
Next Generation Cytogenetics in Myeloid Hematological Neoplasms: Detection of CNVs and Translocations

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. NGS Analysis
2.3. Conventional Cytogenetic Analysis and MLPA
3. Results
3.1. Sequencing Coverage and Quality
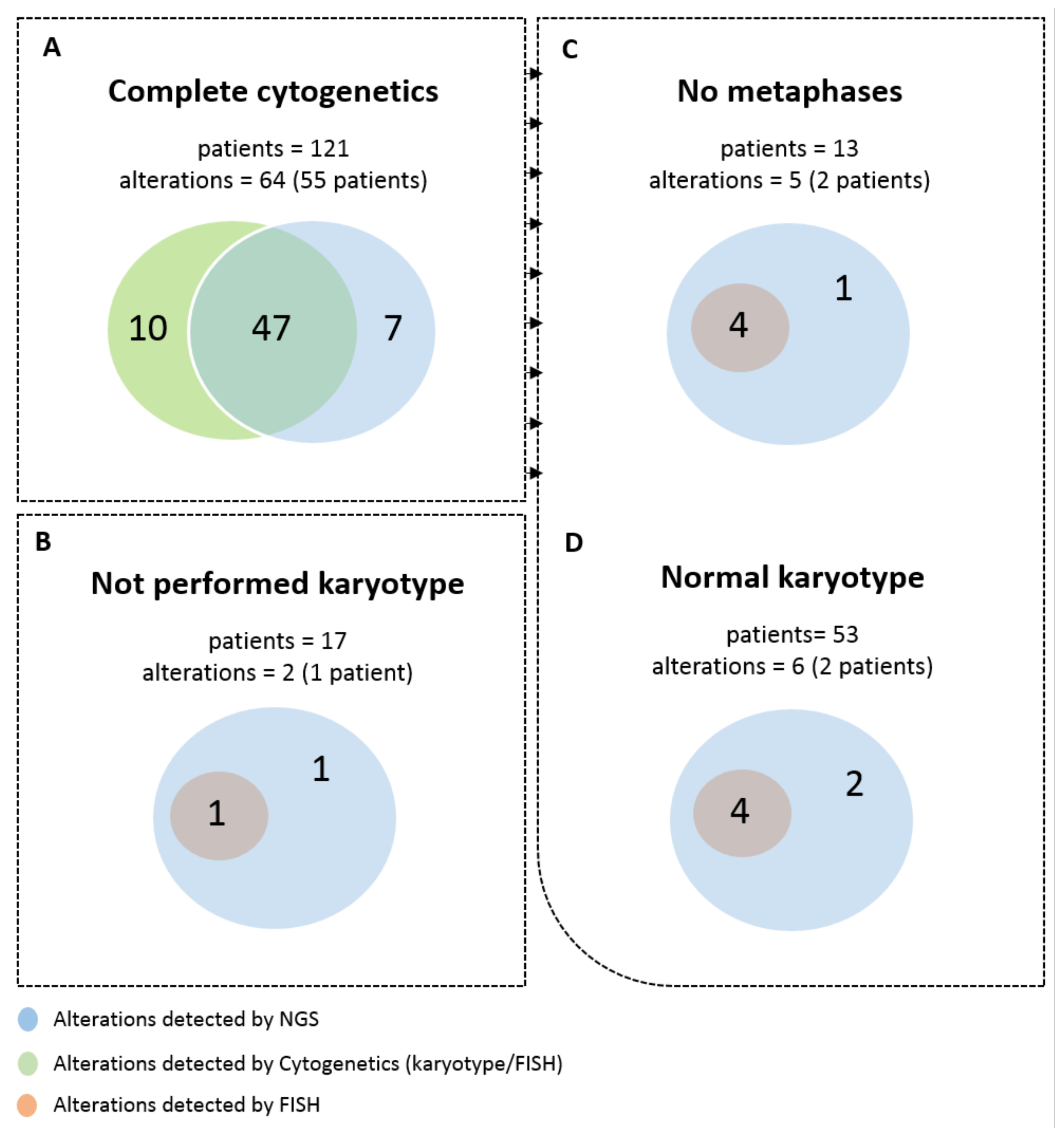
3.2. Detection of Frequent Alterations by Cytogenetics and/or NGS
3.3. Secondary CNV Detected by NGS
3.4. Secondary CNVs Detected by Karyotype
3.5. Possible Impact of CNV Detection by NGS in Prognosis Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Rack, K.A.; van den Berg, E.; Haferlach, C.; Beverloo, H.B.; Costa, D.; Espinet, B.; Foot, N.; Jeffries, S.; Martin, K.; O’Connor, S.; et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia 2019, 33, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, P.R.; Mikhail, F.M. Diagnostic and Prognostic Utility of Fluorescence In situ Hybridization (FISH) Analysis in Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 568–573. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Hodge, J.C.; Tucker, T.; Shetty, S.; Yenamandra, A.; Dixon-McIver, A.; Bryke, C.; Huxley, E.; Lennon, P.A.; Raca, G.; et al. Assessing copy number aberrations and copy neutral loss of heterozygosity across the genome as best practice: An evidence based review of clinical utility from the cancer genomics consortium (CGC) working group for myelodysplastic syndrome, myelodysplastic/myeloproliferative and myeloproliferative neoplasms. Cancer Genet. 2018, 228–229, 197–217. [Google Scholar] [CrossRef]
- Mehrotra, M.; Luthra, R.; Ravandi, F.; Sargent, R.L.; Barkoh, B.A.; Abraham, R.; Mishra, B.M.; Jeffrey Medeiros, L.; Patel, K.P. Identification of clinically important chromosomal aberrations in acute myeloid leukemia by array-based comparative genomic hybridization. Leuk. Lymphoma 2014, 55, 2538–2548. [Google Scholar] [CrossRef][Green Version]
- Zahir, F.R.; Marra, M.A. Use of Affymetrix Arrays in the Diagnosis of Gene Copy-Number Variation. Curr. Protoc. Hum. Genet. 2015, 85, 8–13. [Google Scholar] [CrossRef]
- Praulich, I.; Tauscher, M.; Göhring, G.; Glaser, S.; Hofmann, W.; Feurstein, S.; Flotho, C.; Lichter, P.; Niemeyer, C.M.; Schlegelberger, B.; et al. Clonal heterogeneity in childhood myelodysplastic syndromes-challenge for the detection of chromosomal imbalances by Array-CGH. Genes Chromosom. Cancer 2010, 49, 885–900. [Google Scholar] [CrossRef]
- Rabin, K.R.; Man, T.K.; Yu, A.; Folsom, M.R.; Zhao, Y.J.; Rao, P.H.; Plon, S.E.; Naeem, R.C. Clinical utility of array comparative genomic hybridization for detection of chromosomal abnormalities in pediatric acute lymphoblastic leukemia. Pediatr. Blood Cancer 2008, 51, 171–177. [Google Scholar] [CrossRef]
- Tallman, M.S.; Wang, E.S.; Altman, J.K.; Appelbaum, F.R.; Bhatt, V.R.; Bixby, D.; Coutre, S.E.; De Lima, M.; Fathi, A.T.; Fiorella, M.; et al. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 721–749. [Google Scholar] [CrossRef] [PubMed]
- Duncavage, E.J.; Schroeder, M.C.; O’Laughlin, M.; Wilson, R.; MacMillan, S.; Bohannon, A.; Kruchowski, S.; Garza, J.; Du, F.; Hughes, A.E.O.; et al. Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers. N. Engl. J. Med. 2021, 384, 924–935. [Google Scholar] [CrossRef]
- Mack, E.K.M.; Marquardt, A.; Langer, D.; Ross, P.; Ultsch, A.; Kiehl, M.G.; Mack, H.I.D.; Haferlach, T.; Neubauer, A.; Brendel, C. Comprehensive genetic diagnosis of acute myeloid leukemia by next-generation sequencing. Haematologica 2019, 104, 277–287. [Google Scholar] [CrossRef]
- McKerrell, T.; Moreno, T.; Ponstingl, H.; Bolli, N.; Dias, J.M.L.; Tischler, G.; Colonna, V.; Manasse, B.; Bench, A.; Bloxham, D.; et al. Development and validation of a comprehensive genomic diagnostic tool for myeloid malignancies. Blood 2016, 128, E1–E9. [Google Scholar] [CrossRef]
- Yao, R.; Yu, T.; Qing, Y.; Wang, J.; Shen, Y. Evaluation of copy number variant detection from panel-based next-generation sequencing data. Mol. Genet. Genomic Med. 2019, 7. [Google Scholar] [CrossRef]
- Kluk, M.J.; Lindsley, R.C.; Aster, J.C.; Lindeman, N.I.; Szeto, D.; Hall, D.; Kuo, F.C. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J. Mol. Diagn. 2016, 18, 507–515. [Google Scholar] [CrossRef]
- Vosberg, S.; Herold, T.; Hartmann, L.; Neumann, M.; Opatz, S.; Metzeler, K.H.; Schneider, S.; Graf, A.; Krebs, S.; Blum, H.; et al. Close correlation of copy number aberrations detected by next-generation sequencing with results from routine cytogenetics in acute myeloid leukemia. Genes Chromosom. Cancer 2016, 55, 553–567. [Google Scholar] [CrossRef]
- Neumann, M.; Vosberg, S.; Schlee, C.; Heesch, S.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Graf, A.; Krebs, S.; Bartram, I.; et al. Mutational spectrum of adult T-ALL. Oncotarget 2015, 6, 2754–2766. [Google Scholar] [CrossRef]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; Van De Ven, C.; De Groot-Kruseman, H.A.; De Haas, V.; et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Project, G.; et al. The Sequence Alignment/Map format and SAMtools. Bioinform. Appl. Note 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Picard Tools—By Broad Institute. Available online: https://broadinstitute.github.io/picard/ (accessed on 1 June 2021).
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- International Standing Committee on Human Cytogenomic Nomenclature; McGowan-Jordan, J.; Simons, A.; Schmid, M. ISCN: An International System for Human Cytogenomic Nomenclature; Karger: Basel, Switzerland, 2016; ISBN 9783318058574. [Google Scholar]
- Carbonell, D.; Suárez-González, J.; Chicano, M.; Andrés-Zayas, C.; Triviño, J.C.; Rodríguez-Macías, G.; Bastos-Oreiro, M.; Font, P.; Ballesteros, M.; Muñiz, P.; et al. Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms. Cancers 2019, 11, 1364. [Google Scholar] [CrossRef]
- Maserati, E.; Aprili, F.; Vinante, F.; Locatelli, F.; Amendola, G.; Zatterale, A.; Milone, G.; Minelli, A.; Bernardi, F.; Lo Curto, F.; et al. Trisomy 8 in myelodysplasia and acute leukemia is constitutional in 15–20% of cases. Genes Chromosom. Cancer 2002, 33, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Saumell, S.; Solé, F.; Arenillas, L.; Montoro, J.; Valcárcel, D.; Pedro, C.; Sanzo, C.; Luño, E.; Giménez, T.; Arnan, M.; et al. Trisomy 8, a cytogenetic abnormality in myelodysplastic syndromes, is constitutional or not? PLoS ONE 2015, 10, e0129375. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Lyon, France, 2017; p. 585. [Google Scholar]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Lasho, T.L.; Gangat, N.; Begna, K.H.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A. Revised cytogenetic risk stratification in primary myelofibrosis: Analysis based on 1002 informative patients. Leukemia 2018, 32, 1189–1199. [Google Scholar] [CrossRef]
- Drevon, L.; Marceau, A.; Maarek, O.; Cuccuini, W.; Clappier, E.; Eclache, V.; Cluzeau, T.; Richez, V.; Berkaoui, I.; Dimicoli-Salazar, S.; et al. Myelodysplastic syndrome (MDS) with isolated trisomy 8: A type of MDS frequently associated with myeloproliferative features? A report by the Groupe Francophone des Myélodysplasies. Br. J. Haematol. 2018, 182, 843–850. [Google Scholar] [CrossRef]
- Such, E.; Cervera, J.; Costa, D.; Solé, F.; Vallespí, T.; Luño, E.; Collado, R.; Calasanz, M.J.; Hernández-Rivas, J.M.; Cigudosa, J.C.; et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica 2011, 96, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Braulke, F.; Müller-Thomas, C.; Götze, K.; Platzbecker, U.; Germing, U.; Hofmann, W.-K.; Giagounidis, A.A.N.; Lübbert, M.; Greenberg, P.L.; Bennett, J.M.; et al. Frequency of del(12p) is commonly underestimated in myelodysplastic syndromes: Results from a German diagnostic study in comparison with an international control group. Genes Chromosom. Cancer 2015, 54, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Zhang, C.R.C.; Meyer, C.; Stinson, C.L.; Pham, T.; Bruxner, T.J.C.; Venn, N.C.; Trahair, T.N.; Sutton, R.; Marschalek, R.; et al. Targeted Next-Generation Sequencing for Detecting MLL Gene Fusions in Leukemia. Mol. Cancer Res. 2018, 16, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Shumilov, E.; Joncourt, R.; Porret, N.; Tchinda, J.; Legros, M.; Scarpelli, I.; Hewer, E.; Novak, U.; Schoumans, J.; et al. Detection of rare reciprocal RUNX1 rearrangements by next-generation sequencing in acute myeloid leukemia. Genes Chromosom. Cancer 2020, 59, 268–274. [Google Scholar] [CrossRef]
- Eisfeld, A.-K.; Kohlschmidt, J.; Mrózek, K.; Blachly, J.S.; Nicolet, D.; Kroll, K.; Orwick, S.; Carroll, A.J.; Stone, R.M.; de la Chapelle, A.; et al. Adult acute myeloid leukemia with trisomy 11 as the sole abnormality is characterized by the presence of five distinct gene mutations: MLL-PTD, DNMT3A, U2AF1, FLT3-ITD and IDH2. Leukemia 2016, 30, 2254–2258. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, K.; MRÓzek, K.; Lawrence, D.; Arthur, D.C.; Pettenati, M.J.; Stamberg, J.; Qumsiyeh, M.B.; Verma, R.S.; Maccallum, J.; Schiffer, C.A.; et al. Clinical characteristics of patients with de novo acute myeloid leukaemia and isolated trisomy 11: A Cancer and Leukemia Group B study. Br. J. Haematol. 1998, 101, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Strati, P.; Daver, N.; Ravandi, F.; Pemmaraju, N.; Pierce, S.; Garcia-Manero, G.; Nazha, A.; Kadia, T.; Jabbour, E.; Borthakur, G.; et al. Biological and clinical features of trisomy 21 in adult patients with acute myeloid leukemia. Clin. Lymphoma. Myeloma Leuk. 2013, 13 (Suppl. S2), S276–S281. [Google Scholar] [CrossRef]
- Cavalcante de Andrade Silva, M.; Krepischi, A.C.V.; Kulikowski, L.D.; Zanardo, E.A.; Nardinelli, L.; Leal, A.M.; Costa, S.S.; Muto, N.H.; Rocha, V.; Velloso, E.D.R.P. Deletion of RUNX1 exons 1 and 2 associated with familial platelet disorder with propensity to acute myeloid leukemia. Cancer Genet. 2018, 222–223, 32–37. [Google Scholar] [CrossRef]
- Vormittag-Nocito, E.; Ni, H.; Schmidt, M.L.; Lindgren, V. Thrombocytopenia and Predisposition to Acute Myeloid Leukemia due to Mosaic Ring 21 with Loss of RUNX1: Cytogenetic and Molecular Characterization. Mol. Syndromol. 2019, 9, 306–311. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Gur, H.D.; Wang, S.A.; Tang, Z.; Hu, S.; Li, S.; Medeiros, L.J.; Tang, G. Clinical significance of isolated del(7p) in myeloid neoplasms. Leuk. Res. 2017, 55, 18–22. [Google Scholar] [CrossRef]
- Boudry-Labis, E.; Roche-Lestienne, C.; Nibourel, O.; Boissel, N.; Terre, C.; Perot, C.; Eclache, V.; Gachard, N.; Tigaud, I.; Plessis, G.; et al. Neurofibromatosis-1 gene deletions and mutations in de novo adult acute myeloid leukemia. Am. J. Hematol. 2013, 88, 306–311. [Google Scholar] [CrossRef]
- Haferlach, C.; Grossmann, V.; Kohlmann, A.; Schindela, S.; Kern, W.; Schnittger, S.; Haferlach, T. Deletion of the tumor-suppressor gene NF1 occurs in 5% of myeloid malignancies and is accompanied by a mutation in the remaining allele in half of the cases. Leukemia 2012, 26, 834–839. [Google Scholar] [CrossRef]
- King-Underwood, L.; Pritchard-Jones, K.; Zimmermann, M.; Balgobind, B.V.; Arentsen-Peters, S.T.C.J.M.; Alders, M.; Willasch, A.; Kaspers, G.J.L.; Trka, J.; Baruchel, A.; et al. Wilms’ Tumor (WT1) Gene Mutations Occur Mainly in Acute Myeloid Leukemia and May Confer Drug Resistance. Blood 2009, 91, 2961–2968. [Google Scholar] [CrossRef]
- Xiang, Z.; Abdallah, A.-O.; Govindarajan, R.; Mehta, P.; Emanuel, P.D.; Papenhausen, P.; Schichman, S.A. MYC amplification in multiple marker chromosomes and EZH2 microdeletion in a man with acute myeloid leukemia. Cancer Genet. 2015, 208, 96–100. [Google Scholar] [CrossRef]
- Ning, Y.; Slovak, M.L.; Schultz, R.A.; Gojo, I.; Baer, M.R. Cryptic chromosome abnormalities in a patient with mixed phenotype acute leukemia. Leuk. Lymphoma 2014, 55, 680–682. [Google Scholar] [CrossRef][Green Version]
- Tosello, V.; Mansour, M.R.; Barnes, K.; Paganin, M.; Sulis, M.L.; Jenkinson, S.; Allen, C.G.; Gale, R.E.; Linch, D.C.; Palomero, T.; et al. WT1 mutations in T-ALL. Blood 2009, 114, 1038–1045. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| % of the Alteration by FISH | |||||
|---|---|---|---|---|---|
| PN | Disease | Alteration | Method | Non-Cultured Sample | Cultured Sample |
| 3 | MDS | Monosomy 7 | FISH * | 90% * | NA |
| 12 | MDS/MPN | del(12p) | Karyotype (2/20) | 3.6% | 6.4% |
| 14 | MDS | Trisomy 8 | Karyotype (7/20) | 10.4% | 15.2% |
| 17 | AML | inv(16) | Karyotype (2/6) | 20% | 30% |
| 46 | AML | Trisomy 8 | Karyotype (4/10) | 7.2% | 9.8% |
| 56 | MDS | Trisomy 8 | Karyotype (3/20) | 9.8% | 13.8% |
| 64 | AML | Trisomy 8 | Karyotype (5/11) | 5.4% | 5.2% |
| 72 | MDS | Trisomy 8 | Karyotype (3/13) | 18.4% | 22.4% |
| 97 | MPAL | del(5q) | FISH | 20.50% | 21% |
| 101 | MDS | Trisomy 8 | Karyotype (2/20) | 5% | 7.8% |
| PN | Disease | Alteration Detected by NGS | Karyotype | FISH | Method of Validation |
|---|---|---|---|---|---|
| 23 | AML | del(12p) | 46,XY,t(1;10)(p36;q11),inv(2)(q31q37)(14)/46,XY(6) | NP | MLPA |
| 36 | MPN | Trisomy 19 | NP | del(20q) | MLPA |
| 47 | MPAL | del(5q) | 46,XY(20) | NP | MLPA |
| 49 | MDS | del(12p) | 46,XY,add(7)(q22)(3)/46,XY(9) | del(7q) | MLPA |
| 55 | AML | t(10;11) | 47,X,t(Y;15)(q11;p11),+8,inv(12)(q13q15)(19)/46,XY(1) | MLL rearranged | RT-QPCR |
| 60 | AML | del(12p) | NM | del(7q),del(5q),del(17p), loss of MECOM/RPN1 | MLPA |
| 109 | AML | del(12p) | 43,XX,−1,del(2)(p11),−3,der(3)t(1;3)(p11;p23),−5,−7, add(11)(p14),add(19)(q13),+mar(cp15)/46,XX(5) | −7, del(5q) | MLPA |
| 110 | MDS | del(12p) | 46,XX(15) | NP | MLPA |
| 123 | MDS | del(12p) | 43,XY,del(3)(p13),−5,−7,add(12)(p13),−20, dmin(7)/46,XY(13) | NP | MLPA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chicano, M.; Carbonell, D.; Suárez-González, J.; Lois, S.; Ballesteros-Culebras, M.; Andrés-Zayas, C.; Muñiz, P.; Rodríguez-Macias, G.; Bastos-Oreiro, M.; Font, P.; et al. Next Generation Cytogenetics in Myeloid Hematological Neoplasms: Detection of CNVs and Translocations. Cancers 2021, 13, 3001. https://doi.org/10.3390/cancers13123001
Chicano M, Carbonell D, Suárez-González J, Lois S, Ballesteros-Culebras M, Andrés-Zayas C, Muñiz P, Rodríguez-Macias G, Bastos-Oreiro M, Font P, et al. Next Generation Cytogenetics in Myeloid Hematological Neoplasms: Detection of CNVs and Translocations. Cancers. 2021; 13(12):3001. https://doi.org/10.3390/cancers13123001
Chicago/Turabian StyleChicano, María, Diego Carbonell, Julia Suárez-González, Sergio Lois, Mercedes Ballesteros-Culebras, Cristina Andrés-Zayas, Paula Muñiz, Gabriela Rodríguez-Macias, Mariana Bastos-Oreiro, Patricia Font, and et al. 2021. "Next Generation Cytogenetics in Myeloid Hematological Neoplasms: Detection of CNVs and Translocations" Cancers 13, no. 12: 3001. https://doi.org/10.3390/cancers13123001
APA StyleChicano, M., Carbonell, D., Suárez-González, J., Lois, S., Ballesteros-Culebras, M., Andrés-Zayas, C., Muñiz, P., Rodríguez-Macias, G., Bastos-Oreiro, M., Font, P., Ballesteros, M., Kwon, M., Anguita, J., Díez-Martín, J. L., Buño, I., & Martínez-Laperche, C. (2021). Next Generation Cytogenetics in Myeloid Hematological Neoplasms: Detection of CNVs and Translocations. Cancers, 13(12), 3001. https://doi.org/10.3390/cancers13123001