-T. Forkhead box transcription factor FOXO3a suppresses estrogen-dependent breast cancer cell proliferation and tumorigenesis. Breast Cancer Res. 2008, 10, R21. [Google Scholar] [CrossRef]
- Karadedou, C.T.; Gomes, A.R.; Chen, J.; Petkovic, M.; Ho, K.-K.; Zwolinska, A.K.; Feltes, A.; Wong, S.Y.; Chan, K.Y.K.; Cheung, Y.-N.; et al. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene 2011, 31, 1845–1858. [Google Scholar] [CrossRef]
- Levanon, K.; Sapoznik, S.; Bahar-Shany, K.; Brand, H.; Shapira-Frommer, R.; Korach, J.; Hirsch, M.S.; Roh, M.H.; Miron, A.; Liu, J.F.; et al. FOXO3a loss is a frequent early event in high-grade pelvic serous carcinogenesis. Oncogene 2014, 33, 4424–4432. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; E Greenberg, M. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-Type Ca2+ Channel Inhibition Sensitizes Ovarian Cancer to Carboplatin. Mol. Cancer Ther. 2016, 15, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cai, Q.; Xu, Y. FOXM1 is a downstream target of LPA and YAP oncogenic signaling pathways in high grade serous ovarian cancer. Oncotarget 2015, 6, 27688–27699. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Park, J.-H.; Miyamoto, T.; Takamatsu, N.; Kato, T.; Iwasa, A.; Okabe, S.; Imai, Y.; Fujiwara, K.; Nakamura, Y.; et al. T-LAK Cell-Originated Protein Kinase (TOPK) as a Prognostic Factor and a Potential Therapeutic Target in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 6110–6117. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.K.; Mucaj, V.; Biju, K.M.; Nakazawa, M.S.; Gohil, M.; Cash, T.P.; Yoon, S.S.; Skuli, N.; Park, K.M.; Gerecht, S.; et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E3402–E3411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, P.; Jing, Y.; Yan, Y.; Cai, M.-C.; Zhang, M.; Zhang, S.; Peng, H.; Ji, Z.-L.; Di, W.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating FoxM1. Theranostics 2016, 6, 219–230. [Google Scholar] [CrossRef]
- Momeny, M.; Eyvani, H.; Barghi, F.; Ghaffari, S.H.; Javadikooshesh, S.; Jamadi, R.H.; Esmaeili, F.; Alishahi, Z.; Zaghal, A.; Bashash, D.; et al. Inhibition of bromodomain and extraterminal domain reduces growth and invasive characteristics of chemoresistant ovarian carcinoma cells. Anti-Cancer Drugs 2018, 29, 1011–1020. [Google Scholar] [CrossRef]
- Andrikopoulou, A.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.-A.; Zagouri, F. Clinical perspectives of BET inhibition in ovarian cancer. Cell. Oncol. 2021, 44, 237–249. [Google Scholar] [CrossRef]
- Llauradó, M.; Majem, B.; Castellví, J.; Cabrera, S.; Gil-Moreno, A.; Reventós, J.; Ruiz, A. Analysis of Gene Expression Regulated by the ETV5 Transcription Factor in OV90 Ovarian Cancer Cells Identifies FOXM1 Overexpression in Ovarian Cancer. Mol. Cancer Res. 2012, 10, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.-T.; Wong, S.-T.; Neill, G.W.; Ghali, L.R.; Philpott, M.P.; Quinn, A.G. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002, 62, 4773–4780. [Google Scholar] [PubMed]
- Douard, R.; Moutereau, S.; Pernet, P.; Chimingqi, M.; Allory, Y.; Manivet, P.; Conti, M.; Vaubourdolle, M.; Cugnenc, P.-H.; Loric, S. Sonic Hedgehog–dependent proliferation in a series of patients with colorectal cancer. Surgery 2006, 139, 665–670. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, J.; Ferrandon, S.; Glowacki, K.J.; Houghton, J.A. Targeting GLI by GANT61 involves mechanisms dependent on inhibition of both transcription and DNA licensing. Oncotarget 2016, 7, 80190–80207. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Huang, D.; Lu, N.; Chen, D.; Zhang, M.; Yan, Y.; Dengliang, H.; Lu, Q.; Lu, H.; Luo, S. Aberrantly activated Gli2-KIF20A axis is crucial for growth of hepatocellular carcinoma and predicts poor prognosis. Oncotarget 2016, 7, 26206–26219. [Google Scholar] [CrossRef]
- Blanco-Bose, W.E.; Murphy, M.J.; Ehninger, A.; Offner, S.; Dubey, C.; Huang, W.; Moore, D.D.; Trumpp, A. C-Myc and its target FoxM1 are critical downstream effectors of constitutive androstane receptor (CAR) mediated direct liver hyperplasia. Hepatology 2008, 48, 1302–1311. [Google Scholar] [CrossRef]
- Huynh, K.M.; Soh, J.-W.; Dash, R.; Sarkar, D.; Fisher, P.B.; Kang, D. FOXM1 expression mediates growth suppression during terminal differentiation of HO-1 human metastatic melanoma cells. J. Cell. Physiol. 2011, 226, 194–204. [Google Scholar] [CrossRef]
- Pan, H.; Zhu, Y.; Wei, W.; Shao, S.; Rui, X. Transcription factor FoxM1 is the downstream target of c-Myc and contributes to the development of prostate cancer. World J. Surg. Oncol. 2018, 16, 59. [Google Scholar] [CrossRef]
- Mencalha, A.L.; Binato, R.; Ferreira, G.M.; Du-Rocher, B.; Abdelhay, E. Forkhead Box M1 (FoxM1) Gene Is a New STAT3 Transcriptional Factor Target and Is Essential for Proliferation, Survival and DNA Repair of K562 Cell Line. PLoS ONE 2012, 7, e48160. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Luo, Y.; Gu, X.; Zhan, W.; Wang, X. Twist1 Promotes Gastric Cancer Cell Proliferation through Up-Regulation of FoxM1. PLoS ONE 2013, 8, e77625. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Gartel, A.L. A novel mode of FoxM1 regulation: Positive auto-regulatory loop. Cell Cycle 2009, 8, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-H.; Black, M.; Ustiyan, V.; Le, T.; Fulford, L.; Sridharan, A.; Medvedovic, M.; Kalinichenko, V.V.; Whitsett, J.A.; Kalin, T.V. SPDEF Inhibits Prostate Carcinogenesis by Disrupting a Positive Feedback Loop in Regulation of the Foxm1 Oncogene. PLoS Genet. 2014, 10, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Gong, A.; Jing, Z.; Aldape, K.D.; Kang, S.-H.; Sawaya, R.; Huang, S. Forkhead Box M1 Is Regulated by Heat Shock Factor 1 and Promotes Glioma Cells Survival under Heat Shock Stress. J. Biol. Chem. 2013, 288, 1634–1642. [Google Scholar] [CrossRef]
- Zhao, E.; Ding, J.; Xia, Y.; Liu, M.; Ye, B.; Choi, J.-H.; Yan, C.; Dong, Z.; Huang, S.; Zha, Y.; et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep. 2016, 14, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Carr, J.R.; Wang, Z.; Nogueira, V.; Hay, N.; Tyner, A.L.; Lau, L.F.; Costa, R.H.; Raychaudhuri, P. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009, 28, 2908–2918. [Google Scholar] [CrossRef]
- Chen, P.-M.; Wu, T.-C.; Shieh, S.-H.; Wu, Y.-H.; Li, M.-C.; Sheu, G.-T.; Cheng, Y.-W.; Chen, C.-Y.; Lee, H. MnSOD Promotes Tumor Invasion via Upregulation of FoxM1–MMP2 Axis and Related with Poor Survival and Relapse in Lung Adenocarcinomas. Mol. Cancer Res. 2012, 11, 261–271. [Google Scholar] [CrossRef]
- Xia, L.; Mo, P.; Huang, W.; Zhang, L.; Wang, Y.; Zhu, H.; Tian, D.; Liu, J.; Chen, Z.; Zhang, Y.; et al. The TNF-α/ROS/HIF-1-induced Upregulation of FoxMI Expression Promotes HCC Proliferation and Resistance to Apoptosis. Carcinogenesis 2012, 33, 2250–2259. [Google Scholar] [CrossRef]
- Xia, L.-M.; Huang, W.-J.; Wang, B.; Liu, M.; Zhang, Q.; Yan, W.; Zhu, Q.; Luo, M.; Zhou, Z.-Z.; Tian, D.-A. Transcriptional up-regulation of FoxM1 in response to hypoxia is mediated by HIF-1. J. Cell. Biochem. 2009, 106, 247–256. [Google Scholar] [CrossRef]
- Lu, J.; Wang, Z.; Cao, J.; Chen, Y.; Dong, Y. A novel and compact review on the role of oxidative stress in female reproduction. Reprod. Biol. Endocrinol. 2018, 16, 80. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Razavi, Z.S.; Tajiknia, V.; Majidi, S.; Ghandali, M.; Mirzaei, H.R.; Rahimian, N.; Hamblin, M.R.; Mirzaei, H. Gynecologic cancers and non-coding RNAs: Epigenetic regulators with emerging roles. Crit. Rev. Oncol. 2021, 157, 103192. [Google Scholar] [CrossRef]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, J.; He, Y.; Wang, Y. Hsa_circ_0061140 Knockdown Reverses FOXM1-Mediated Cell Growth and Metastasis in Ovarian Cancer through miR-370 Sponge Activity. Mol. Ther. Nucleic Acids 2018, 13, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.; Hou, M.; Yuan, J.; Yang, L.; Zeng, X.; Xi, M.; Chen, J. LncRNA PVT1 epigenetically stabilizes and post-transcriptionally regulates FOXM1 by acting as a microRNA sponge and thus promotes malignant behaviors of ovarian cancer cells. Am. J. Transl. Res. 2020, 12, 2860–2874. [Google Scholar]
- Duan, N.; Hu, X.; Yang, X.; Cheng, H.; Zhang, W. MicroRNA-370 directly targets FOXM1 to inhibit cell growth and metastasis in osteosarcoma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 10250–10260. [Google Scholar]
- Feng, Y.; Wang, L.; Zeng, J.; Shen, L.; Liang, X.; Yu, H.; Liu, S.; Liu, Z.; Sun, Y.; Li, W.; et al. FoxM1 is Overexpressed in Helicobacter pylori–Induced Gastric Carcinogenesis and Is Negatively Regulated by miR-370. Mol. Cancer Res. 2013, 11, 834–844. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, J.; Zhou, M.; Li, B.; Zhang, Y.; Huang, T.; Wang, L.; Jia, J.; Chen, C. The tumor suppressive role of miRNA-370 by targeting FoxM1 in acute myeloid leukemia. Mol. Cancer 2012, 11, 56. [Google Scholar] [CrossRef]
- Xu, M.-d.; Wang, Y.; Weng, W.; Wei, P.; Qi, P.; Zhang, Q.; Tan, C.; Ni, S.-j.; Dong, L.; Yang, Y.; et al. A Positive Feedback Loop of lncRNA-PVT1 and FOXM1 Facilitates Gastric Cancer Growth and Invasion. Clin. Cancer Res. 2017, 23, 2071–2080. [Google Scholar] [CrossRef]
- Saleembhasha, A.; Mishra, S. Long non-coding RNAs as pan-cancer master gene regulators of associated protein-coding genes: A systems biology approach. PeerJ 2019, 7, e6388. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Zheng, Q.; Bao, C.; He, J.; Chen, B.; Lyu, D.; Zheng, B.; Xu, Y.; Long, Z.; et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2017, 388, 208–219. [Google Scholar] [CrossRef]
- Li, M.; Chi, C.; Zhou, L.; Chen, Y.; Tang, X. Circular PVT1 regulates cell proliferation and invasion via miR-149-5p/FOXM1 axis in ovarian cancer. J. Cancer 2021, 12, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Zhao, W.; Xiong, J.; Cao, R. miR-149 Inhibits Non-Small-Cell Lung Cancer Cells EMT by Targeting FOXM1. Biochem. Res. Int. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wang, G.-H.; Bian, Z.-L.; Li, X.-W.; Zhu, B.-Y.; Jin, C.-J.; Ju, S.-Q. Long non-coding RNA CCAL/miR-149/FOXM1 axis promotes metastasis in gastric cancer. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Luo, J.; Fu, Z.; Ying, J.; Yu, Y.; Yu, W. miR-134 inhibits epithelial to mesenchymal transition by targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett. 2012, 586, 3761–3765. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Z.-Y.; Pan, D.-D. Penicillin-binding protein 1A mutation-positive Helicobacter pylori promotes epithelial-mesenchymal transition in gastric cancer via the suppression of microRNA. Int. J. Onco. L. 2019, 54, 916–928. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Li, J.; Sang, X. Diallyl disulfide suppresses FOXM1-mediated proliferation and invasion in osteosarcoma by upregulating miR. J. Cell. Biochem. 2019, 120, 7286–7296. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Cao, F.; Han, B.; Xu, L. MiRNA-134 suppresses esophageal squamous cell carcinoma progression by targeting FOXM1. Int. J. Clin. Exp. Pathol. 2019, 12, 2130–2138. [Google Scholar]
- Wei, Y.; Wang, Z.; Zong, Y.; Deng, D.; Chen, P.; Lu, J. LncRNA MFI2-AS1 promotes HCC progression and metastasis by acting as a competing endogenous RNA of miR-134 to upregulate FOXM1 expression. Biomed. Pharmacother. 2020, 125, 109890. [Google Scholar] [CrossRef]
- He, S.; Liao, B.; Deng, Y.; Su, C.; Tuo, J.; Liu, J.; Yao, S.; Xu, L. MiR-216b inhibits cell proliferation by targeting FOXM1 in cervical cancer cells and is associated with better prognosis. BMC Cancer 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wang, X.; Tu, C.; Wang, S.; Qu, J.; Xiao, S. microRNA-216b inhibits cell proliferation and migration in human melanoma by targeting FOXM1 in vitro and in vivo. Cell Biol. Int. 2017, 41, 1272–1282. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, G.; Zhang, Y.; Huo, H.; Zhao, Y. miR-216b inhibits glioma cell migration and invasion through suppression of FoxM1. Oncol. Rep. 2017, 38, 1751–1759. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Du, X.; Yao, Y.; Wang, L.; Jia, Y. MiR-216b suppresses cell proliferation, migration, invasion, and epithelial–mesenchymal transition by regulating FOXM1 expression in human non-small cell lung cancer. OncoTargets Ther. 2019, 12, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Sener, E.F.; Taheri, S.; Nalbantoglu, U.; Kokcu, N.D.; Tahtasakal, R.; Cınar, V.; Guler, A.; Ozkul, Y.; Dönmez-Altuntas, H.; et al. MicroRNA profiling identifies Forkhead box transcription factor M1 (FOXM1) regulated miR-186 and miR-200b alterations in triple negative breast cancer. Cell. Signal. 2021, 83, 109979. [Google Scholar] [CrossRef]
- Chan, D.W.; Hui, W.W.Y.; Cai, P.C.H.; Liu, M.X.; Yung, M.M.H.; Mak, C.S.L.; Leung, T.H.Y.; Chan, K.K.L.; Ngan, H.Y.S. Targeting GRB7/ERK/FOXM1 Signaling Pathway Impairs Aggressiveness of Ovarian Cancer Cells. PLoS ONE 2012, 7, e52578. [Google Scholar] [CrossRef]
- Lok, G.T.M.; Chan, D.W.; Liu, V.W.S.; Hui, W.W.Y.; Leung, T.H.Y.; Yao, K.M.; Ngan, H.Y.S. Aberrant Activation of ERK/FOXM1 Signaling Cascade Triggers the Cell Migration/Invasion in Ovarian Cancer Cells. PLoS ONE 2011, 6, e23790. [Google Scholar] [CrossRef]
- Zhang, Y.; Goodfellow, R.; Li, Y.; Yang, S.; Winters, C.J.; Thiel, K.W.; Leslie, K.K.; Yang, B. NEDD4 ubiquitin ligase is a putative oncogene in endometrial cancer that activates IGF-1R/PI3K/Akt signaling. Gynecol. Oncol. 2015, 139, 127–133. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, S.; Li, J.; Xu, X.; Weng, Y.; Zheng, M.; Ouyang, L.; Li, F. CXCL12-induced upregulation of FOXM1 expression promotes human glioblastoma cell invasion. Biochem. Biophys. Res. Commun. 2014, 447, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Cui, W.; Jiang, X.; Zhang, Z.; Gnosa, S.; Ali, Z.; Jensen, L.; Jönsson, J.-I.; Blockhuys, S.; Lam, E.W.-F.; et al. The Critical Role of Dysregulated RhoB Signaling Pathway in Radioresistance of Colorectal Cancer. Int. J. Radiat. Oncol. 2019, 104, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sun, Y.; Ji, P.; Li, X.; Cogdell, D.; Yang, D.; Parker, K.B.C.; Shmulevich, I.; Chen, K.; Sood, A.K.; et al. MiR-506 suppresses proliferation and induces senescence by directly targeting the CDK4/6–FOXM1 axis in ovarian cancer. J. Pathol. 2014, 233, 308–318. [Google Scholar] [CrossRef]
- Weichert, W.; Denkert, C.; Schmidt, M.; Gekeler, V.; Wolf, G.; Köbel, M.; Dietel, M.; Hauptmann, S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br. J. Cancer 2004, 90, 815–821. [Google Scholar] [CrossRef]
- Zhang, R.; Shi, H.; Ren, F.; Liu, H.; Zhang, M.; Deng, Y.; Li, X. Misregulation of polo-like protein kinase 1, P53 and P21WAF1 in epithelial ovarian cancer suggests poor prognosis. Oncol. Rep. 2015, 33, 1235–1242. [Google Scholar] [CrossRef]
- Saldana, M.; Vandervorst, K.; Berg, A.L.; Lee, H.; Carraway, K.L. Otubain 1: A non-canonical deubiquitinase with an emerging role in cancer. Endocr. Relat. Cancer 2019, 26, R1–R14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, X.; Xu, M.; Weng, W.; Zhang, Q.; Yang, Y.; Wei, P.; Du, X. OTUB1-catalyzed deubiquitination of FOXM1 facilitates tumor progression and predicts a poor prognosis in ovarian cancer. Oncotarget 2016, 7, 36681–36697. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Arceci, A.; Bonacci, T.; Wang, X.; Stewart, K.; Damrauer, J.S.; Hoadley, K.A.; Emanuele, M.J. FOXM1 Deubiquitination by USP21 Regulates Cell Cycle Progression and Paclitaxel Sensitivity in Basal-like Breast Cancer. Cell Rep. 2019, 26, 3076–3086.e6. [Google Scholar] [CrossRef]
- Raychaudhuri, P.; Park, H.J. FoxM1: A Master Regulator of Tumor Metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef]
- Aytes, A.; Mitrofanova, A.; Lefebvre, C.; Alvarez, M.J.; Castillo-Martin, M.; Zheng, T.; Eastham, J.A.; Gopalan, A.; Pienta, K.J.; Shen, M.M.; et al. Cross-Species Regulatory Network Analysis Identifies a Synergistic Interaction between FOXM1 and CENPF that Drives Prostate Cancer Malignancy. Cancer Cell 2014, 25, 638–651. [Google Scholar] [CrossRef]
- Halasi, M.; Gartel, A.L. FOX(M1) news–it is cancer. Mol. Cancer Ther. 2013, 12, 245–254. [Google Scholar] [CrossRef]
- Fernández, M.A.; Medema, R.H. Novel functions of FoxM1: From molecular mechanisms to cancer therapy. Front. Oncol. 2013, 3, 30. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Tassi, R.A.; Todeschini, P.; Siegel, E.R.; Calza, S.; Cappella, P.E.; Ardighieri, L.; Cadei, M.; Bugatti, M.; Romani, C.; Bandiera, E.; et al. FOXM1 expression is significantly associated with chemotherapy resistance and adverse prognosis in non-serous epithelial ovarian cancer patients. J. Exp. Clin. Cancer Res. 2017, 36, 1–18. [Google Scholar] [CrossRef]
- Li, Y.; Guo, H.; Wang, Z.; Bu, H.; Wang, S.; Wang, H.; Fang, H.; Liu, Z.; Kong, B. Cyclin F and KIF20A, FOXM1 target genes, increase proliferation and invasion of ovarian cancer cells. Exp. Cell Res. 2020, 395, 112212. [Google Scholar] [CrossRef]
- Chan, D.W.; Hui, W.W.Y.; Wang, J.J.; Yung, M.M.H.; Hui, L.M.N.; Qin, Y.; Liang, R.R.; Leung, T.H.Y.; Xu, D.; Chan, K.K.L.; et al. DLX1 acts as a crucial target of FOXM1 to promote ovarian cancer aggressiveness by enhancing TGF-β/SMAD4 signaling. Oncogene 2017, 36, 1404–1416. [Google Scholar] [CrossRef]
- Jin, C.; Liu, Z.; Li, Y.; Bu, H.; Wang, Y.; Xu, Y.; Qiu, C.; Yan, S.; Yuan, C.; Li, R.; et al. PCNA-associated factor P15PAF, targeted by FOXM1, predicts poor prognosis in high-grade serous ovarian cancer patients. Int. J. Cancer 2018, 143, 2973–2984. [Google Scholar] [CrossRef]
- Zhang, Z.; Tu, K.; Liu, F.; Liang, M.; Yu, K.; Wang, Y.; Luo, Y.; Yang, B.; Qin, Y.; He, D.; et al. FoxM1 promotes the migration of ovarian cancer cell through KRT5 and KRT7. Gene 2020, 757, 144947. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.Y.; Janzen, D.M.; Schafenacker, A.M.; Velasco, V.S.; Shung, M.S.; Cheng, D.; Huang, J.; Witte, O.N.; Memarzadeh, S. Stem-Like Epithelial Cells Are Concentrated in the Distal End of the Fallopian Tube: A Site for Injury and Serous Cancer Initiation. Stem Cells 2012, 30, 2487–2497. [Google Scholar] [CrossRef]
- An, Q.; Liu, T.; Wang, M.Y.; Yang, Y.J.; Zhang, Z.D.; Liu, Z.J.; Yang, B. KRT7 promotes epithelial-mesenchymal transition in ovarian cancer via the TGF-β/Smad2/3 signaling pathway. Oncol. Rep. 2021, 45, 481–492. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Divya, S.P.; Parvathareddy, S.K.; Alhoshani, N.M.; Al-Badawi, I.A.; Tulbah, A.; Al-Dayel, F.; Siraj, A.K.; Al-Kuraya, K.S. FoxM1 and β-catenin predicts aggressiveness in Middle Eastern ovarian cancer and their co-targeting impairs the growth of ovarian cancer cells. Oncotarget 2017, 9, 3590–3604. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.L.; Hough, R.; Bernaudo, S.; Peng, C. Wnt/β-catenin signalling in ovarian cancer: Insights into its hyperactivation and function in tumorigenesis. J. Ovarian Res. 2019, 12, 1–17. [Google Scholar] [CrossRef]
- Parashar, D.; Nair, B.; Geethadevi, A.; George, J.; Nair, A.; Tsaih, S.-W.; Kadamberi, I.P.; Nair, G.K.G.; Lu, Y.; Ramchandran, R.; et al. Peritoneal Spread of Ovarian Cancer Harbors Therapeutic Vulnerabilities Regulated by FOXM1 and EGFR/ERBB2 Signaling. Cancer Res. 2020, 80, 5554–5568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, Y.; Wang, Y.; Yin, X.; He, Y.; Chen, L.; Wang, W.; Liu, T.; Di, W. FOXM1 Modulates Cisplatin Sensitivity by Regulating EXO1 in Ovarian Cancer. PLoS ONE 2014, 9, e96989. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Madden, J.; Neums, L.; Moulder, R.K.; Forrest, M.L.; Chien, J. Olaparib-induced Adaptive Response Is Disrupted by FOXM1 Targeting that Enhances Sensitivity to PARP Inhibition. Mol. Cancer Res. 2018, 16, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Brückner, L.; Reinshagen, A.; Hoang, N.; Höhn, A.; Lordick, F.; Bechmann, I.; Aktas, B.; Nel, I.; Kallendrusch, S. FOXM1 Inhibition in Ovarian Cancer Tissue Cultures Affects Individual Treatment Susceptibility Ex Vivo. Cancers 2021, 13, 956. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.; Leung, M.; Qin, J.; Qin, Y.; Wang, J.; Lee, Y.; Yao, K.-M. The Forkhead box transcription factor FOXM1 is required for the maintenance of cell proliferation and protection against oxidative stress in human embryonic stem cells. Stem Cell Res. 2016, 16, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Ning, G.; Howitt, B.E.; Mehra, K.; Wu, L.; Wang, X.; Hong, Y.; Kern, F.; Wei, T.S.; Zhang, T.; et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J. Pathol. 2016, 238, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Wen, N.; Wang, Y.; Wen, L.; Zhao, S.-H.; Ai, Z.-H.; Wang, Y.; Wu, B.; Lu, H.-X.; Yang, H.; Liu, W.-C.; et al. Overexpression of FOXM1 predicts poor prognosis and promotes cancer cell proliferation, migration and invasion in epithelial ovarian cancer. J. Transl. Med. 2014, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Meng, H.; Li, S.; Shen, Y.; Wang, H.; Shan, W.; Qiu, J.; Zhang, J.; Cheng, W. Identification of Potential Biomarkers in Association with Progression and Prognosis in Epithelial Ovarian Cancer by Integrated Bioinformatics Analysis. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Zhao, F.; Siu, M.K.Y.; Jiang, L.; Tam, K.F.; Ngan, H.Y.S.; Le, X.F.; Wong, O.G.W.; Wong, E.S.Y.; Gomes, A.R.; Bella, L.; et al. Overexpression of Forkhead Box Protein M1 (FOXM1) in Ovarian Cancer Correlates with Poor Patient Survival and Contributes to Paclitaxel Resistance. PLoS ONE 2014, 9, e113478. [Google Scholar] [CrossRef]
- Chiu, W.-T.; Huang, Y.-F.; Tsai, H.-Y.; Chen, C.-C.; Chang, C.-H.; Huang, S.-C.; Hsu, K.-F.; Chou, C.-Y. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 2014, 6, 2349–2365. [Google Scholar] [CrossRef]
- Mekhdjian, A.H.; Kai, F.; Rubashkin, M.G.; Prahl, L.; Przybyla, L.M.; McGregor, A.L.; Bell, E.S.; Barnes, J.M.; Dufort, C.C.; Ou, G.; et al. Integrin-mediated traction force enhances paxillin molecular associations and adhesion dynamics that increase the invasiveness of tumor cells into a three-dimensional extracellular matrix. Mol. Biol. Cell 2017, 28, 1467–1488. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.-W.; Lee, S.-S.; Chang, C.-Y.; Lang, Y.-D.; Jou, Y.-S. A New Switch for TGFβ in Cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.-T.; Lee, H.-T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 Promotes β-Catenin Nuclear Localization and Controls Wnt Target-Gene Expression and Glioma Tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef]
- Wierstra, I. The transcription factor FOXM1c binds to and transactivates the promoter of the tumor suppressor gene E-cadherin. Cell Cycle 2011, 10, 760–766. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z. E-cadherin’s dark side: Possible role in tumor progression. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2012, 1826, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Roque, R.J.; Sousa, F.C.; Figueiredo-Dias, M. Epithelial-mesenchymal interconversions in ovarian cancer: The levels and functions of E-cadherin in intraabdominal dissemination. Oncol. Rev. 2020, 14, 475. [Google Scholar] [CrossRef] [PubMed]
- Trillsch, F.; Kuerti, S.; Eulenburg, C.; Burandt, E.; Woelber, L.; Prieske, K.; Eylmann, K.; Oliveira-Ferrer, L.; Milde-Langosch, K.; Mahner, S. E-Cadherin fragments as potential mediators for peritoneal metastasis in advanced epithelial ovarian cancer. Br. J. Cancer 2016, 114, 213–220. [Google Scholar] [CrossRef]
- Wells, A.; Yates, C.; Shepard, C.R. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 2008, 25, 621–628. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef]
- Balli, D.; Ren, X.; Chou, F.-S.; Cross, E.; Zhang, Y.; Kalinichenko, V.V.; Kalin, T.V. Foxm1 transcription factor is required for macrophage migration during lung inflammation and tumor formation. Oncogene 2011, 31, 3875–3888. [Google Scholar] [CrossRef]
- Galbo, P.M.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Wang, L.; Yao, B.; Chen, T.; Li, Q.; Liu, Z.; Liu, R.; Niu, Y.; Song, T.; et al. Resolvin D1 prevents epithelial-mesenchymal transition and reduces the stemness features of hepatocellular carcinoma by inhibiting paracrine of cancer-associated fibroblast-derived COMP. J. Exp. Clin. Cancer Res. 2019, 38, 1–17. [Google Scholar] [CrossRef]
- Maniati, E.; Berlato, C.; Gopinathan, G.; Heath, O.; Kotantaki, P.; Lakhani, A.; McDermott, J.; Pegrum, C.; Delaine-Smith, R.M.; Pearce, O.M.; et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020, 30, 525–540.e7. [Google Scholar] [CrossRef]
- Iyer, S.; Zhang, S.; Yucel, S.; Horn, H.; Smith, S.G.; Reinhardt, F.; Hoefsmit, E.; Assatova, B.; Casado, J.; Meinsohn, M.-C.; et al. Genetically Defined Syngeneic Mouse Models of Ovarian Cancer as Tools for the Discovery of Combination Immunotherapy. Cancer Discov. 2021, 11, 384–407. [Google Scholar] [CrossRef]
- Zona, S.; Bella, L.; Burton, M.J.; de Moraes, G.N.; Lam, E.W.-F. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Barger, C.J.; Chee, L.; Albahrani, M.; Munoz-Trujillo, C.; Boghean, L.; Branick, C.; Odunsi, K.; Drapkin, R.; Zou, L.; Karpf, A.R. Author response: Co-regulation and function of FOXM1/RHNO1 bidirectional genes in cancer. eLife 2021, 10, e55070. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xia, L.; Guo, Q.; Zhu, J.; Deng, Y.; Wu, X. Identification of Chemoresistance-Associated Key Genes and Pathways in High-Grade Serous Ovarian Cancer by Bioinformatics Analyses. Cancer Manag. Res. 2020, 12, 5213–5223. [Google Scholar] [CrossRef]
- Gartel, A.L. FoxM1 inhibitors as potential anticancer drugs. Expert Opin. Ther. Targets 2008, 12, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.M.-M.; Myatt, S.S.; Marson, C.M.; Coombes, R.C.; Constantinidou, D.; Lam, E.W.-F. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 2008, 7, 2022–2032. [Google Scholar] [CrossRef]
- Westhoff, G.L.; Chen, Y.; Teng, N.N. Targeting Foxm1 Improves Cytotoxicity of Paclitaxel and Cisplatinum in Platinum-Resistant Ovarian Cancer. Int. J. Gynecol. Cancer 2017, 27, 887–894. [Google Scholar] [CrossRef]
- Bhat, U.G.; Halasi, M.; Gartel, A.L. FoxM1 Is a General Target for Proteasome Inhibitors. PLoS ONE 2009, 4, e6593. [Google Scholar] [CrossRef] [PubMed]
- Hardy, L.R.; Pergande, M.; Esparza, K.; Heath, K.N.; Önyüksel, H.; Cologna, S.M.; Burdette, J.E. Proteomic analysis reveals a role for PAX8 in peritoneal colonization of high grade serous ovarian cancer that can be targeted with micelle encapsulated thiostrepton. Oncogene 2019, 38, 6003–6016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ging, N.C.; Komoda, T.; Hanada, T.; Suzuki, T.; Watanabe, K. Antibiotic susceptibility of mammalian mitochondrial translation. FEBS Lett. 2005, 579, 6423–6427. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.R.; Park, H.J.; Wang, Z.; Kiefer, M.M.; Raychaudhuri, P. FoxM1 Mediates Resistance to Herceptin and Paclitaxel. Cancer Res. 2010, 70, 5054–5063. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Wang, C.; Wang, J.; Wang, Z.; Huang, D.; Yan, M.; Kamran, M.; Liu, Q.; Xu, B. Aurora kinase A stabilizes FOXM1 to enhance paclitaxel resistance in triple-negative breast cancer. J. Cell. Mol. Med. 2019, 23, 6442–6453. [Google Scholar] [CrossRef]
- Okada, K.; Fujiwara, Y.; Takahashi, T.; Nakamura, Y.; Takiguchi, S.; Nakajima, K.; Miyata, H.; Yamasaki, M.; Kurokawa, Y.; Mori, M.; et al. Overexpression of Forkhead Box M1 Transcription Factor (FOXM1) is a Potential Prognostic Marker and Enhances Chemoresistance for Docetaxel in Gastric Cancer. Ann. Surg. Oncol. 2013, 20, 1035–1043. [Google Scholar] [CrossRef]
- Li, X.; Qiu, W.; Liu, B.; Yao, R.; Liu, S.; Yao, Y.; Liang, J. Forkhead box transcription factor 1 expression in gastric cancer: FOXM1 is a poor prognostic factor and mediates resistance to docetaxel. J. Transl. Med. 2013, 11, 204. [Google Scholar] [CrossRef]
- Li, X.; Yao, R.; Yue, L.; Qiu, W.; Qi, W.; Liu, S.; Yao, Y.; Liang, J. FOXM 1 mediates resistance to docetaxel in gastric cancer via up-regulating Stathmin. J. Cell. Mol. Med. 2014, 18, 811–823. [Google Scholar] [CrossRef]
- Huang, X.; Qin, J.; Lu, S. Up-regulation of miR-877 induced by paclitaxel inhibits hepatocellular carcinoma cell proliferation though targeting FOXM1. Int. J. Clin. Exp. Pathol. 2015, 8, 1515–1524. [Google Scholar]
- Yuan, B.; Liu, Y.; Yu, X.; Yin, L.; Peng, Y.; Gao, Y.; Zhu, Q.; Cao, T.; Yang, Y.; Fan, X.; et al. FOXM1 contributes to taxane resistance by regulating UHRF1-controlled cancer cell stemness. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Hou, Y.; Zhu, Q.; Li, Z.; Peng, Y.; Yu, X.; Yuan, B.; Liu, Y.; Liu, Y.; Yin, L.; Peng, Y.; et al. The FOXM1–ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017, 8, e2659. [Google Scholar] [CrossRef]
- Ahmed, M.; Uddin, S.; Hussain, A.R.; Alyan, A.; Jehan, Z.; Al-Dayel, F.; Al-Nuaim, A.; Al-Sobhi, S.; Amin, T.; Bavi, P.; et al. FoxM1 and Its Association with Matrix Metalloproteinases (MMP) Signaling Pathway in Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E1–E13. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, P.; Chen, L.; Chen, H. Down-regulation of FoxM1 by thiostrepton or small interfering RNA inhibits proliferation, transformation ability and angiogenesis, and induces apoptosis of nasopharyngeal carcinoma cells. Int. J. Clin. Exp. Pathol. 2014, 7, 5450–5460. [Google Scholar]
- de Nestal, M.G.; Delbue, D.; Silva, K.L.; Robaina, M.C.; Khongkow, P.; Gomes, A.R.; Zona, S.; Crocamo, S.; Mencalha, A.L.; Magalhães, L.M.; et al. FOXM1 targets XIAP and Survivin to modulate breast cancer survival and chemoresistance. Cell. Signal. 2015, 27, 2496–2505. [Google Scholar] [CrossRef]
- Jiang, L.; Wu, X.; Wang, P.; Wen, T.; Yu, C.; Wei, L.; Chen, H. Targeting FoxM1 by thiostrepton inhibits growth and induces apoptosis of laryngeal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2014, 141, 971–981. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sabini, C.; Sorbi, F.; Cunnea, P.; Fotopoulou, C. Ovarian cancer stem cells: Ready for prime time? Arch. Gynecol. Obstet. 2020, 301, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Galván, S.; Carnero, A. Targeting Cancer Stem Cells to Overcome Therapy Resistance in Ovarian Cancer. Cells 2020, 9, 1402. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.-T. FOXM1 coming of age: Time for translation into clinical benefits? Front. Oncol. 2012, 2, 146. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tan, G.; Ding, M.; Dong, D.; Chen, T.; Meng, X.; Huang, X.; Tan, Y. Foxm1 transcription factor is required for maintenance of pluripotency of P19 embryonal carcinoma cells. Nucleic Acids Res. 2010, 38, 8027–8038. [Google Scholar] [CrossRef]
- Kelleher, F.C.; O’Sullivan, H. FOXM1 in sarcoma: Role in cell cycle, pluripotency genes and stem cell pathways. Oncotarget 2016, 7, 42792–42804. [Google Scholar] [CrossRef]
- Wang, Z.; Park, H.J.; Carr, J.R.; Chen, Y.-J.; Zheng, Y.; Li, J.; Tyner, A.L.; Costa, R.H.; Bagchi, S.; Raychaudhuri, P. FoxM1 in Tumorigenicity of the Neuroblastoma Cells and Renewal of the Neural Progenitors. Cancer Res. 2011, 71, 4292–4302. [Google Scholar] [CrossRef]
- Ning, Y.-X.; Li, Q.-X.; Ren, K.-Q.; Quan, M.-F.; Cao, J.-G. 7-difluoromethoxyl-5,4′-di-n-octyl genistein inhibits ovarian cancer stem cell characteristics through the downregulation of FOXM1. Oncol. Lett. 2014, 8, 295–300. [Google Scholar] [CrossRef]
- Young, M.-J.; Wu, Y.-H.; Chiu, W.-T.; Weng, T.-Y.; Huang, Y.-F.; Chou, C.-Y. All-trans retinoic acid downregulates ALDH1-mediated stemness and inhibits tumour formation in ovarian cancer cells. Carcinogenesis 2015, 36, 498–507. [Google Scholar] [CrossRef]
- Li, M.; Yang, J.; Zhou, W.; Ren, Y.; Wang, X.; Chen, H.; Zhang, J.; Chen, J.; Sun, Y.; Cui, L.; et al. Activation of an AKT/FOXM1/STMN1 pathway drives resistance to tyrosine kinase inhibitors in lung cancer. Br. J. Cancer 2017, 117, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Cao, X.; Yang, Y.; Song, Z.; Zhang, J.; Wang, Z. Upregulation of FoxM1 by MnSOD Overexpression Contributes to Cancer Stem-Like Cell Characteristics in the Lung Cancer H460 Cell Line. Technol. Cancer Res. Treat. 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Wen, M.; Xu, C.; Chen, A.; Qiu, Y.-B.; Cao, J.-G.; Zhang, J.-S.; Song, Z.-W. 8-bromo-7-methoxychrysin targets NF-κB and FoxM1 to inhibit lung cancer stem cells induced by pro-inflammatory factors. J. Cancer 2019, 10, 5244–5255. [Google Scholar] [CrossRef] [PubMed]
- Kopanja, D.; Pandey, A.; Kiefer, M.; Wang, Z.; Chandan, N.; Carr, J.R.; Franks, R.; Yu, D.-Y.; Guzman, G.; Maker, A.; et al. Essential roles of FoxM1 in Ras-induced liver cancer progression and in cancer cells with stem cell features. J. Hepatol. 2015, 63, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Liu, L.; Yuan, Q.; Li, X.; Cui, Y.; Ren, K.; Zou, C.; Chen, A.; Xu, C.; Qiu, Y.; et al. Isovitexin reduces carcinogenicity and stemness in hepatic carcinoma stem-like cells by modulating MnSOD and FoxM1. J. Exp. Clin. Cancer Res. 2019, 38, 1–18. [Google Scholar] [CrossRef]
- Nakano, I. Transcription factors as master regulator for cancer stemness: Remove milk from fox? Expert Rev. Anticancer. Ther. 2014, 14, 873–875. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, Y.; Kim, K.H.; Kim, D.G.; Cho, H.J.; Kim, Y.; Rheey, J.; Shin, K.; Seo, Y.J.; Choi, Y.-S.; Lee, J.-I.; et al. FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PLoS ONE 2015, 10, e0137703. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jin, D.; Guan, L.; Zhang, C.-C.; Wu, T.; Wang, Y.-J.; Gao, D. CEP55 promoted the migration, invasion and neuroshpere formation of the glioma cell line. UNeurosci. Lett. 2019, 705, 80–86. [Google Scholar] [CrossRef]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777. [Google Scholar] [CrossRef]
- Song, I.-S.; Jeong, Y.J.; Jeong, S.H.; Heo, H.J.; Kim, H.K.; Bae, K.B.; Park, Y.-H.; Kim, S.U.; Kim, J.-M.; Kim, N.; et al. FOXM1-Induced PRX3 Regulates Stemness and Survival of Colon Cancer Cells via Maintenance of Mitochondrial Function. Gastroenterology 2015, 149, 1006–1016.e9. [Google Scholar] [CrossRef]
- Song, I.-S.; Jeong, Y.J.; Seo, Y.J.; Byun, J.M.; Kim, Y.N.; Jeong, D.H.; Han, J.; Kim, K.T.; Jang, S.-W. Peroxiredoxin 3 maintains the survival of endometrial cancer stem cells by regulating oxidative stress. Oncotarget 2017, 8, 92788–92800. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, L.; Yu, Q.; Dong, D.; Tan, G.; Huang, X.; Tan, Y. The miR-134 attenuates the expression of transcription factor FOXM1 during pluripotent NT2/D1 embryonal carcinoma cell differentiation. Exp. Cell Res. 2015, 330, 442–450. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Buccitelli, C.; Salgueiro, L.; Rowald, K.; Sotillo, R.; Mardin, B.R.; Korbel, J.O. Pan-cancer analysis distinguishes transcriptional changes of aneuploidy from proliferation. Genome Res. 2017, 27, 501–511. [Google Scholar] [CrossRef]
- Zhang, W.; Klinkebiel, D.; Barger, C.J.; Pandey, S.; Guda, C.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability. Cancers 2020, 12, 764. [Google Scholar] [CrossRef]
- Karpf, A.R.; Matsui, S.-i. Genetic Disruption of Cytosine DNA Methyltransferase Enzymes Induces Chromosomal Instability in Human Cancer Cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal Instability and Tumors Promoted by DNA Hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Gemenetzidis, E.; Bose, A.; Riaz, A.M.; Chaplin, T.; Young, B.D.; Ali, M.; Sugden, D.; Thurlow, J.K.; Cheong, S.-C.; Teo, S.-H.; et al. FOXM1 Upregulation Is an Early Event in Human Squamous Cell Carcinoma and it Is Enhanced by Nicotine during Malignant Transformation. PLoS ONE 2009, 4, e4849. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.-T.; Gemenetzidis, E.; Patel, D.; Tariq, R.; Nadir, A.; Bahta, A.W.; Waseem, A.; Hutchison, I.L. FOXM1 Induces a Global Methylation Signature That Mimics the Cancer Epigenome in Head and Neck Squamous Cell Carcinoma. PLoS ONE 2012, 7, e34329. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.-T.; Gemenetzidis, E.; Chaplin, T.; Young, B.D.; Philpott, M.P. Upregulation of FOXM1 induces genomic instability in human epidermal keratinocytes. Mol. Cancer 2010, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Merchut-Maya, J.M.; Bartek, J.; Maya-Mendoza, A. Regulation of replication fork speed: Mechanisms and impact on genomic stability. DNA Repair 2019, 81, 102654. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, D.S.; Doetsch, P.W.; Werner, E. Replication stress and FOXM1 drive radiation induced genomic instability and cell transformation. PLoS ONE 2020, 15, e0235998. [Google Scholar] [CrossRef]
- Kyuno, T.; Kohno, T.; Konno, T.; Yamaguchi, H.; Kyuno, D.; Imamura, M.; Kimura, Y.; Kojima, T.; Takemasa, I. Glucose-Dependent FOXM1 Promotes Epithelial-to-Mesenchymal Transition Via Cellular Metabolism and Targeting Snail in Human Pancreatic Cancer. Pancreas 2020, 49, 273–280. [Google Scholar] [CrossRef]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 Promotes the Warburg Effect and Pancreatic Cancer Progression via Transactivation of LDHA Expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhou, F.; Li, N.; Li, Q.; Wang, L. FOXM1-LDHA signaling promoted gastric cancer glycolytic phenotype and progression. Int. J. Clin. Exp. Pathol. 2015, 8, 6756–6763. [Google Scholar] [PubMed]
- Shang, R.; Pu, M.; Li, Y.; Wang, D. FOXM1 regulates glycolysis in hepatocellular carcinoma by transactivating glucose transporter 1 expression. Oncol. Rep. 2017, 37, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Bala, B.R.K.; Katragunta, K.; Sarma, U.M.; Jain, N. Abundance of d-2-hydroxyglutarate in G2/M is determined by FOXM1 in mutant IDH1-expressing cells. FEBS Lett. 2019, 593, 2177–2193. [Google Scholar] [CrossRef]
- Zand, B.; Previs, R.A.; Zacharias, N.M.; Rupaimoole, R.; Mitamura, T.; Nagaraja, A.; Guindani, M.; Dalton, H.J.; Yang, L.; Baddour, J.; et al. Role of Increased n-acetylaspartate Levels in Cancer. J. Natl. Cancer Inst. 2016, 108, djv426. [Google Scholar] [CrossRef]
- Wang, Y.; Yun, Y.; Wu, B.; Wen, L.; Wen, M.; Yang, H.; Zhao, L.; Liu, W.; Huang, S.; Wen, N.; et al. FOXM1 promotes reprogramming of glucose metabolism in epithelial ovarian cancer cells via activation of GLUT1 and HK2 transcription. Oncotarget 2016, 7, 47985–47997. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Park, H.J.; Gusarova, G.; Wang, Z.; Carr, J.R.; Li, J.; Kim, K.-H.; Qiu, J.; Park, Y.-D.; Williamson, P.R.; Hay, N.; et al. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol. Med. 2011, 3, 21–34. [Google Scholar] [CrossRef]
- Chen, Y.; Ruben, E.A.; Rajadas, J.; Teng, N.N. In silico investigation of FOXM1 binding and novel inhibitors in epithelial ovarian cancer. Bioorganic Med. Chem. 2015, 23, 4576–4582. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.; Ngan, A.; Leung, M.-H.; Kwok, D.; Liu, V.; Chan, D.; Leung, W.Y.; Yao, K.-M. FOXM1b, which is present at elevated levels in cancer cells, has a greater transforming potential than FOXM1c. Front. Oncol. 2013, 3, 11. [Google Scholar] [PubMed]
- Dai, J.; Yang, L.; Wang, J.; Xiao, Y.; Ruan, Q. Prognostic Value of FOXM1 in Patients with Malignant Solid Tumor: A Meta-Analysis and System Review. Dis. Markers 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, D.; Yu, Q.; Li, L.; Wu, P. Prognostic value of FOXM1 in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 32298–32308. [Google Scholar] [CrossRef]
- Engqvist, H.; Parris, T.Z.; Kovács, A.; Rönnerman, E.W.; Sundfeldt, K.; Karlsson, P.; Helou, K. Validation of Novel Prognostic Biomarkers for Early-Stage Clear-Cell, Endometrioid and Mucinous Ovarian Carcinomas Using Immunohistochemistry. Front. Oncol. 2020, 10, 162. [Google Scholar] [CrossRef]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The costs of using unauthenticated, over-passaged cell lines: How much more data do we need? Biotechniques 2007, 43, 575–586. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Zakarya, R.; Howell, V.M.; Colvin, E.K. Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches. Int. J. Mol. Sci. 2020, 21, 4806. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of Bevacizumab in the Primary Treatment of Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef]
- Cheon, D.-J.; Tong, Y.; Sim, M.-S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A Collagen-Remodeling Gene Signature Regulated by TGF-β Signaling Is Associated with Metastasis and Poor Survival in Serous Ovarian Cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.-L.; Leung, C.S.; Wong, K.-K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β Modulates Ovarian Cancer Invasion by Upregulating CAF-Derived Versican in the Tumor Microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef]
- Turner, T.B.; Buchsbaum, D.J.; Straughn, J.M.; Randall, T.D.; Arend, R.C. Ovarian cancer and the immune system—The role of targeted therapies. Gynecol. Oncol. 2016, 142, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef]
- Odunsi, K.; Matsuzaki, J.; James, S.R.; Mhawech-Fauceglia, P.; Tsuji, T.; Miller, A.; Zhang, W.; Akers, S.N.; Griffiths, E.A.; Miliotto, A.; et al. Epigenetic Potentiation of NY-ESO-1 Vaccine Therapy in Human Ovarian Cancer. Cancer Immunol. Res. 2014, 2, 37–49. [Google Scholar] [CrossRef]
- Sehouli, J.; Senyuva, F.; Fotopoulou, C.; Neumann, U.; Denkert, C.; Werner, L.; Gülten, O. Özcelik Intra-abdominal tumor dissemination pattern and surgical outcome in 214 patients with primary ovarian cancer. J. Surg. Oncol. 2009, 99, 424–427. [Google Scholar] [CrossRef]
- Gerber, S.A.; Rybalko, V.Y.; Bigelow, C.E.; Lugade, A.A.; Foster, T.H.; Frelinger, J.G.; Lord, E.M. Preferential Attachment of Peritoneal Tumor Metastases to Omental Immune Aggregates and Possible Role of a Unique Vascular Microenvironment in Metastatic Survival and Growth. Am. J. Pathol. 2006, 169, 1739–1752. [Google Scholar] [CrossRef]
- Flesken-Nikitin, A.; Choi, K.-C.; Eng, J.P.; Shmidt, E.N.; Nikitin, A.Y. Induction of Carcinogenesis by Concurrent Inactivation of p53 and Rb1 in the Mouse Ovarian Surface Epithelium. Cancer Res. 2003, 63, 3459–3463. [Google Scholar]
- Emerling, B.M.; Weinberg, F.; Liu, J.-L.; Mak, T.W.; Chandel, N.S. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc. Natl. Acad. Sci. USA 2008, 105, 2622–2627. [Google Scholar] [CrossRef]
- Mishra, R.; Hanker, A.; Garrett, J.T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget 2017, 8, 114371–114392. [Google Scholar] [CrossRef]
- Lam, E.W.-F.; Francis, R.E.; Myatt, S.S.; Krol, J.; Hartman, J.; Peck, B.; McGovern, U.B.; Wang, J.; Guest, S.K.; Filipovic, A.; et al. FoxM1 is a downstream target and marker of HER2 overexpression in breast cancer. Int. J. Oncol. 2009, 35, 57–68. [Google Scholar] [CrossRef][Green Version]
- Momeny, M.; Zarrinrad, G.; Moghaddaskho, F.; Poursheikhani, A.; Sankanian, G.; Zaghal, A.; Mirshahvaladi, S.; Esmaeili, F.; Eyvani, H.; Barghi, F.; et al. Dacomitinib, a pan-inhibitor of ErbB receptors, suppresses growth and invasive capacity of chemoresistant ovarian carcinoma cells. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Momeny, M.; Esmaeili, F.; Hamzehlou, S.; Yousefi, H.; Javadikooshesh, S.; Vahdatirad, V.; Alishahi, Z.; Mousavipak, S.H.; Bashash, D.; Dehpour, A.R.; et al. The ERBB receptor inhibitor dacomitinib suppresses proliferation and invasion of pancreatic ductal adenocarcinoma cells. Cell. Oncol. 2019, 42, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Du, J.; Xie, K. FOXM1 and its oncogenic signaling in pancreatic cancer pathogenesis. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2014, 1845, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Noack, S.; Raab, M.; Matthess, Y.; Sanhaji, M.; Kramer, A.; Győrffy, B.; Kaderali, L.; El-Balat, A.; Becker, S.; Strebhardt, K. Synthetic lethality in CCNE1-amplified high grade serous ovarian cancer through combined inhibition of Polo-like kinase 1 and microtubule dynamics. Oncotarget 2018, 9, 25842–25859. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Blay, J.-Y.; De Greve, J.; Brain, E.; Machiels, J.-P.; Soria, J.-C.; Sleijfer, S.; Wolter, P.; Ray-Coquard, I.; Fontaine, C.; et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network of Core Institutes (NOCI). Eur. J. Cancer 2010, 46, 2206–2215. [Google Scholar] [CrossRef]
- Demeure, M.J.; Armaghany, T.; Ejadi, S.; Ramanathan, R.K.; Elfiky, A.; Strosberg, J.R.; Smith, D.C.; Whitsett, T.; Liang, W.S.; Sekar, S.; et al. A phase I/II study of TKM-080301, a PLK1-targeted RNAi in patients with adrenocortical cancer (ACC). J. Clin. Oncol. 2016, 34, 2547. [Google Scholar] [CrossRef]
- Sun, L.; Ren, X.; Wang, I.-C.; Pradhan, A.; Zhang, Y.; Flood, H.; Han, B.; Whitsett, J.A.; Kalin, T.V.; Kalinichenko, V.V. The FOXM1 inhibitor RCM-1 suppresses goblet cell metaplasia and prevents IL-13 and STAT6 signaling in allergen-exposed mice. Sci. Signal. 2017, 10, eaai8583. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.S.; Sanders, D.A.; Rodriguez, R.; Balasubramanian, S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat. Chem. 2011, 3, 725–731. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Bhat, U.G.; Hughes, D.E.; Wang, I.-C.; Costa, R.H.; Gartel, A.L. Identification of a Chemical Inhibitor of the Oncogenic Transcription Factor Forkhead Box M1. Cancer Res. 2006, 66, 9731–9735. [Google Scholar] [CrossRef]
- Wang, M.; Gartel, A.L. Micelle-Encapsulated Thiostrepton as an Effective Nanomedicine for Inhibiting Tumor Growth and for Suppressing FOXM1 in Human Xenografts. Mol. Cancer Ther. 2011, 10, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. Thiazole Antibiotics Siomycin a and Thiostrepton Inhibit the Transcriptional Activity of FOXM1. Front. Oncol. 2013, 3, 150. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Pandit, B.; Wang, M.; Nogueira, V.; Hay, N.; Gartel, A.L. Combination of Oxidative Stress and FOXM1 Inhibitors Induces Apoptosis in Cancer Cells and Inhibits Xenograft Tumor Growth. Am. J. Pathol. 2013, 183, 257–265. [Google Scholar] [CrossRef]
- Ketola, K.; Munuganti, R.S.; Davies, A.; Nip, K.M.; Bishop, J.L.; Zoubeidi, A. Targeting Prostate Cancer Subtype 1 by Forkhead Box M1 Pathway Inhibition. Clin. Cancer Res. 2017, 23, 6923–6933. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kim, K.-Y.; Yu, S.-N.; Park, S.-G.; Yu, H.-S.; Seo, Y.-K.; Ahn, S.-C. Monensin Induces PC-3 Prostate Cancer Cell Apoptosis via ROS Production and Ca2+ Homeostasis Disruption. Anticancer. Res. 2016, 36, 5835–5843. [Google Scholar]
- Nagaraju, G.P.; Zafar, S.F.; El-Rayes, B.F. Pleiotropic effects of genistein in metabolic, inflammatory, and malignant diseases. Nutr. Rev. 2013, 71, 562–572. [Google Scholar] [CrossRef]
- Wang, Z.; Ahmad, A.; Banerjee, S.; Azmi, A.; Kong, D.; Li, Y.; Sarkar, F.H. FoxM1 is a Novel Target of a Natural Agent in Pancreatic Cancer. Pharm. Res. 2010, 27, 1159–1168. [Google Scholar] [CrossRef]
- Cao, J.; Ning, Y.; Li, Q.; Xiang, H.; Liu, F. Apoptosis induced by 7-difluoromethoxyl-5,4’-di-n-octyl genistein via the inactivation of FoxM1 in ovarian cancer cells. Oncol. Rep. 2012, 27, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Ren, K.; Song, Z.; Li, D.; Quan, M.; Zheng, Y.; Cao, J.; Zeng, W.; Zou, H. 7-Difluoromethoxyl-5,4′-di-n-octyl genistein inhibits the stem-like characteristics of gastric cancer stem-like cells and reverses the phenotype of epithelial-mesenchymal transition in gastric cancer cells. Oncol. Rep. 2016, 36, 1157–1165. [Google Scholar] [CrossRef]
- Gormally, M.; Dexheimer, T.S.; Marsico, G.; Sanders, D.A.; Lowe, C.; Matak-Vinković, D.; Michael, S.; Jadhav, A.; Rai, G.; Maloney, D.J.; et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Xiang, Q.; Tan, G.; Jiang, X.; Wu, K.; Tan, W.; Tan, Y. Suppression of FOXM1 Transcriptional Activities via a Single-Stranded DNA Aptamer Generated by SELEX. Sci. Rep. 2017, 7, 45377. [Google Scholar] [CrossRef]
- Ziegler, Y.; Laws, M.J.; Guillen, V.S.; Kim, S.H.; Dey, P.; Smith, B.P.; Gong, P.; Bindman, N.; Zhao, Y.; Carlson, K.; et al. Suppression of FOXM1 activities and breast cancer growth in vitro and in vivo by a new class of compounds. NPJ Breast Cancer 2019, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Lee, W.; Kwon, M.; Lee, H.N. Dual inhibition of FOXM1 and its compensatory signaling pathway decreased the survival of ovarian cancer cells. Oncol. Rep. 2020, 45, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Xu, M.; Cao, X.; Chen, X.; Luo, X. Inactivation of AKT, ERK and NF-κB by genistein derivative, 7-difluoromethoxyl-5,4′-di-n-octylygenistein, reduces ovarian carcinoma oncogenicity. Oncol. Rep. 2017, 38, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Branigan, T.B.; Kozono, D.; Schade, A.E.; Deraska, P.; Rivas, H.G.; Sambel, L.; Reavis, H.D.; Shapiro, G.I.; D’Andrea, A.D.; DeCaprio, J.A. MMB-FOXM1-driven premature mitosis is required for CHK1 inhibitor sensitivity. Cell Rep. 2021, 34, 108808. [Google Scholar] [CrossRef]
- Diab, A.; Gem, H.; Swanger, J.; Kim, H.Y.; Smith, K.; Zou, G.; Raju, S.; Kao, M.; Fitzgibbon, M.; Loeb, K.R.; et al. FOXM1 drives HPV+ HNSCC sensitivity to WEE1 inhibition. Proc. Natl. Acad. Sci. USA 2020, 117, 28287–28296. [Google Scholar] [CrossRef]
- Chung, S.; Vail, P.J.; Witkiewicz, A.K.; Knudsen, E.S. Coordinately Targeting Cell-Cycle Checkpoint Functions in Integrated Models of Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 2290–2304. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J.H. Targeting transcription factors in cancer—from undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- van den Brand, D.; Mertens, V.; Massuger, L.F.A.G.; Brock, R. siRNA in ovarian cancer–Delivery strategies and targets for therapy. J. Control. Release 2018, 283, 45–58. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Zhou, X.; Zhao, R.; Wang, H. Applications of CRISPR-Cas9 in gynecological cancer research. Clin. Genet. 2020, 97, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Áyen, Á.; Martínez, Y.J.; Marchal, J.A.; Boulaiz, H. Recent Progress in Gene Therapy for Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 1930. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
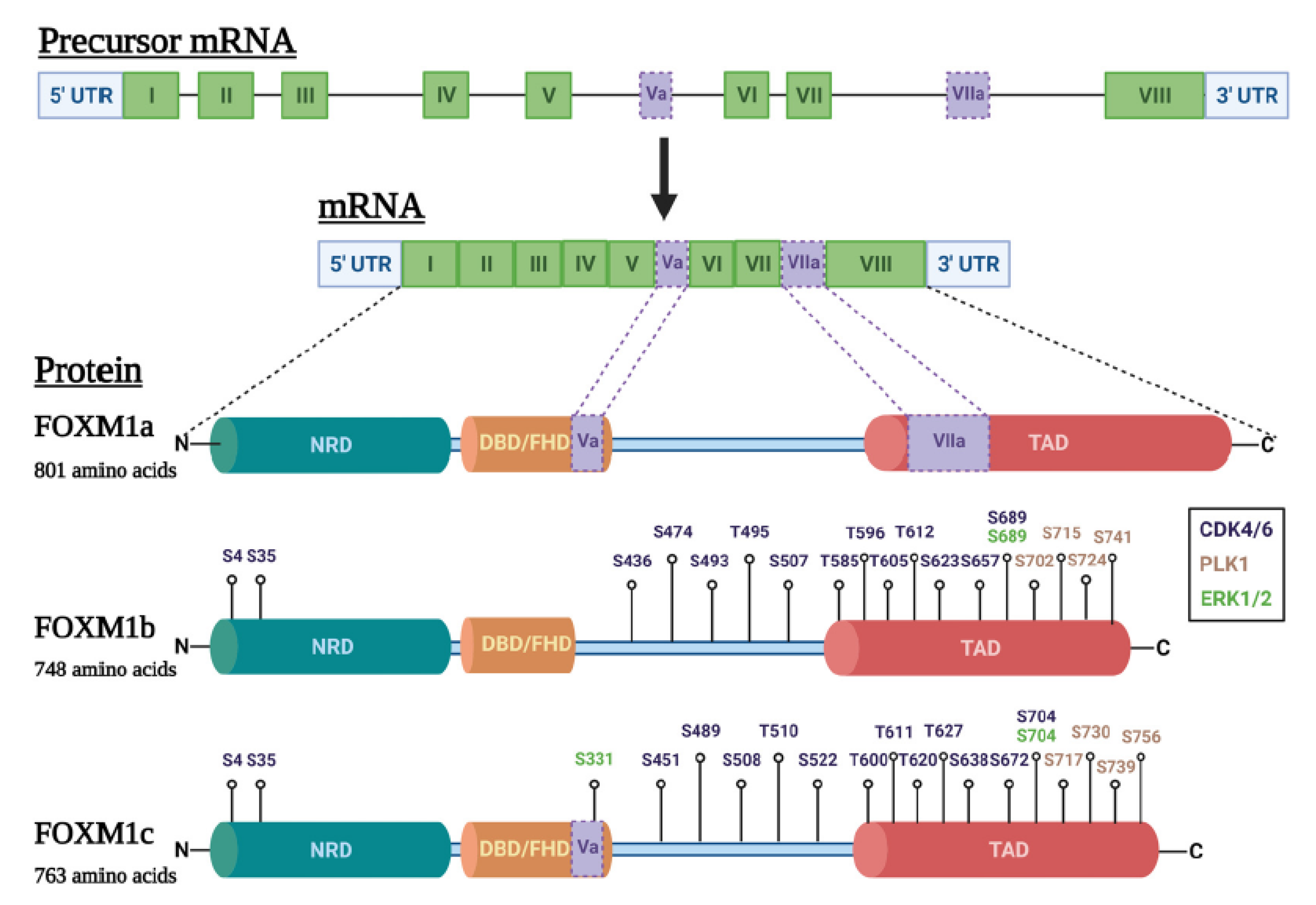{kind=link}
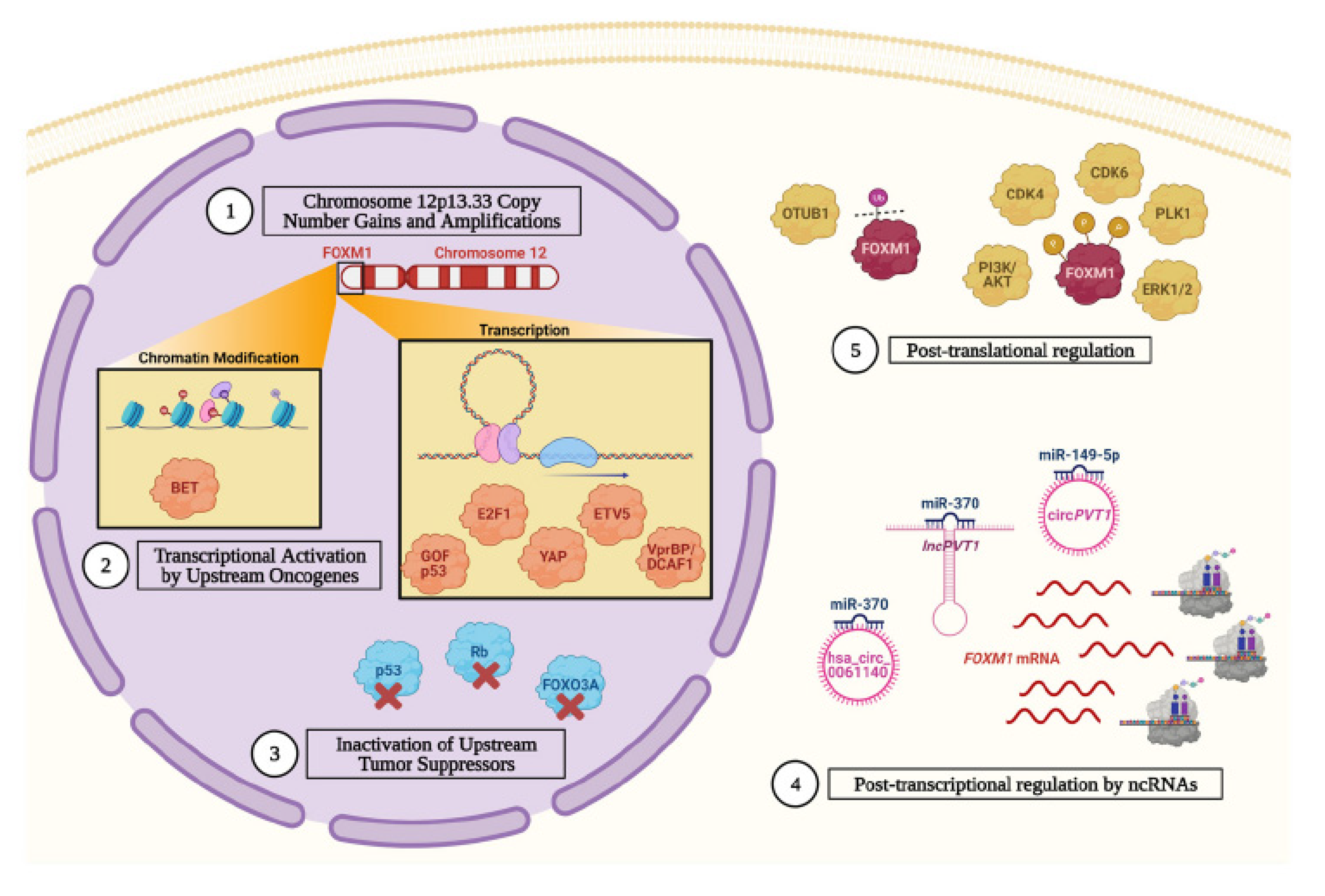{kind=link}
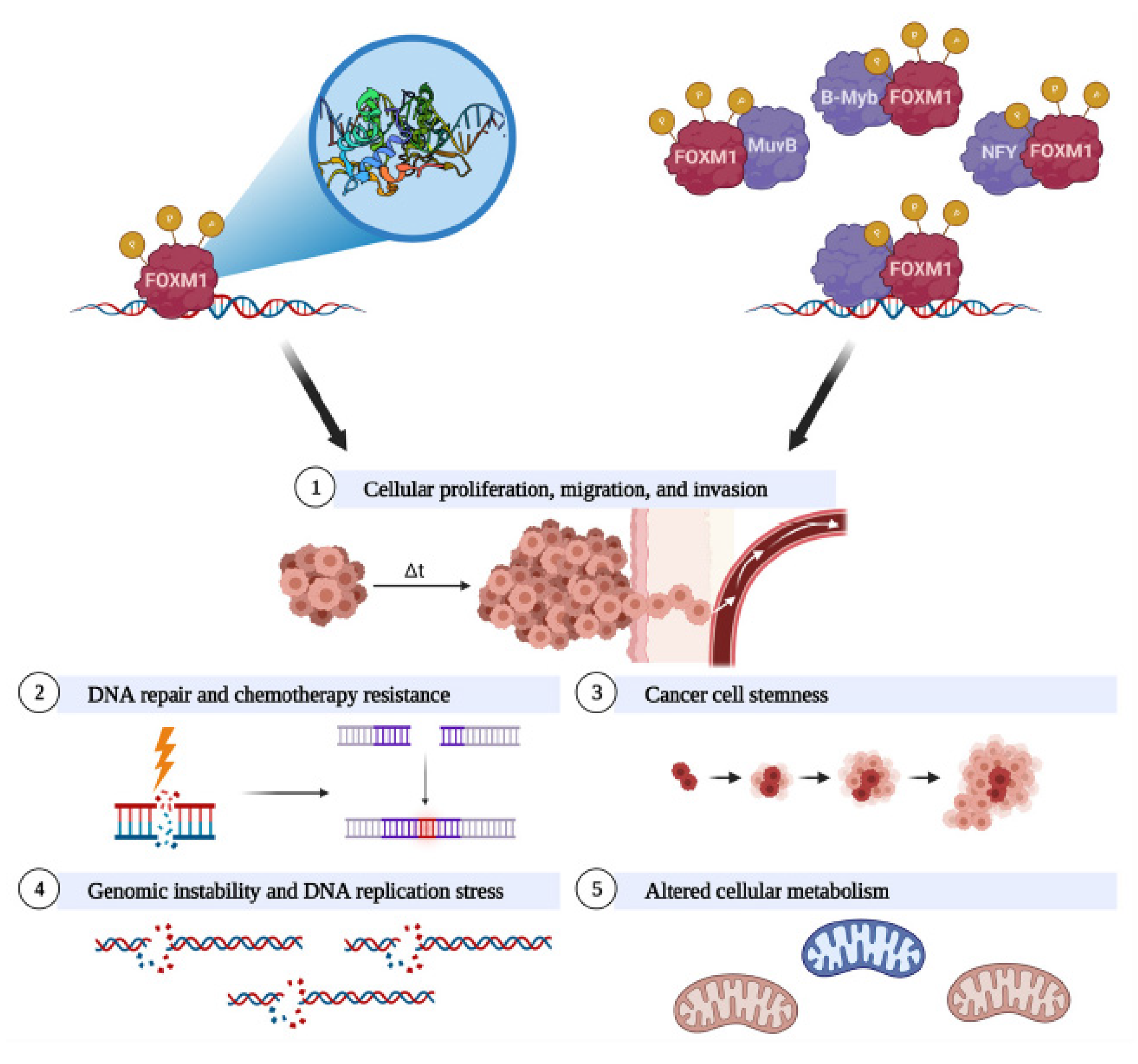{kind=link}
| Isoform Name | Structure | Known Function | Size | RefSeq ID | Ensembl ID | UniProt ID | References |
|---|---|---|---|---|---|---|---|
| Well-Characterized FOXM1 Isoforms | |||||||
| FOXM1a | Includes alternative exons Va and VIIa | Not transcriptionally active | 801 aa | NM_202002 | ENST00000342628 | Q08050-3 | [13,14,28,29] |
| FOXM1b | Omits alternative exons Va and VIIa | Transcriptionally active | 748 aa | NM_202003 | ENST00000361953 | Q08050-2 | [13,14,28,29] |
| FOXM1c | Includes alternative exon Va; omits alternative exon VIIa | Transcriptionally active | 763 aa | NM_021953 | ENST00000359843 | Q08050-1 | [13,14,28,29] |
| Other Reported FOXM1 Isoforms | |||||||
| FOXM1b1 | Omits alternative exons Va and VIIa; omits alanine residue at the beginning of exon III; includes glutamine residue at the end of exon V | Transcriptionally active with functions similar to FOXM1b | 748 aa | NM_001243088 | ENST00000627656 | A0A0D9SFF0 | [33] |
| FOXM1b2 | Omits alternative exons Va and VIIa; omits alanine residue at the beginning of exon III | Transcriptionally active with functions similar to FOXM1b | 747 aa | NM_001243089 | N/A | N/A | [33] |
| FOXM1d | Includes alternative exon VIIa; omits alternative exon Va | Not transcriptionally active; binds directly to oncogenic proteins | 786 aa | N/A | N/A | A0A2P9DTZ0-1 | [31,34,35] |
| FOXM1 variant gAug10 | No evidence at the protein level | N/A | N/A | N/A | ENST00000536066 | N/A | [36] |
| FOXM1 variant lAug10 | No evidence at the protein level | N/A | N/A | N/A | N/A | N/A | [36] |
| FOXM1 Target (Gene) | Known Oncogenic Mechanism | References |
|---|---|---|
| Cellular Proliferation | ||
| Cyclin B1 (CCNB1) | Cyclin protein that promotes mitosis | [127,222] |
| S-phase kinase-associated protein 2 (SKP2) | F-box protein that mediates cell cycle entry and G1/S transitions | [127] |
| Polo-like kinase 1 (PLK1) | Protein kinase that mediates mitosis and cytokinesis | [127] |
| Cell division cycle 25B (CDC25B) | Tyrosine protein phosphatase that mediates cell cycle progression and mitosis | [222] |
| Centrosomal protein 55 (CEP55) | Mitotic phosphoprotein that mediates cytokinesis | [222] |
| Centrometere protein F (CENPF) | Microtubule-binding protein and mediates cell division | [222] |
| DNA topoisomerase II Alpha (TOP2A) | DNA topoisomerase that mediates DNA transcription and replication and chromosome condensation and segregation | [222] |
| Cyclin F (CCNF) | F-box protein that mediates the stability of proteins that regulate cell cycle and genome stability | [223] |
| Protein regulator of cytokinesis 1 (PRC1) | Microtubule-associated protein essential for cytokinesis (related to mitosis-related genes in ovarian cancer) | [223] |
| Homeobox DLX-1 (DLX1) | Transcription factor that modulates the TGF-β1/SMAD4 signaling pathway in ovarian cancer | [224] |
| Proliferation cell nuclear antigen clamp-associated factor (PCLAF) | PCNA-binding protein that regulates DNA repair, cell cycle progression, and proliferation (and activates the PI3K/AKT/mTOR signaling pathways in ovarian cancer) | [225] |
| Kinesin-like protein KIF20A (KIF20A) | Kinesin protein that participates in cytokinesis and intracellular transportation | [223] |
| Cellular Migration and Invasion | ||
| Cyclin F (CCNF) | F-box protein that mediates the stability of proteins that regulate cell cycle and genome stability | [223] |
| Protein regulator of cytokinesis 1 (PRC1) | Microtubule-associated protein essential for cytokinesis (related to mitosis-related genes in ovarian cancer) | [223] |
| Matrix metalloproteinase 2 (MMP2) | Metalloproteinase that mediates extracellular matrix degradation | [222] |
| Homeobox DLX-1 (DLX1) | Transcription factor that modulates the TGF-β1/SMAD4 signaling pathway in ovarian cancer | [224] |
| Proliferation cell nuclear antigen clamp-associated factor (PCLAF) | PCNA-binding protein that regulates DNA repair, cell cycle progression, and proliferation (and activates the PI3K/AKT/mTOR signaling pathways in ovarian cancer) | [225] |
| Kinesin-like protein KIF20A (KIF20A) | Kinesin protein that participates in cytokinesis and intracellular transportation | [223] |
| Cytokeratin-5 (KRT5) | Filament protein that is found in FTE stem cells and serous ovarian cancer (may promote stemness) | [226,227] |
| Cytokeratin-7 (KRT7) | Filament protein that promotes cell–matrix adhesion and extracellular matrix degradation in ovarian cancer | [226,228] |
| β-catenin (CTNNB1) | Transcriptional co-regulator protein and adaptor protein for cell adhesion that contributes to ovarian cancer metastasis, stemness, chemoresistance, angiogenesis, and immune evasion | [229,230] |
| Integrin β1 (ITGB1) | Integrin protein that facilitates the adhesion of ovarian cancer spheroids | [231] |
| Integrin αV (ITGAV) | Integrin protein that facilitates the adhesion of ovarian cancer spheroids | [231] |
| Integrin α5 (ITGA5) | Integrin protein that facilitates the adhesion of ovarian cancer spheroids | [231] |
| Lamin B1 (LMNB1) | Nuclear lamina protein that facilitates the adhesion of ovarian cancer spheroids | [231] |
| Fibronectin 1 (FN1) | Extracellular matrix glycoprotein that facilitates the adhesion of ovarian cancer spheroids | [231] |
| Chemotherapy Resistance and DNA Repair | ||
| Exonuclease 1 (EXO1) | Homologous DNA damage repair protein | [232] |
| Proliferation cell nuclear antigen clamp-associated factor (PCLAF) | PCNA-binding protein that regulates DNA repair, cell cycle progression, and proliferation (and activates the PI3K/AKT/mTOR signaling pathways in ovarian cancer) | [225] |
| Protein regulator of cytokinesis 1 (PRC1) | Microtubule-associated protein essential for cytokinesis (related to mitosis-related genes in ovarian cancer) | [223] |
| Cyclin B1 (CCNB1) | Cyclin protein that promotes mitosis | [233] |
| BRCA1 (BRCA1) | Homologous DNA damage repair protein | [233,234] |
| BRCA2 (BRCA2) | Homologous DNA damage repair protein | [234] |
| RAD51 (RAD51) | Homologous DNA damage repair protein | [233,234] |
| Fanconi anemia group F protein (FANCF) | Homologous DNA damage repair protein | [233] |
| RAD51 paralog D (RAD51D) | Homologous DNA damage repair protein | [233] |
| Fanconi anemia group D2 protein (FANCD2) | Homologous DNA damage repair protein | [233] |
| Altered Cellular Metabolism | ||
| Glucose transporter 1 (GLUT1) | Glucose transport protein that promotes aerobic glycolysis in ovarian cancer | [235] |
| Hexokinase 2 (HK2) | Glycolytic enzyme that promotes aerobic glycolysis in ovarian cancer | [235] |
| Effect on Ovarian Cancer Cell Phenotype | Concentration | Assays | References |
|---|---|---|---|
| Thiostrepton | |||
| Reduced cellular proliferation/viability | 0.1–20 µM | XTT, AlamarBlue, sulforhodamine B, MTT | [136,205,229,233,260] |
| Reduced cellular proliferation/viability of patient ascites cells ex vivo when used alone and in combination with paclitaxel and cisplatin | 1–20 µM | Sulfohodamine B | [260] |
| Reduced cellular proliferation/viability synergistically when used in combination with 1 µM cisplatin | 2.5–10 µM | AlamarBlue | [136] |
| Reduced cellular proliferation/viability by sensitizing cisplatin-resistant cells to cisplatin | 0.5–1 µM | MTT | [240] |
| Reduced cellular proliferation/viability by sensitizing rucaparib-resistant cells to rucaparib | 0.1–1.25 µM | Sulfohodamine B | [233] |
| Reduced cellular migration | 5–20 µM | Transwell | [154,205,206,229] |
| Reduced cellular invasion | 5–20 µM | Matrigel transwell | [154,205,206,229] |
| Reduced colony formation | 5–10 µM | Clonogenic | [229] |
| Reduced colony formation synergistically when used in combination with 2.5 µM FH535 (β-catenin inhibitor) | 5 µM | Clonogenic | [229] |
| Reduced colony formation by sensitizing PARPi-resistant cells to PARPi | 0.5–1 µM | Clonogenic | [233] |
| Slowed wound closure rate | 5–10 µM | Wound healing | [206] |
| Induced apoptosis | 1–10 µM | qRT-PCR, western blot, annexin-V/propidium iodide flow cytometry, caspase-3 activity | [136,229,233,260] |
| Induced apoptosis synergistically when used in combination with 2.5 µM FH535 (β-catenin inhibitor) | 5 µM | Annexin-V/propidium iodide flow cytometry | [229] |
| Induced DNA damage | 7.5–10 µM | Alkaline comet | [233] |
| Induced PARP1 trapping onto chromatin when combined with Olaparib | 5–10 µM | PARP trapping | [233] |
| Reduced sphere formation | 1 µM | Spheroid formation | [295] |
| Decreased HUVEC tube formation and VEFG secretion | 5–10 µM | HUVEC tube formation, ELISA | [229] |
| Decreased MMP-9 and PLAUR gene expression levels | 5–10 µM | Sem-quantitative RT-PCR | [206] |
| Decreased NOTCH1 protein expression levels | 1 µM | Western blot | [295] |
| Decreased active β-catenin, overall β-catenin, TCF4, cyclin D1, cMYC, uPAR, VEGF, MMP-9, and MMP-2 protein expression levels when used alone and in combination with FH535 (β-catenin inhibitor) | 5 µM | Western blot | [229] |
| Reduced tumor size in mice | 200–300 µM/kg, 20–50 mg/kg | Cell line-derived xenograft | [205,229,231,240] |
| Reduced tumor size in mice when used in combination with cisplatin | 50 mg/kg | Cell line-derived xenograft | [240] |
| Reduced tumor size in mice when used in combination with latanib | 20 mg/kg | Cell line-derived xenograft | [231] |
| Reduced tumor size in mice when used in combination with FH535 (β-catenin inhibitor) | 20 mg/kg | Cell line-derived xenograft | [229] |
| Increased overall survival in mice when used in combination with latanib | 20 mg/kg | Cell line-derived xenograft | [231] |
| Reduced number of tumor spheroids in the peritoneal fluid in mice when used alone and used in combination with latanib | 20 mg/kg | Cell line-derived xenograft | [231] |
| Reduced cellular proliferation and induced apoptosis in patient tumors grown ex vivo alone, in combination with olaparib, and in combination with carboplatin | 3 µM | Immunofluorescence on fixated tissue | [234] |
| FDI-6 | |||
| Reduced cellular proliferation/viability | 1–30 µM | Not specified, cell counting kit-8 and microscopic imaging analysis | [362,365] |
| Reduced cellular proliferation/viability when used in combination with tipifarnib, sapatinib, or rottlerin | 3–10 µM | Cell counting kit-8 and microscopic imaging analysis | [365] |
| Increased N-Ras protein expression | 1–10 µM | Western blot | [365] |
| Decreased p-PKCδ and HER3 protein expression | 1–10 µM | Western blot | [365] |
| 7-difluoromethoxyl-5,4-di-n-octyl genistein (DFOG) | |||
| Reduced cellular proliferation/viability | 1–10 µM | MTT | [360] |
| Reduced colony formation | 1–10 µM | Clonogenic | [360,366] |
| Induced G2/M-phase cell cycle arrest | 1–10 µM | Cell cycle analysis | [360] |
| Induced apoptosis | 1–10 µM | Histone/DNA ELISA, propidium iodide flow cytometry | [360] |
| Reduced sphere formation | 1–10 µM | Spheroid formation | [366] |
| Decreased CD133, CD44, ALDH1, and NF-κBp65 protein expression levels | 1–10 µM | Western blot | [366] |
| Decreased phosphorylation of AKT, ERK1/2, and FOXO3A | 3–10 µM | Western blot | [366] |
| N-phenylphenanthren-9-amine | |||
| Reduced cellular proliferation/viability | 0.01–10 µM | Sulforhodamine B | [321] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Barger, C.J.; Karpf, A.R. FOXM1: A Multifunctional Oncoprotein and Emerging Therapeutic Target in Ovarian Cancer. Cancers 2021, 13, 3065. https://doi.org/10.3390/cancers13123065
Liu C, Barger CJ, Karpf AR. FOXM1: A Multifunctional Oncoprotein and Emerging Therapeutic Target in Ovarian Cancer. Cancers. 2021; 13(12):3065. https://doi.org/10.3390/cancers13123065
Chicago/Turabian StyleLiu, Cassie, Carter J. Barger, and Adam R. Karpf. 2021. "FOXM1: A Multifunctional Oncoprotein and Emerging Therapeutic Target in Ovarian Cancer" Cancers 13, no. 12: 3065. https://doi.org/10.3390/cancers13123065
APA StyleLiu, C., Barger, C. J., & Karpf, A. R. (2021). FOXM1: A Multifunctional Oncoprotein and Emerging Therapeutic Target in Ovarian Cancer. Cancers, 13(12), 3065. https://doi.org/10.3390/cancers13123065