
A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth

 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. SDS-PAGE and Western Blotting
2.3. Cloning Strategy
2.4. Lentivirus Production
2.5. Flow Cytometry Screening
2.6. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.7. AlamarBlue
2.8. Animals
2.9. Tumor Growth Experiments
2.10. Matrigel Plug Assays
2.11. Statistics
3. Results
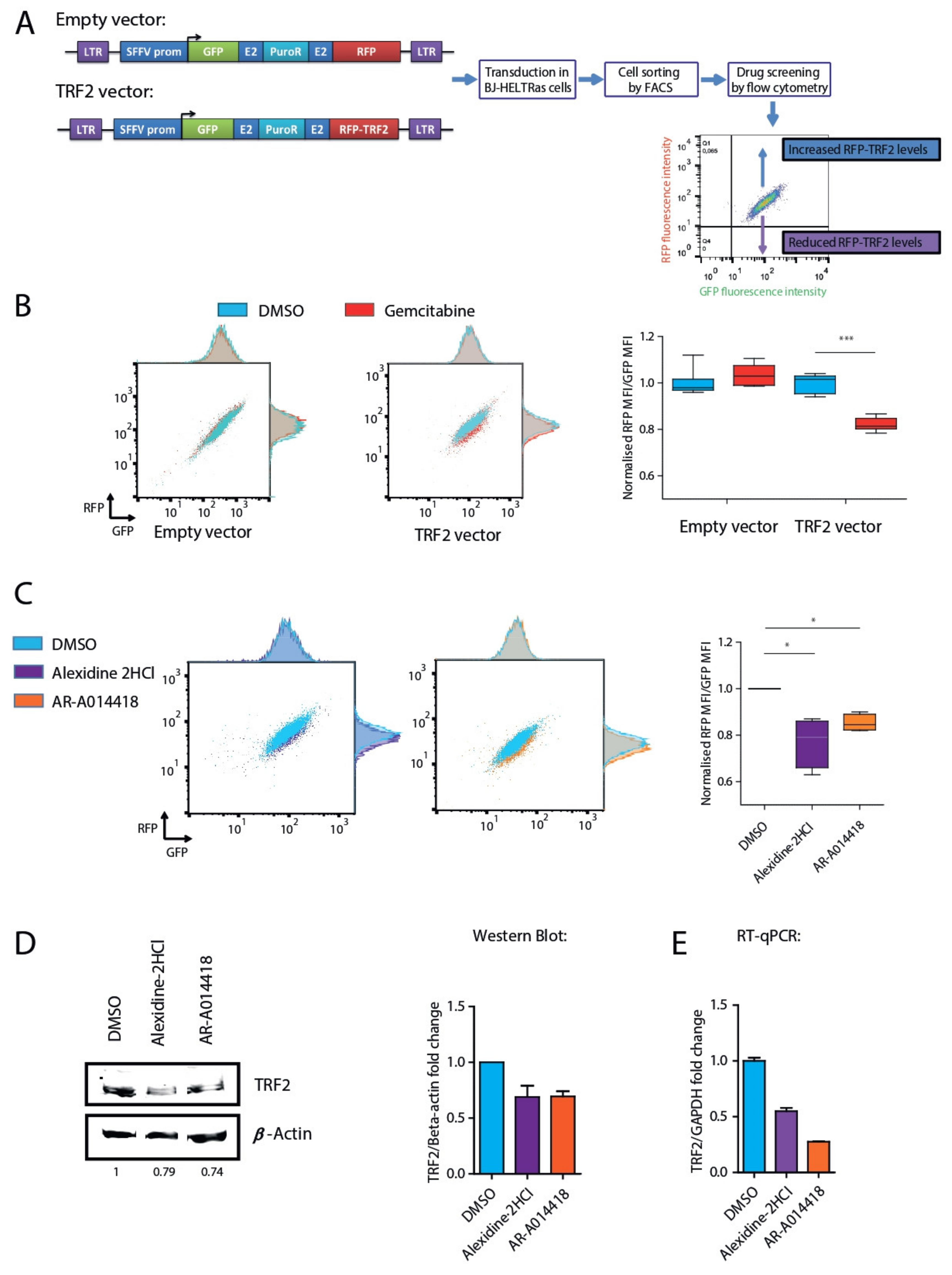
3.1. Identification of TRF2 Inhibitory Molecules
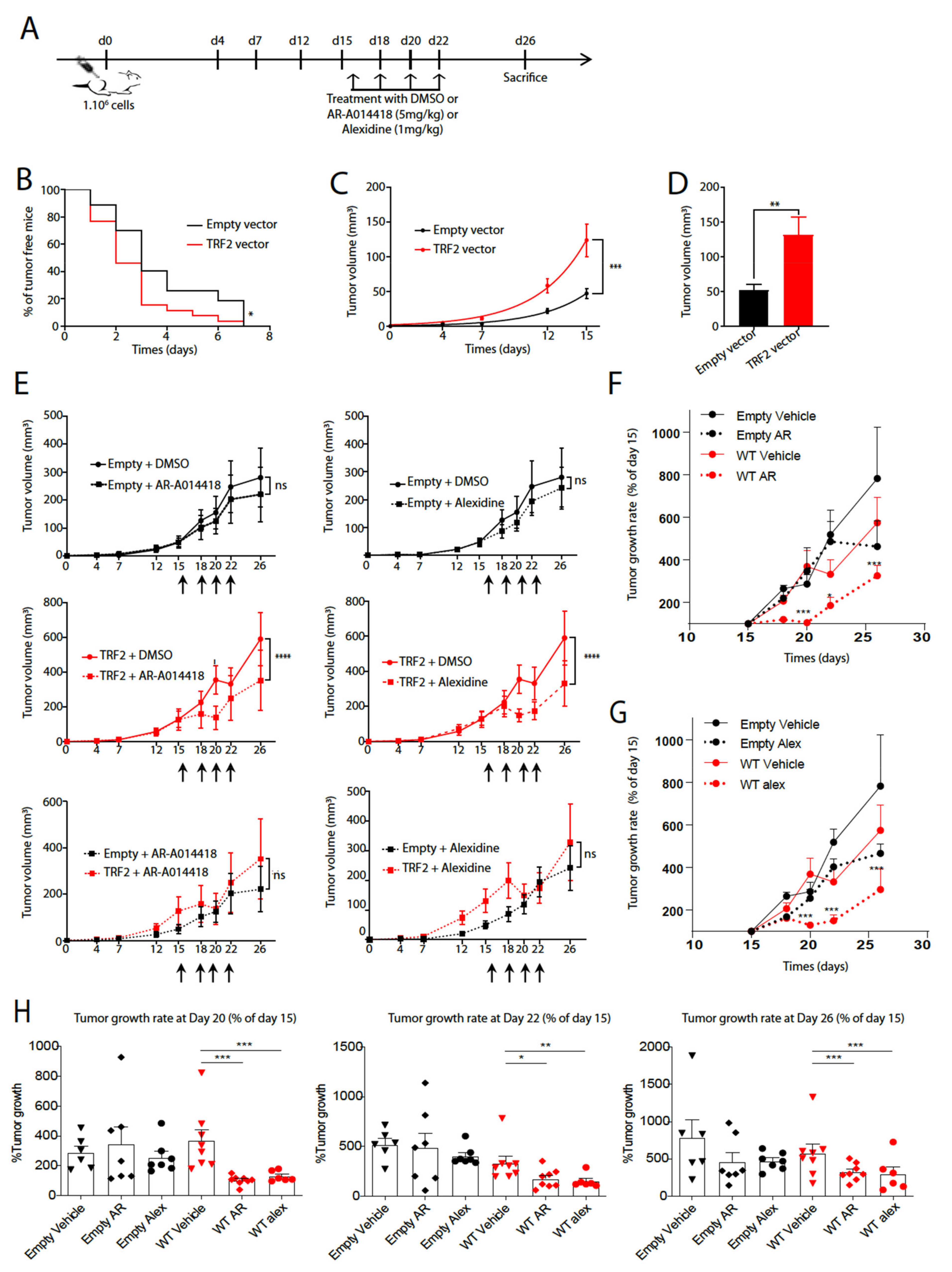
3.2. AR-A014418 and Alexidine·2HCl Reversed the Tumorigenicity Conferred by High TRF2 Expression Levels
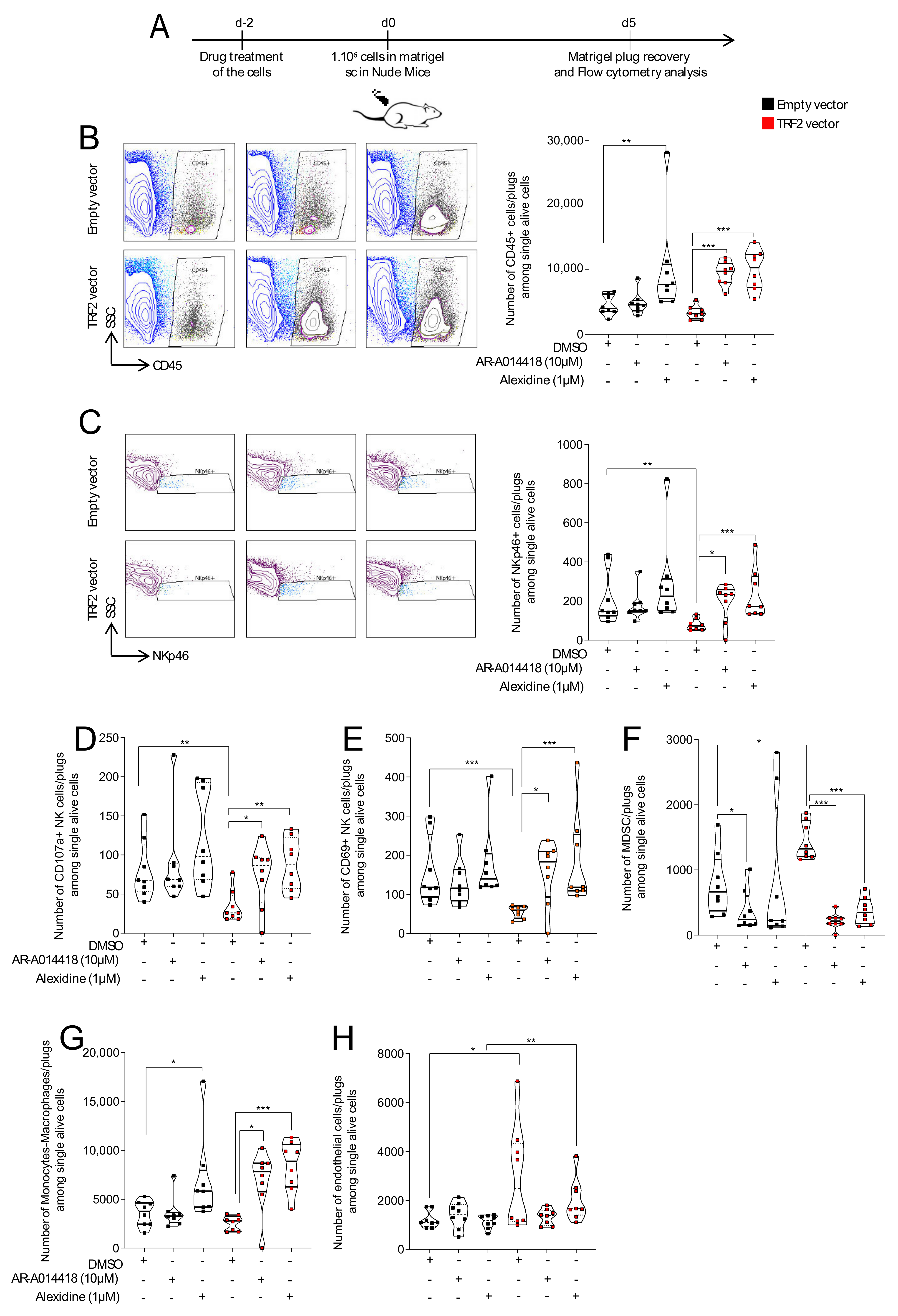
3.3. AR-A014418 and Alexidine·2HCl Counteracted TRF2-Dependent Immunosuppression and Neo-Angiogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giraud-Panis, M.-J.; Pisano, S.; Benarroch-Popivker, D.; Pei, B.; Le Du, M.-H.; Gilson, E. One Identity or More for Telomeres? Front. Oncol. 2013, 3, 48. [Google Scholar] [CrossRef]
- Gilson, E.; Géli, V. How telomeres are replicated. Nat. Rev. Mol. Cell Biol. 2007, 8, 825–838. [Google Scholar] [CrossRef]
- Armanios, M.; Blackburn, E.H. The telomere syndromes. Nat. Rev. Genet. 2012, 13, 693–704. [Google Scholar] [CrossRef]
- Kordinas, V.; Ioannidis, A.; Chatzipanagiotou, S. The Telomere/Telomerase System in Chronic Inflammatory Diseases. Cause or Effect? Genes 2016, 7, 60. [Google Scholar] [CrossRef]
- Kim, N.; Piatyszek, M.; Prowse, K.; Harley, C.; West, M.; Ho, P.; Coviello, G.; Wright, W.; Weinrich, S.; Shay, J. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Sugarman, E.T.; Zhang, G.; Shay, J.W. In perspective: An update on telomere targeting in cancer. Mol. Carcinog. 2019, 58, 1581–1588. [Google Scholar] [CrossRef]
- Artandi, S.E.; Chang, S.; Lee, S.-L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nat. Cell Biol. 2000, 406, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.A.; Maser, R.S.; Xia, H.; McNamara, K.; Protopopov, A.; Chen, L.; Hezel, A.F.; Kim, C.F.; Bronson, R.T.; Castrillon, D.H.; et al. Telomere dysfunction promotes genome instability and metastatic potential in a K-ras p53 mouse model of lung cancer. Carcinogenesis 2008, 29, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase Therapy Provokes ALT and Mitochondrial Adaptive Mechanisms in Cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef]
- Fernandes, S.G.; Dsouza, R.; Pandya, G.; Kirtonia, A.; Tergaonkar, V.; Lee, S.Y.; Garg, M.; Khattar, E. Role of Telomeres and Telomeric Proteins in Human Malignancies and Their Therapeutic Potential. Cancers 2020, 12, 1901. [Google Scholar] [CrossRef]
- Augereau, A.; De Roodenbeke, C.T.; Simonet, T.; Bauwens, S.; Horard, B.; Callanan, M.; Leroux, M.; Jallades, L.; Salles, G.; Gilson, E.; et al. Telomeric damage in early stage of chronic lymphocytic leukemia correlates with shelterin dysregulation. Blood 2011, 118, 1316–1322. [Google Scholar] [CrossRef]
- Biroccio, A.; Cherfils-Vicini, J.; Augereau, A.; Pinte, S.; Bauwens, S.; Ye, J.; Simonet, T.; Horard, B.; Jamet, K.; Cervera, L.; et al. TRF2 inhibits a cell-extrinsic pathway through which natural killer cells eliminate cancer cells. Nat. Cell Biol. 2013, 15, 818–828. [Google Scholar] [CrossRef] [PubMed]
- El Maï, M.; Wagner, K.-D.; Michiels, J.-F.; Ambrosetti, D.; Borderie, A.; Destree, S.; Renault, V.; Djerbi, N.; Giraud-Panis, M.-J.; Gilson, E.; et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014, 9, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.J.; Quesada, V.; Foronda, M.; Conde, L.; Martínez-Trillos, A.; Villamor, N.; Rodríguez, D.; Kwarciak, A.; Garabaya, C.; Gallardo, M.; et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 526–530. [Google Scholar] [CrossRef]
- Bejarano, L.; Schuhmacher, A.J.; Méndez, M.; Megías, D.; Blanco-Aparicio, C.; Martínez, S.; Pastor, J.; Squatrito, M.; Blasco, M.A. Inhibition of TRF1 Telomere Protein Impairs Tumor Initiation and Progression in Glioblastoma Mouse Models and Patient-Derived Xenografts. Cancer Cell 2017, 32, 590–607. [Google Scholar] [CrossRef]
- Benhamou, Y.; Picco, V.; Raybaud, H.; Sudaka, A.; Chamorey, E.; Brolih, S.; Monteverde, M.; Merlano, M.; Nigro, C.L.; Ambrosetti, D.; et al. Telomeric repeat-binding factor 2: A marker for survival and anti-EGFR efficacy in oral carcinoma. Oncotarget 2016, 7, 44236–44251. [Google Scholar] [CrossRef] [PubMed]
- Cherfils-Vicini, J.; Zizza, P.; Gilson, E.; Biroccio, A. A novel pathway links telomeres to NK-cell activity. OncoImmunology 2014, 3, e27358. [Google Scholar] [CrossRef]
- Cherfils-Vicini, J.; Gilson, E. Inhibiting TRF 1 upstream signaling pathways to target telomeres in cancer cells. EMBO Mol. Med. 2019, 11, 11. [Google Scholar] [CrossRef]
- Zizza, P.; Dinami, R.; Porru, M.; Cingolani, C.; Salvati, E.; Rizzo, A.; D’Angelo, C.; Petti, E.; Amoreo, C.A.; Mottolese, M.; et al. TRF2 positively regulates SULF2 expression increasing VEGF-A release and activity in tumor microenvironment. Nucleic Acids Res. 2019, 47, 3365–3382. [Google Scholar] [CrossRef] [PubMed]
- Cherfils-Vicini, J.; Iltis, C.; Cervera, L.; Pisano, S.; Croce, O.; Sadouni, N.; Győrffy, B.; Collet, R.; Renault, V.M.; Rey-Millet, M.; et al. Cancer cells induce immune escape via glycocalyx changes controlled by the telomeric protein TRF 2. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Di Maro, S.; Zizza, P.; Salvati, E.; DE Luca, V.; Capasso, C.; Fotticchia, I.; Pagano, B.; Marinelli, L.; Gilson, E.; Novellino, E.; et al. Shading the TRF2 Recruiting Function: A New Horizon in Drug Development. J. Am. Chem. Soc. 2014, 136, 16708–16711. [Google Scholar] [CrossRef]
- Roisin, A.; Buchsbaum, S.; Mocquet, V.; Jalinot, P. The fluorescent protein stability assay: An efficient method for monitoring intracellular protein stability. Biotechniques 2021, 70, btn-2021-0032. [Google Scholar] [CrossRef]
- Su, C.-H.; Chu, W.-C.; Lan, K.-H.; Li, C.-P.; Chao, Y.; Lin, H.-C.; Lee, S.-D.; Tsai, Y.-C.; Lee, W.-P. Gemcitabine causes telomere attrition by stabilizing TRF2. Eur. J. Cancer 2012, 48, 3465–3474. [Google Scholar] [CrossRef]
- Fujita, K.; Horikawa, I.; Mondal, A.M.; Jenkins, L.M.M.; Appella, E.; Vojtesek, B.; Bourdon, J.-C.; Lane, D.P.; Harris, C.C. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat. Cell Biol. 2010, 12, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Shakoori, A.; Mai, W.; Miyashita, K.; Yasumoto, K.; Takahashi, Y.; Ooi, A.; Kawakami, K.; Minamoto, T. Inhibition of GSK-3β activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 2007, 98, 1388–1393. [Google Scholar] [CrossRef]
- Kenny, H.A.; Lal-Nag, M.; White, E.A.; Shen, M.; Chiang, C.-Y.; Mitra, A.K.; Zhang, Y.; Curtis, M.W.; Schryver, E.M.; Bettis, S.; et al. Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.-C.; Jerning, E.; Markgren, P.-O.; Borgegård, T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [PubMed]
- Diala, I.; Wagner, N.; Magdinier, F.; Shkreli, M.; Sirakov, M.; Bauwens, S.; Schluth-Bolard, C.; Simonet, T.; Renault, V.M.; Ye, J.; et al. Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep. 2013, 14, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.-D.; Ying, Y.; Leong, W.; Jiang, J.; Hu, X.; Chen, Y.; Michiels, J.-F.; Lu, Y.; Gilson, E.; Wagner, N.; et al. The differential spatiotemporal expression pattern of shelterin genes throughout lifespan. Aging 2017, 9, 1219–1232. [Google Scholar] [CrossRef]
- Robin, J.D.; Burbano, M.J.; Peng, H.; Croce, O.; Thomas, J.L.; Laberthonniere, C.; Renault, V.; Lototska, L.; Pousse, M.; Tessier, F.; et al. Mitochondrial function in skeletal myofibers is controlled by a TRF2-SIRT3 axis over lifetime. Aging Cell 2020, 19, e13097. [Google Scholar] [CrossRef]
- Muñoz, P.; Blanco, R.; Flores, J.M.; Blasco, M.A. XPF nuclease-dependent telomere loss and increased DNA damage in mice overexpressing TRF2 result in premature aging and cancer. Nat. Genet. 2005, 37, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.M.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.G.; Walker, A.E.; Trott, D.W.; Machin, D.R.; Henson, G.D.; Reihl, K.D.; Cawthon, R.M.; Denchi, E.L.; Liu, Y.; Bloom, S.I.; et al. Induced Trf2 deletion leads to aging vascular phenotype in mice associated with arterial telomere uncapping, senescence signaling, and oxidative stress. J. Mol. Cell. Cardiol. 2019, 127, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.J.; Gray, K.; Kumar, S.; Clarke, M.C.H.; Bennett, M.R. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Specificity | Company | Clone | Species | Isotype | Fluorochrome | Reference |
|---|---|---|---|---|---|---|
| anti CD107a FITC | BD Biosciences | 1D4B | Rat | IgG2a/k | FITC | 553793 |
| anti CD11b | BD Biosciences | M1/70 | Rat | IgG2b | APC-H7 | 550993 |
| Anti CD69 PE-Cy7 | BD Biosciences | H1.2F3 | Hamster | IgG1/K | PE-Cy7 | 552879 |
| Anti NKp46 CD335 | BD Biosciences | 29A1.4 | Rat | IgG2a | Alexa 647 | 560755 |
| Anti-CD3e | Biolegend | 145-C11 | Armenin Hamster | IgG | PerCP | 100302 |
| Anti-CD45 | BD Biosciences | 30-F11 | Rat | IgG2b, κ | AF700 | 560510 |
| Anti-F4/80 | Biolegend | BM8 | Rat | IgG2a, κ | BV510 | 123135 |
| Anti-Ly6C/Ly6G (Gr-1) | BD Biosciences | RB6-8C5 | Rat | IgG2b, κ | PE | 553128 |
| anti-CD31 | BD Biosciences | MEC 13.3 | Rat | IgG2a, κ | BV421 | 562939 |
| TRF2 | Imgenex | 4A794.15 | Mouse | IgG1, κ | N.A. | IMG-124A |
| Beta-Actin | Abcam | Rabbit | IgG | N.A. | Ab8227 | |
| Anti-mouse | Licor | Goat | IgG | IRDye 680 | 926-32220 | |
| Anti-Rabbit | Licor | Goat | IgG | IRDye 800CW | 926-32211 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Maï, M.; Janho dit Hreich, S.; Gaggioli, C.; Roisin, A.; Wagner, N.; Ye, J.; Jalinot, P.; Cherfils-Vicini, J.; Gilson, E. A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth. Cancers 2021, 13, 2998. https://doi.org/10.3390/cancers13122998
El Maï M, Janho dit Hreich S, Gaggioli C, Roisin A, Wagner N, Ye J, Jalinot P, Cherfils-Vicini J, Gilson E. A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth. Cancers. 2021; 13(12):2998. https://doi.org/10.3390/cancers13122998
Chicago/Turabian StyleEl Maï, Mounir, Serena Janho dit Hreich, Cedric Gaggioli, Armelle Roisin, Nicole Wagner, Jing Ye, Pierre Jalinot, Julien Cherfils-Vicini, and Eric Gilson. 2021. "A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth" Cancers 13, no. 12: 2998. https://doi.org/10.3390/cancers13122998
APA StyleEl Maï, M., Janho dit Hreich, S., Gaggioli, C., Roisin, A., Wagner, N., Ye, J., Jalinot, P., Cherfils-Vicini, J., & Gilson, E. (2021). A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth. Cancers, 13(12), 2998. https://doi.org/10.3390/cancers13122998