
Genome-Wide Transcriptomic Analysis of Non-Tumorigenic Tissues Reveals Aging-Related Prognostic Markers and Drug Targets in Renal Cell Carcinoma

 ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Overview of the Proposed Approach
2.2. Materials
2.3. Data Pre-Processing
2.4. Identifying Aging-Related Genes and miRNAs
2.5. Finding DEGs in Cancer
2.6. Building Modules Using Protein–Protein Interaction Networks
2.7. Experimental Validation
2.7.1. Experimental Reagents
2.7.2. Cell Culture
2.7.3. Collection of Conditioned Media
2.7.4. siRNA-Mediated Gene Knockdown
2.7.5. Real-Time Quantitative PCR
2.7.6. Invasion Assay
2.7.7. Immunocytochemistry
2.7.8. The Enzyme-Linked Immunosorbent Assay (ELISA)
2.7.9. Animals
2.7.10. Zebrafish–Human Cancer Xenograft Model
2.7.11. Statistics for Cell-Based Analysis
3. Results
3.1. Characteristics of the Data
3.1.1. Demographics
3.1.2. Survival Analysis with Chronological Age
3.2. Tissue-Specific Aging-Related Genes and miRNAs
3.3. Association between Survival and Aging-Related Genes in BLCA, BRCA, and THCA
3.4. Analysis of KIRC
3.4.1. Association between Survival and Age-Related Genes in TCGA-KIRC
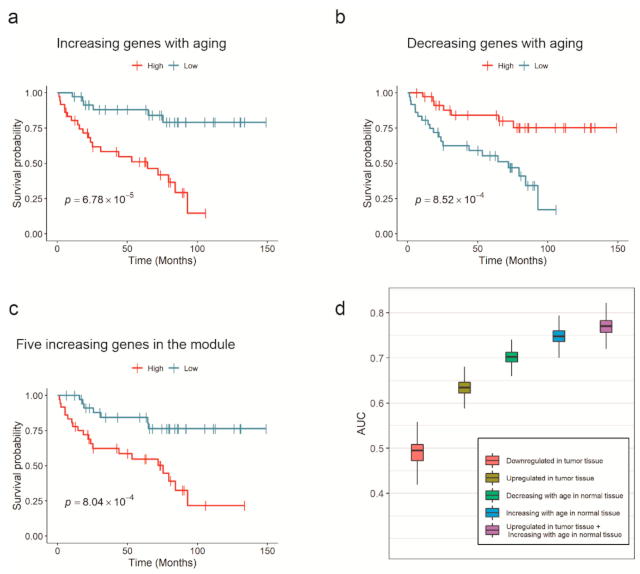
3.4.2. Module Analysis
3.4.3. Validation of Survival Significance in Kidney Renal Cells with an Independent Dataset
3.4.4. Biological Roles of Aging-Related miRNAs in the Kidney
3.4.5. Deferentially Expressed Genes (DEGs) in Kidney Cancer
3.4.6. Survival Prediction Models
- The average expression of downregulated cancer DEGs in tumor tissue;
- The average expression of upregulated cancer DEGs in tumor tissue;
- Decreasing index in normal tissue;
- Increasing index in normal tissue;
- The combination of 2 and 4.
3.5. Experimental Validation
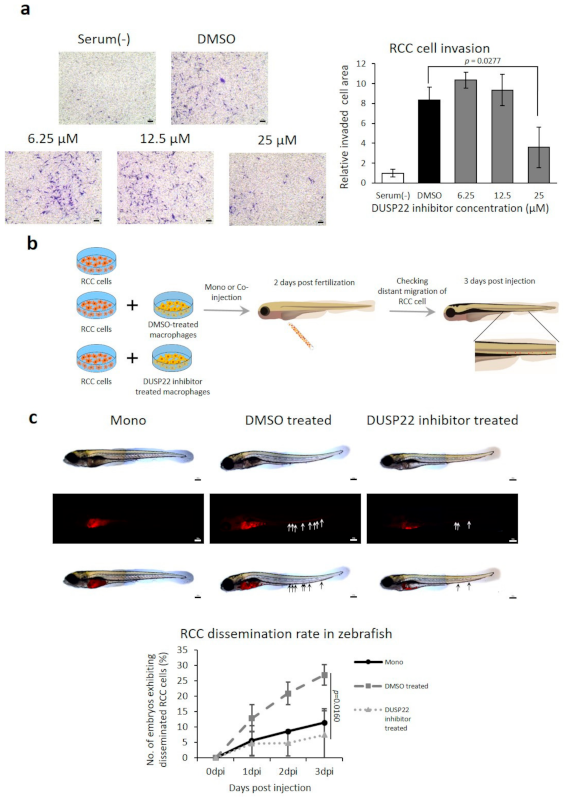
3.5.1. Upregulated Expression of DUSP22, MAPK14, MAPKAPK3, STAT1, and VCP Genes in Aged Mouse Primary Bone Marrow-Derived Macrophages (BMDM)
3.5.2. Inhibited RCC Invasion by DUSP22 Knockdown in Macrophages Derived from Old Mice
3.5.3. DUSP22 Promotes Macrophage-Induced RCC Metastasis In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

Appendix A.1. Bootstrapping
- It is an aging-related gene in more than 50 bootstrap runs; and
- It is an aging-related gene in all samples
Appendix A.2. Survival Analysis
Appendix A.3. Diffusion Kernel
Appendix A.4. Association between Survival and Downregulation of Age-Related Genes in BLCA, BRCA, and THCA
References
- Aunan, J.R.; Cho, W.C.; Søreide, K. The biology of aging and cancer: A brief overview of shared and divergent molecular hallmarks. Aging Dis. 2017, 8, 628. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Cho, W.C.; Ben-Yehuda, A. Cancer and aging-the inflammatory connection. Aging Dis. 2017, 8, 611. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.D.; Sivarapatna, A.; Gress, R.E. The aging immune system and its relationship with cancer. Aging Health 2011, 7, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Levi, N.; Papismadov, N.; Solomonov, I.; Sagi, I.; Krizhanovsky, V. The ECM path of senescence in aging: Components and modifiers. FEBS J. 2020, 287, 2636–2646. [Google Scholar] [CrossRef]
- Harper, E.I.; Sheedy, E.F.; Stack, M.S. With great age comes great metastatic ability: Ovarian cancer and the appeal of the aging peritoneal microenvironment. Cancers (Basel) 2018, 10, 230. [Google Scholar] [CrossRef]
- Yang, J.; Huang, T.; Petralia, F.; Long, Q.; Zhang, B.; Argmann, C.; Zhao, Y.; Mobbs, C.V.; Schadt, E.E.; Zhu, J.; et al. Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci. Rep. 2015, 5, 15145. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Consortium, G. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Brandt, J.; Garne, J.P.; Tengrup, I.; Manjer, J. Age at diagnosis in relation to survival following breast cancer: A cohort study. World J. Surg. Oncol. 2015, 13, 1–11. [Google Scholar] [CrossRef]
- Ganly, I.; Nixon, I.J.; Wang, L.Y.; Palmer, F.L.; Migliacci, J.C.; Aniss, A.; Sywak, M.; Eskander, A.E.; Freeman, J.L.; Campbell, M.J. Survival from differentiated thyroid cancer: What has age got to do with it? Thyroid 2015, 25, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Bechis, S.K.; Carroll, P.R.; Cooperberg, M.R. Impact of age at diagnosis on prostate cancer treatment and survival. J. Clin. Oncol. 2011, 29, 235. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519. [Google Scholar]
- Network, C.G.A.R. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43. [Google Scholar]
- Network, C.G.A.R. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202. [Google Scholar]
- Network, C.G.A.R. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315. [Google Scholar]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543. [Google Scholar]
- Patil, S.; Manola, J.; Elson, P.; Negrier, S.; Escudier, B.; Eisen, T.; Atkins, M.; Bukowski, R.; Motzer, R.J. Improvement in overall survival of patients with advanced renal cell carcinoma: Prognostic factor trend analysis from an international data set of clinical trials. J. Urol. 2012, 188, 2095–2100. [Google Scholar] [CrossRef]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef]
- Glass, D.; Viñuela, A.; Davies, M.N.; Ramasamy, A.; Parts, L.; Knowles, D.; Brown, A.A.; Hedman, A.K.; Small, K.S.; Buil, A.; et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013, 14, R75. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Cox, D.R. Regression Models and Life-Tables. J. R. Stat. Soc. Ser. B 1972, 34, 187–220. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef]
- Lee, H.; Tu, Z.; Deng, M.; Sun, F.; Chen, T. Diffusion kernel-based logistic regression models for protein function prediction. OMICS 2006, 10, 40–55. [Google Scholar] [CrossRef]
- Zhang, C.; Fu, L.; Fu, J.; Hu, L.; Yang, H.; Rong, T.-H.; Li, Y.; Liu, H.; Fu, S.-B.; Zeng, Y.-X. Fibroblast growth factor receptor 2–positive fibroblasts provide a suitable microenvironment for tumor development and progression in esophageal carcinoma. Clin. Cancer Res. 2009, 15, 4017–4027. [Google Scholar] [CrossRef]
- Jung, D.-W.; Oh, E.-S.; Park, S.-H.; Chang, Y.-T.; Kim, C.-H.; Choi, S.-Y.; Williams, D.R. A novel zebrafish human tumor xenograft model validated for anti-cancer drug screening. Mol. Biosyst. 2012, 8, 1930–1939. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Na, J.C.; Nagaya, N.; Rha, K.H.; Han, W.K.; Kim, I.Y. DNA damage response pathway alteration in locally advanced clear-cell renal-cell carcinoma is associated with a poor outcome. Clin. Genitourin. Cancer 2019, 17, 299–305.e1. [Google Scholar] [CrossRef]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.-F.; Leng, N.; Abbas, A.R. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell 2020, 38, 803–817.e4. [Google Scholar] [CrossRef]
- Bai, X.Y.; Ma, Y.; Ding, R.; Fu, B.; Shi, S.; Chen, X.M. miR-335 and miR-34a Promote renal senescence by suppressing mitochondrial antioxidative enzymes. J. Am. Soc. Nephrol. 2011, 22, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhao, L.; Pan, X.; Jin, L.; Lin, C.; Xu, W.; Xu, J.; Guan, X.; Wu, X.; Wang, Y.; et al. Tumor suppressor microRNA-136-5p regulates the cellular function of renal cell carcinoma. Oncol. Lett. 2018, 15, 5995–6002. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiang, Y.-C.; Sun, C.-K.; Chen, Q.-M. Role of the tumor microenvironment in tumor progression and the clinical applications. Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef] [PubMed]
- Frisch, B.; Bartl, R.; Burkhardt, R. Bone marrow biopsy in clinical medicine: An overview. Haematology (Budap.) 1982, 15, 245–285. [Google Scholar] [PubMed]
- Ju, A.; Cho, Y.-C.; Kim, B.R.; Park, S.G.; Kim, J.-H.; Kim, K.; Lee, J.; Park, B.C.; Cho, S. Scaffold role of DUSP22 in ASK1-MKK7-JNK signaling pathway. PLoS ONE 2016, 11, e0164259. [Google Scholar] [CrossRef]
- Ebelt, N.D.; Cantrell, M.A.; Van Den Berg, C.L. c-Jun N-terminal kinases mediate a wide range of targets in the metastatic cascade. Genes Cancer 2013, 4, 378–387. [Google Scholar] [CrossRef]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef]
- Massari, F.; Di Nunno, V.; Santoni, M.; Gatto, L.; Caserta, C.; Morelli, F.; Zafarana, E.; Carrozza, F.; Mosca, A.; Mollica, V. Toward a genome-based treatment landscape for renal cell carcinoma. Crit. Rev. Oncol. Hematol. 2019, 142, 141–152. [Google Scholar] [CrossRef]
- Rini, B.; Goddard, A.; Knezevic, D.; Maddala, T.; Zhou, M.; Aydin, H.; Campbell, S.; Elson, P.; Koscielny, S.; Lopatin, M. A 16-gene assay to predict recurrence after surgery in localised renal cell carcinoma: Development and validation studies. Lancet Oncol. 2015, 16, 676–685. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Martini, J.-F.; Magheli, A.; Svedman, C.; Lopatin, M.; Knezevic, D.; Goddard, A.D.; Febbo, P.G.; Li, R. Validation of the 16-gene recurrence score in patients with locoregional, high-risk renal cell carcinoma from a phase III trial of adjuvant sunitinib. Clin. Cancer Res. 2018, 24, 4407–4415. [Google Scholar] [CrossRef]
- Sonouchi, K.; Hamilton, T.A.; Tannenbaum, C.S.; Tubbs, R.R.; Bukowski, R.; Finke, J.H. Chemokine gene expression in the murine renal cell carcinoma, RENCA, following treatment in vivo with interferon-alpha and interleukin-2. Am. J. Pathol. 1994, 144, 747. [Google Scholar]
- Cho, H.; Seo, Y.; Loke, K.M.; Kim, S.-W.; Oh, S.-M.; Kim, J.-H.; Soh, J.; Kim, H.S.; Lee, H.; Kim, J. Cancer-stimulated CAFs enhance monocyte differentiation and protumoral TAM activation via IL6 and GM-CSF secretion. Clin. Cancer Res. 2018, 24, 5407–5421. [Google Scholar] [CrossRef] [PubMed]
- Feulner, L.; Najafabadi, H.S.; Tanguay, S.; Rak, J.; Riazalhosseini, Y. Age-related variations in gene expression patterns of renal cell carcinoma. Urol. Oncol. 2019, 37, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Ravi, P.; Bakouny, Z.; Schmidt, A.; Choueiri, T.K. Novel Therapeutic Approaches and the Evolution of Drug Development in Advanced Kidney Cancer. Cancer J. 2020, 26, 464–470. [Google Scholar] [CrossRef]
- Meng, F.; Xiao, Y.; Xie, L.; Liu, Q.; Qian, K. Diagnostic and prognostic value of ABC transporter family member ABCG1 gene in clear cell renal cell carcinoma. Channels (Austin) 2021, 15, 375–385. [Google Scholar] [CrossRef]
- Ju, A.; Cho, S. Inhibition of Dual-specificity Phosphatase 22 (DUSP22) by PRL-3 inhibitor I. Bull. Korean Chem. Soc. 2012, 33, 3142–3144. [Google Scholar] [CrossRef]
- Thu Le, H.T.; Cho, Y.C.; Lee, S.; Kim, Y.G.; Kim, K.; Park, S.G.; Park, B.C.; Cho, S. PTP Inhibitor XIX Inhibits DUSP22 by Conformational Change. Bull. Korean Chem. Soc. 2019, 40, 118–121. [Google Scholar]
- Spadaccino, F.; Netti, G.S.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Ranieri, E. [Diagnostic and prognostic markers of renal cell carcinoma]. G. Ital. Nefrol. 2020, 37. [Google Scholar]
- Ljungberg, B. Prognostic markers in renal cell carcinoma. Curr. Opin. Urol. 2007, 17, 303–308. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, W.; Sun, R.; Huang, Z. Novel insights into clear cell renal cell carcinoma prognosis by comprehensive characterization of aberrant alternative splicing signature: A study based on large-scale sequencing data. Bioengineered 2021, 12, 1091–1110. [Google Scholar] [CrossRef]
- Joosten, S.; Odeh, S.; Koch, A.; Buekers, N.; Aarts, M.; Baldewijns, M.; Van Neste, L.; van Kuijk, S.; Schouten, L.; van den Brandt, P. Development of a prognostic risk model for clear cell renal cell carcinoma by systematic evaluation of DNA methylation markers. Clin. Epigenetics 2021, 13, 1–11. [Google Scholar] [CrossRef]
- Farber, N.J.; Kim, C.J.; Modi, P.K.; Hon, J.D.; Sadimin, E.T.; Singer, E.A. Renal cell carcinoma: The search for a reliable biomarker. Transl. Cancer Res. 2017, 6, 620–632. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Sample Size (Survival/Deceased) | Mean Age | Aging Genes | Aging microRNAs | |||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | microRNA | Increasing | Decreasing | Total | Increasing | Decreasing | Total | ||
| BLCA | 19 (8/11) | 19 (8/11) | 70.32 | 43 | 94 | 137 | 2 | 3 | 5 |
| BRCA | 113 (69/44) | 104 (61/43) | 57.98 | 772 | 1678 | 2450 | 45 | 42 | 87 |
| HNSC | 44 (11/33) | 44 (11/33) | 62.63 | 52 | 62 | 114 | 5 | 9 | 14 |
| KICH | 24 (20/4) | 25 (21/4) | 54.55 | 202 | 139 | 341 | 5 | 10 | 15 |
| KIRC | 72 (45/27) | 71 (45/26) | 62.96 | 162 | 87 | 249 | 17 | 15 | 32 |
| KIRP | 32 (25/7) | 34 (26/8) | 62.40 | 137 | 215 | 352 | 9 | 20 | 29 |
| LIHC | 50 (16/34) | 50 (16/34) | 61.53 | 13 | 65 | 78 | 8 | 18 | 26 |
| LUAD | 59 (33/26) | 46 (33/13) | 65.83 | 339 | 289 | 628 | 12 | 11 | 23 |
| LUSC | 49 (19/30) | 45 (22/23) | 69.25 | 21 | 36 | 57 | 8 | 13 | 21 |
| STAD | 32 (23/9) | 45 (33/12) | 69.25 | 14 | 0 | 14 | 4 | 3 | 7 |
| THCA | 58 (54/4) | 59 (55/4) | 46.03 | 386 | 205 | 591 | 34 | 36 | 70 |
| UCEC | 35 (20/3, 12 Not Available) | 33 (19/3, 11 Not Available) | 59.87 | 15 | 6 | 21 | 12 | 16 | 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, E.; Kim, J.-H.; Um, J.; Jung, D.-W.; Williams, D.R.; Lee, H. Genome-Wide Transcriptomic Analysis of Non-Tumorigenic Tissues Reveals Aging-Related Prognostic Markers and Drug Targets in Renal Cell Carcinoma. Cancers 2021, 13, 3045. https://doi.org/10.3390/cancers13123045
Oh E, Kim J-H, Um J, Jung D-W, Williams DR, Lee H. Genome-Wide Transcriptomic Analysis of Non-Tumorigenic Tissues Reveals Aging-Related Prognostic Markers and Drug Targets in Renal Cell Carcinoma. Cancers. 2021; 13(12):3045. https://doi.org/10.3390/cancers13123045
Chicago/Turabian StyleOh, Euiyoung, Jun-Hyeong Kim, JungIn Um, Da-Woon Jung, Darren R. Williams, and Hyunju Lee. 2021. "Genome-Wide Transcriptomic Analysis of Non-Tumorigenic Tissues Reveals Aging-Related Prognostic Markers and Drug Targets in Renal Cell Carcinoma" Cancers 13, no. 12: 3045. https://doi.org/10.3390/cancers13123045
APA StyleOh, E., Kim, J.-H., Um, J., Jung, D.-W., Williams, D. R., & Lee, H. (2021). Genome-Wide Transcriptomic Analysis of Non-Tumorigenic Tissues Reveals Aging-Related Prognostic Markers and Drug Targets in Renal Cell Carcinoma. Cancers, 13(12), 3045. https://doi.org/10.3390/cancers13123045