
Transcriptomic and Genetic Associations between Alzheimer’s Disease, Parkinson’s Disease, and Cancer

 , ,
, ,  , ,
, ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Differential Gene Expression Meta-Analyses, Gene Set Enrichment Analyses, Weighted Co-Expression Network Analyses, and Measures of Transcriptomic Association between NDG Disorders and Cancer
2.2. Human Interactome-Based Overlaps and Cross-Trait LD Score Regression Analyses
2.3. Identification of Drugs Indicated for the Treatment of AD, PD, and Cancer as Potential Modulators of their Comorbidities through LINCS L1000 Analysis
3. Results
3.1. Results of Differential Gene Expression Meta-Analyses
3.2. Transcriptomic Associations between Neurodegenerative Disorders and Cancers
3.2.1. Transcriptomic Associations between AD and Cancer
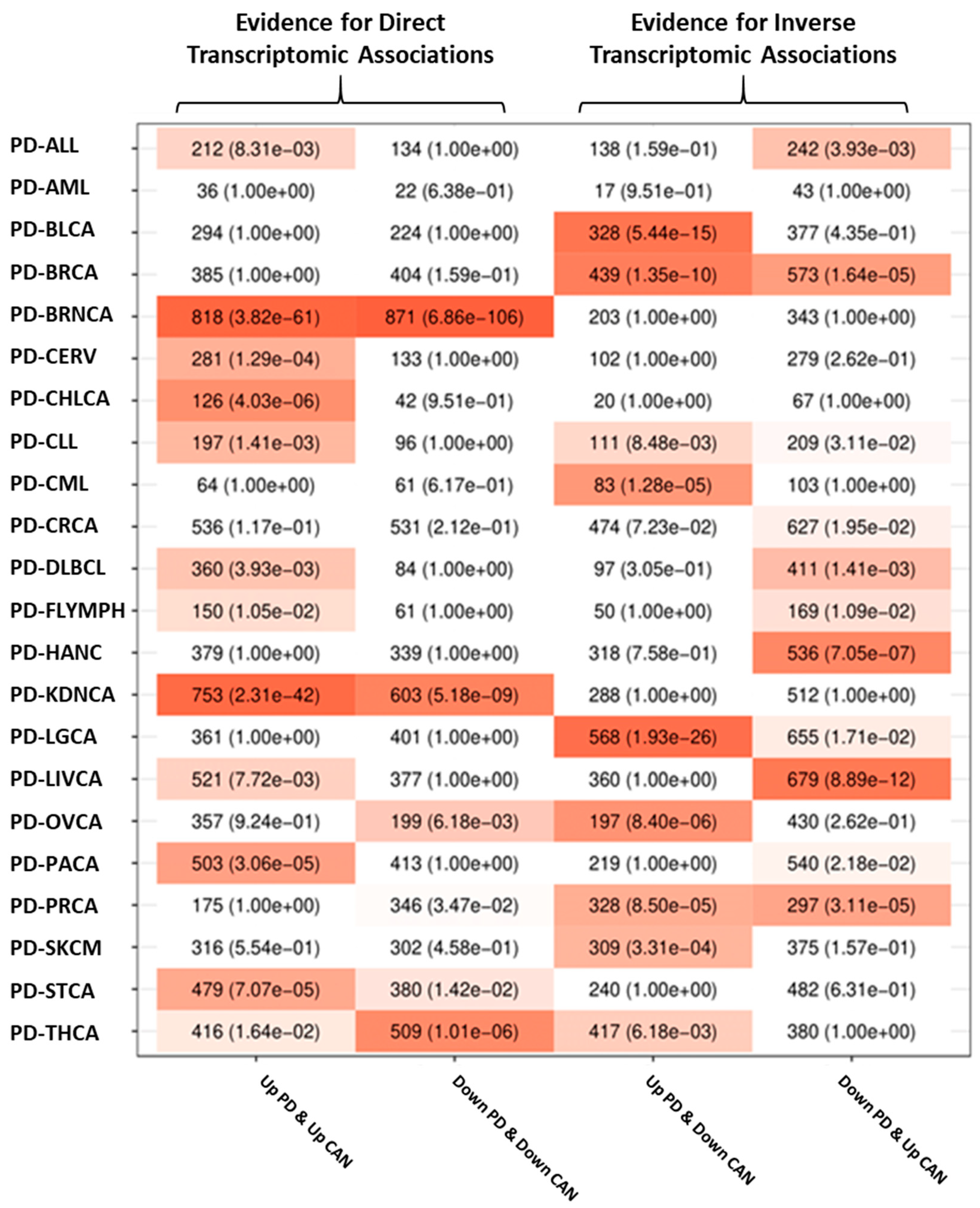
3.2.2. Transcriptomic Associations between PD and Cancer
3.3. Validation of the Intersection Analyses Using an Alternative Set of Cancer Data
3.4. Pathways and Consensus Co-Expression Modules Deregulated in Both Neurodegenerative Disorders and Cancer
3.4.1. Gene Set Enrichment Analyses (GSEA) Results
3.4.2. Consensus Weighted Gene Co-Expression Network Analysis (WGCNA) Analysis Results
3.5. Interactome-Based Overlaps and Genetic Correlation Analysis Results
3.5.1. Interactome-Based Overlap Analyses Results
3.5.2. Cross-Trait LD Score Analyses Results
3.6. Transcriptomic Effects of Drugs Indicated for the Treatment of Neurodegenerative Disorders and Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catala-Lopez, F.; Hutton, B.; Driver, J.A.; Page, M.J.; Ridao, M.; Valderas, J.M.; Alonso-Arroyo, A.; Fores-Martos, J.; Martinez, S.; Genova-Maleras, R.; et al. Cancer and central nervous system disorders: Protocol for an umbrella review of systematic reviews and updated meta-analyses of observational studies. Syst. Rev. 2017, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Catala-Lopez, F.; Suarez-Pinilla, M.; Suarez-Pinilla, P.; Valderas, J.M.; Gomez-Beneyto, M.; Martinez, S.; Balanza-Martinez, V.; Climent, J.; Valencia, A.; McGrath, J.; et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: A meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother. Psychosom. 2014, 83, 89–105. [Google Scholar] [CrossRef]
- Catala-Lopez, F.; Genova-Maleras, R.; Vieta, E.; Tabares-Seisdedos, R. The increasing burden of mental and neurological disorders. Eur. Neuropsychopharmacol. 2013, 23, 1337–1339. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.N. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar]
- Diseases, G.B.D.C. Injuries, Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar]
- Roe, C.M.; Behrens, M.I.; Xiong, C.; Miller, J.P.; Morris, J.C. Alzheimer disease and cancer. Neurology 2005, 64, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.M.; Fitzpatrick, A.L.; Xiong, C.; Sieh, W.; Kuller, L.; Miller, J.P.; Williams, M.M.; Kopan, R.; Behrens, M.I.; Morris, J.C. Cancer linked to Alzheimer disease but not vascular dementia. Neurology 2010, 74, 106–112. [Google Scholar] [CrossRef]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef]
- Musicco, M.; Adorni, F.; Di Santo, S.; Prinelli, F.; Pettenati, C.; Caltagirone, C.; Palmer, K.; Russo, A. Inverse occurrence of cancer and Alzheimer disease: A population-based incidence study. Neurology 2013, 81, 322–328. [Google Scholar] [CrossRef]
- Ou, S.-M.; Lee, Y.-J.; Hu, Y.-W.; Liu, C.-J.; Chen, T.-J.; Fuh, J.-L.; Wang, S.-J. Does Alzheimer’s Disease Protect against Cancers? A Nationwide Population-Based Study. Neuroepidemiology 2013, 40, 42–49. [Google Scholar] [CrossRef]
- Freedman, D.M.; Wu, J.; Chen, H.; A Engels, E.; Enewold, L.R.; Freedman, N.D.; Goedert, J.J.; Kuncl, R.W.; Gail, M.H.; Pfeiffer, R.M. Associations between cancer and Parkinson’s disease in U.S. elderly adults. Int. J. Epidemiol. 2016, 45, 741–751. [Google Scholar] [CrossRef]
- Beard, C.M.; Kokmen, E.; Sigler, C.; Smith, G.E.; Petterson, T.; O’Brien, P.C. Cause of death in Alzheimer’s disease. Ann. Epidemiol. 1996, 6, 195–200. [Google Scholar] [CrossRef]
- Ganguli, M.; Dodge, H.H.; Shen, C.; Pandav, R.S.; DeKosky, S.T. Alzheimer disease and mortality: A 15-year epidemiological study. Arch. Neurol. 2005, 62, 779–784. [Google Scholar] [CrossRef]
- Chamandy, N.; Wolfson, C. Underlying cause of death in demented and non-demented elderly Canadians. Neuroepidemiology 2005, 25, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.P.; Benito-Leon, J.; Louis, E.D.; Bermejo-Pareja, F. Alzheimer’s disease is associated with decreased risk of cancer-specific mortality: A prospective study (NEDICES). J. Alzheimers Dis. 2014, 40, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, A.; Joly, J.G. Parkinsonism and cancer. Union Med. Can. 1963, 92, 169–174. [Google Scholar] [PubMed]
- Jansson, B.; Jankovic, J. Low cancer rates among patients with Parkinson’s disease. Ann. Neurol. 1985, 17, 505–509. [Google Scholar] [CrossRef]
- Guttman, M.; Slaughter, P.M.; Theriault, M.E.; DeBoer, D.P.; Naylor, C.D. Parkinsonism in Ontario: Comorbidity associated with hospitalization in a large cohort. Mov. Disord. 2004, 19, 49–53. [Google Scholar] [CrossRef]
- Becker, C.; Brobert, G.P.; Johansson, S.; Jick, S.S.; Meier, C.R. Cancer risk in association with Parkinson disease: A population-based study. Parkinsonism Relat. Disord. 2010, 16, 186–190. [Google Scholar] [CrossRef]
- Fois, A.F.; Wotton, C.J.; Yeates, D.; Turner, M.R.; Goldacre, M.J. Cancer in patients with motor neuron disease, multiple sclerosis and Parkinson’s disease: Record linkage studies. J. Neurol. Neurosurg. Psychiatry 2010, 81, 215–221. [Google Scholar] [CrossRef]
- Rugbjerg, K.; Friis, S.; Lassen, C.F.; Ritz, B.; Olsen, J.H. Malignant melanoma, breast cancer and other cancers in patients with Parkinson’s disease. Int. J. Cancer 2012, 131, 1904–1911. [Google Scholar] [CrossRef]
- Ong, E.L.; Goldacre, R.; Goldacre, M. Differential risks of cancer types in people with Parkinson’s disease: A national record-linkage study. Eur. J. Cancer 2014, 50, 2456–2462. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Weibull, C.E.; Chen, H.; Kamel, F.; Lundholm, C.; Fang, F.; Ye, W. Parkinson’s disease and cancer: A register-based family study. Am. J. Epidemiol. 2014, 179, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Lin, G.-M.; Wu, Y.-C.; Tseng, W.-S.; Kuo, D.-J.; Chu, C.-I.; Li, Y.-H. Does Parkinsons Disease Dementia Reduce Cancer Risk more than Alzheimers Disease Alone? Neuropsychiatry 2018, 7, 354–361. [Google Scholar] [CrossRef]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression and mortality. Neurology 1967, 17, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Koretz, D.; Merikangas, K.R.; Rush, A.J.; Walters, E.E.; Wang, P.S. National Comorbidity Survey, The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003, 289, 3095–3105. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A. Parkinson’s disease and its comorbid disorders: An analysis of Michigan mortality data, 1970 to 1990. Neurology 1994, 44, 1865–1868. [Google Scholar] [CrossRef] [PubMed]
- Vanacore, N.; Spila-Alegiani, S.; Raschetti, R.; Meco, G. Mortality cancer risk in parkinsonian patients: A population-based study. Neurology 1999, 52, 395–398. [Google Scholar] [CrossRef]
- Shi, H.B.; Tang, B.; Liu, Y.W.; Wang, X.F.; Chen, G.J. Alzheimer disease and cancer risk: A meta-analysis. J. Cancer Res. Clin. Oncol. 2015, 141, 485–494. [Google Scholar] [CrossRef]
- Tabares-Seisdedos, R.; Rubenstein, J.L. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: Implications for schizophrenia, autism and cancer. Mol. Psychiatry 2009, 14, 563–589. [Google Scholar] [CrossRef]
- Tabarés-Seisdedos, R.; Dumont, N.; Baudot, A.; Valderas, J.M.; Climent, J.; Valencia, A.; Crespo-Facorro, B.; Vieta, E.; Gómez-Beneyto, M.; Martinez, S.; et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol. 2011, 12, 604–608. [Google Scholar] [CrossRef]
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013, 14, 293–304. [Google Scholar] [CrossRef]
- Freedman, D.M.; Wu, J.; Chen, H.; Kuncl, R.W.; Enewold, L.R.; Engels, E.A.; Freedman, N.D.; Pfeiffer, R.M. Associations between cancer and Alzheimer’s disease in a U.S. Medicare population. Cancer Med. 2016, 5, 2965–2976. [Google Scholar] [CrossRef]
- Peretz, C.; Gurel, R.; Rozani, V.; Gurevich, T.; El-Ad, B.; Tsamir, J.; Giladi, N. Cancer incidence among Parkinson’s disease patients in a 10-yrs time-window around disease onset: A large-scale cohort study. Parkinsonism Relat. Disord. 2016, 28, 68–72. [Google Scholar] [CrossRef]
- Jespersen, C.G.; Nørgaard, M.; Borre, M. Parkinson’s disease and risk of prostate cancer: A Danish population-based case-control study, 1995–2010. Cancer Epidemiol. 2016, 45, 157–161. [Google Scholar] [CrossRef]
- Ganguli, M. Cancer and Dementia: It’s Complicated. Alzheimer Dis. Assoc. Disord. 2015, 29, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.-X. Parkinson’s disease and colorectal cancer risk—A nested case control study. Cancer Epidemiol. 2016, 43, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.H.; Friis, S.; Frederiksen, K. Malignant Melanoma and Other Types of Cancer Preceding Parkinson Disease. Epidemiology 2006, 17, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Ajdacic-Gross, V.; Rodgers, S.; Aleksandrowicz, A.; Mütsch, M.; Steinemann, N.; von Wyl, V.; von Känel, R.; Bopp, M. Cancer co-occurrence patterns in Parkinson’s disease and multiple sclerosis—Do they mirror immune system imbalances? Cancer Epidemiol. 2016, 44, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.D.; Cai, W.; Chen, X. The associations between Parkinson’s disease and cancer: The plot thickens. Transl. Neurodegener. 2015, 4, 1–14. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Chang, S.-N.; Hsiao, T.-H.; Huang, B.-T.; Lin, C.-H.; Yang, P.-C. Association Between Parkinson Disease and Risk of Cancer in Taiwan. JAMA Oncol. 2015, 1, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.F.; Lu, M.K.; Muo, C.H.; Tsai, C.H.; Kao, C.H. Increased risk of brain tumor in patients with Parkinson’s disease: A nationwide cohort study in Taiwan. Acta Neurol. Scand. 2016, 134, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, R.; Elm, J.; Auinger, P.; Sharma, S.; Augustine, E.F.; Khadim, L.; Kieburtz, K. Investigators, Malignant melanoma in early-treated Parkinson’s disease: The NET-PD trial. Mov. Disord. 2014, 29, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Kareus, S.A.; Figueroa, K.P.; Cannon-Albright, L.A.; Pulst, S.M. Shared predispositions of parkinsonism and cancer: A population-based pedigree-linked study. Arch. Neurol. 2012, 69, 1572–1577. [Google Scholar] [CrossRef]
- Tacik, P.; Curry, S.; Fujioka, S.; Strongosky, A.; Uitti, R.J.; van Gerpen, J.A.; Diehl, N.N.; Heckman, M.G.; Wszolek, Z.K. Cancer in Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 31, 28–33. [Google Scholar] [CrossRef]
- Stoyanov, A.; Pamphlett, R. Is the risk of motor neuron disease increased or decreased after cancer? An. Australian case-control study. PLoS ONE 2014, 9, e103572. [Google Scholar] [CrossRef]
- Ahn, H.K.; Bae, J.H.; Hwang, I.C. Risk of cancer among patients with depressive disorder: A meta-analysis and implications. Psycho-Oncology 2016, 25, 1393–1399. [Google Scholar] [CrossRef]
- Heflin, M.T.; Oddone, E.Z.; Pieper, C.F.; Burchett, B.M.; Cohen, H.J. The Effect of Comorbid Illness on Receipt of Cancer Screening by Older People. J. Am. Geriatr. Soc. 2002, 50, 1651–1658. [Google Scholar] [CrossRef]
- Catts, V.S.; Catts, S.V.; O’Toole, B.I.; Frost, A.D.J. Cancer incidence in patients with schizophrenia and their first-degree relatives—A meta-analysis. Acta Psychiatr. Scand. 2008, 117, 323–336. [Google Scholar] [CrossRef]
- Kilian, R.; Becker, T.; Krüger, K.; Schmid, S.; Frasch, K. Health behavior in psychiatric in-patients compared with a German general population sample. Acta Psychiatr. Scand. 2006, 114, 242–248. [Google Scholar] [CrossRef]
- Sanchez-Valle, J.; Tejero, H.; Ibanez, K.; Portero, J.L.; Krallinger, M.; Al-Shahrour, F.; Tabares-Seisdedos, R.; Baudot, A.; Valencia, A. A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer’s Disease, Glioblastoma and Lung cancer. Sci. Rep. 2017, 7, 4474. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, K.; Boullosa, C.; Tabares-Seisdedos, R.; Baudot, A.; Valencia, A. Molecular Evidence for the Inverse Comorbidity between Central Nervous System Disorders and Cancers Detected by Transcriptomic Meta-analyses. PLoS Genet. 2014, 10, e1004173. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-C.A.; Cho, K.; Lindstrom, S.; Kraft, P.; Cormack, J.; Liang, L.; Driver, J.A.; Discovery, B.; ELLIPSE. Transdisciplinary Research in Cancer of the Lung (TRICL) Investigating the genetic relationship between Alzheimer’s disease and cancer using GWAS summary statistics. Qual. Life Res. 2017, 136, 1341–1351. [Google Scholar]
- Fiala, K.H.; Whetteckey, J.; Manyam, B.V. Malignant melanoma and levodopa in Parkinson’s disease: Causality or coincidence? Parkinsonism Relat. Disord. 2003, 9, 321–327. [Google Scholar] [CrossRef]
- Lazarevic-Pasti, T.; Leskovac, A.; Momic, T.; Petrovic, S.; Vasic, V. Modulators of Acetylcholinesterase Activity: From Alzheimer’s Disease to Anti-Cancer Drugs. Curr. Med. Chem. 2017, 24, 3283–3309. [Google Scholar] [CrossRef]
- Moran, L.B.; Graeber, M.B. Towards a pathway definition of Parkinson’s disease: A complex disorder with links to cancer, diabetes and inflammation. Neurogenetics 2008, 9, 1–13. [Google Scholar] [CrossRef]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.-L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3; Duncan, L.; et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef]
- Wang, X.; Kang, D.D.; Shen, K.; Song, C.; Lu, S.; Chang, L.-C.; Liao, S.G.; Huo, Z.; Tang, S.; Ding, Y.; et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics 2012, 28, 2534–2536. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Moreno-Grau, S.; de Rojas, I.; Hernandez, I.; Quintela, I.; Montrreal, L.; Alegret, M.; Hernandez-Olasagarre, B.; Madrid, L.; Gonzalez-Perez, A.; Maronas, O.; et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: The GR@ACE project. Alzheimers Dement. 2019, 15, 1333–1347. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef]
- Zhang, H.; Ahearn, T.U.; Lecarpentier, J.; Barnes, D.; Beesley, J.; Qi, G.; Jiang, X.; O’Mara, T.A.; Zhao, N.; Bolla, M.K.; et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet. 2020, 52, 572–581. [Google Scholar] [CrossRef]
- Michailidou, K.; Collaborators, N.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Phelan, C.M.; AOCS Study Group; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.; Dennis, J.; Pirie, A.; Riggan, M.J.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef]
- Bishop, D.T.; Demenais, F.; Iles, M.M.; Harland, M.; Taylor, J.C.; Corda, E.; Randerson-Moor, J.; Aitken, J.; Avril, M.-F.; Azizi, E.; et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 2009, 41, 920–925. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H.G. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011, 53, 565–576. [Google Scholar]
- Esteras, N.; Alquezar, C.; Bartolomé, F.; De La Encarnación, A.; Bermejo-Pareja, F.; Molina, J.A.; Martín-Requero, Á. G1/S Cell Cycle Checkpoint Dysfunction in Lymphoblasts from Sporadic Parkinson’s Disease Patients. Mol. Neurobiol. 2015, 52, 386–398. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Perry, G.; Smith, M.A. Alzheimer’s disease: The two-hit hypothesis. Lancet Neurol. 2004, 3, 219–226. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef]
- Ignácio, Z.M.; Réus, G.Z.; Arent, C.O.; Abelaira, H.M.; Pitcher, M.R.; Quevedo, J. New perspectives on the involvement of mTOR in depression as well as in the action of antidepressant drugs. Br. J. Clin. Pharmacol. 2016, 82, 1280–1290. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Tokutake, T.; Kasuga, K.; Yajima, R.; Sekine, Y.; Tezuka, T.; Nishizawa, M.; Ikeuchi, T. Hyperphosphorylation of Tau induced by naturally secreted amyloid-beta at nanomolar concentrations is modulated by insulin-dependent Akt-GSK3beta signaling pathway. J. Biol. Chem. 2012, 287, 35222–35233. [Google Scholar] [CrossRef]
- Luo, S.; Kang, S.S.; Wang, Z.H.; Liu, X.; Day, J.X.; Wu, Z.; Peng, J.; Xiang, D.; Springer, W.; Ye, K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing Its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019, 39, 7291–7305. [Google Scholar] [CrossRef]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020, 94, e1592–e1604. [Google Scholar] [CrossRef]
- Wang, G.; Pan, J.; Chen, S.D. Kinases and kinase signaling pathways: Potential therapeutic targets in Parkinson’s disease. Prog. Neurobiol. 2012, 98, 207–221. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707. [Google Scholar] [CrossRef]
- Lee, J.; Chang, J.Y.; Kang, Y.E.; Yi, S.; Lee, M.H.; Joung, K.H.; Kim, K.S.; Shong, M. Mitochondrial Energy Metabolism and Thyroid Cancers. Endocrinol. Metab. 2015, 30, 117–123. [Google Scholar] [CrossRef]
- Zimmermann, F.A.; Neureiter, D.; Sperl, W.; Mayr, J.A.; Kofler, B. Alterations of Oxidative Phosphorylation Complexes in Papillary Thyroid Carcinoma. Cells 2018, 7, 40. [Google Scholar] [CrossRef]
- Mussazhanova, Z.; Shimamura, M.; Kurashige, T.; Ito, M.; Nakashima, M.; Nagayama, Y. Causative role for defective expression of mitochondria-eating protein in accumulation of mitochondria in thyroid oncocytic cell tumors. Cancer Sci. 2020, 111, 2814–2823. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Aiderus, A.; Black, M.A.; Dunbier, A.K. Fatty acid oxidation is associated with proliferation and prognosis in breast and other cancers. BMC Cancer 2018, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 2017, 109, 68–77. [Google Scholar] [CrossRef]
- Ueno, S.I.; Saiki, S.; Fujimaki, M.; Takeshige-Amano, H.; Hatano, T.; Oyama, G.; Ishikawa, K.I.; Yamaguchi, A.; Nojiri, S.; Akamatsu, W.; et al. Zonisamide Administration Improves Fatty Acid beta-Oxidation in Parkinson’s Disease. Cells 2018, 8, 14. [Google Scholar] [CrossRef]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.I.; Mori, A.; Oji, Y.; Okuzumi, A.; Fukuhara, T.; Koinuma, T.; Imamichi, Y.; et al. Decreased long-chain acylcarnitines from insufficient beta-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef]
- Szybinska, A.; Lesniak, W. P53 Dysfunction in Neurodegenerative Diseases—The Cause or Effect of Pathological Changes? Aging Dis. 2017, 8, 506–518. [Google Scholar] [CrossRef]
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell Mol. Life Sci. 2020, 77, 2659–2680. [Google Scholar] [CrossRef]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J. Cell Mol. Med. 2008, 12, 987–994. [Google Scholar] [CrossRef]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1997, 232, 418–421. [Google Scholar] [CrossRef]
- Ohyagi, Y.; Asahara, H.; Chui, D.H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Abeta42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005, 19, 255–257. [Google Scholar] [CrossRef]
- Mogi, M.; Kondo, T.; Mizuno, Y.; Nagatsu, T. p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci. Lett. 2007, 414, 94–97. [Google Scholar] [CrossRef]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutierrez, N.C. Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads Between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2018, 11, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Doty, K.R.; Guillot-Sestier, M.-V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J. Immunol. Res. 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Szende, B.; Magyar, K.; Szegedi, Z. Apoptotic and antiapoptotic effect of (-)deprenyl and (-)-desmethyl-deprenyl on human cell lines. Neurobiology 2000, 8, 249–255. [Google Scholar] [PubMed]
- Ryu, I.; Ryu, M.; Han, J.; Kim, S.; Lee, M.; Ju, X.; Yoo, B.; Lee, Y.; Jang, Y.; Song, I.; et al. L-Deprenyl exerts cytotoxicity towards acute myeloid leukemia through inhibition of mitochondrial respiration. Oncol. Rep. 2018, 40, 3869–3878. [Google Scholar] [CrossRef]
- Branigan, G.L.; Soto, M.; Neumayer, L.; Rodgers, K.; Brinton, R.D. Association between Hormone-Modulating Breast Cancer Therapies and Incidence of Neurodegenerative Outcomes for Women With Breast Cancer. JAMA Netw. Open 2020, 3, e201541. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Manassero, G.; Vasciaveo, V.; Venezia, M.; Tabaton, M.; Tamagno, E. Estrogens Inhibit Amyloid-beta-Mediated Paired Helical Filament-Like Conformation of Tau Through Antioxidant Activity and miRNA 218 Regulation in hTau Mice. J. Alzheimers Dis. 2020, 77, 1339–1351. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Included Studies | No. Samples (Case/Control) | DEGs 0.05/Tested Genes, % | Up 0.05/Down 0.05 | DEGs 0.01/Tested Genes, % | Up 0.01/Down 0.01 | Mean Q | Mean Tau |
|---|---|---|---|---|---|---|---|---|
| AD | 7 | 226 (124/102) | 3341/11,536 (28.96%) | 1364/1977 | 1641/11,536 (14.23%) | 563/1078 | 10.59 | 0.33 |
| PD | 6 | 149 (83/66) | 3473/11,714 (29.65%) | 1626/1847 | 2267/11,714 (19.35%) | 949/1318 | 7.24 | 0.31 |
| ALL | 5 | 1500 (1406/94) | 2315/12,436 (18.62%) | 1373/942 | 1387/12,436 (11.15%) | 920/467 | 16.67 | 0.63 |
| AML | 8 | 1162 (949/213) | 581/16,271 (3.57%) | 415/166 | 217/16,271 (1.33%) | 163/54 | 39.52 | 0.47 |
| BCLA | 5 | 228 (199/29) | 5277/16,054 (32.87%) | 3209/2068 | 3069/16,054 (19.12%) | 1983/1086 | 10.41 | 0.85 |
| BRCA | 8 | 1396 (1164/232) | 8362/20,536 (40.72%) | 4700/3662 | 6662/20,536 (32.44%) | 3781/2881 | 33.97 | 0.46 |
| BRNCA | 7 | 1218 (1124/94) | 9757/15,401 (63.35%) | 5161/4596 | 8228/15,401 (53.43%) | 4437/3791 | 19.89 | 0.35 |
| CERV | 4 | 139 (82/57) | 3612/15,683 (23.03%) | 2215/1397 | 2344/15,683 (14.95%) | 1517/827 | 11.73 | 0.64 |
| CHLCA | 4 | 152 (130/22) | 1234/16,111 (7.66%) | 766/468 | 535/16,111 (3.32%) | 346/189 | 15.6 | 1.55 |
| CLL | 6 | 1021 (884/137) | 2547/16,634 (15.31%) | 1670/877 | 1488/16,634 (8.95%) | 996/492 | 62.94 | 1.24 |
| CML | 3 | 226 (128/98) | 1593/15,125 (10.53%) | 1115/478 | 740/15,125 (4.89%) | 461/279 | 6.88 | 0.33 |
| CRCA | 7 | 1261 (968/293) | 9041/13,595 (66.5%) | 4793/4248 | 7872/13,595 (57.9%) | 4235/3637 | 27.98 | 0.3 |
| DLBCL | 3 | 246 (173/73) | 4673/20,503 (22.79%) | 3284/1389 | 3184/20,503 (15.53%) | 2339/845 | 32.7 | 1.67 |
| FLYMPH | 6 | 214 (147/67) | 1945/15,456 (12.58%) | 1195/750 | 854/15,456 (5.53%) | 542/312 | 26.71 | 1.22 |
| HANC | 9 | 560 (436/124) | 6964/14,760 (47.18%) | 3617/3347 | 5280/14,760 (35.77%) | 2874/2406 | 24.94 | 0.5 |
| KDNCA | 8 | 650 (396/254) | 9575/13,828 (69.24%) | 4974/4601 | 8193/13,828 (59.25%) | 4426/3767 | 31.65 | 0.38 |
| LGCA | 5 | 850 (522/328) | 9468/14,718 (64.33%) | 5637/3831 | 7954/14,718 (54.04%) | 4695/3259 | 26.39 | 0.23 |
| LIVCA | 8 | 839 (383/456) | 6624/10,648 (62.21%) | 3567/3057 | 5401/10,648 (50.72%) | 2996/2405 | 26.91 | 0.23 |
| OVCA | 6 | 222 (154/68) | 5241/18,544 (28.26%) | 3704/1537 | 3415/18,544 (18.42%) | 2496/919 | 21.1 | 0.9 |
| PACA | 9 | 463 (318/145) | 8306/14,257 (58.26%) | 3938/4368 | 6270/14,257 (43.98%) | 3061/3209 | 39.27 | 0.79 |
| PRCA | 6 | 1019 (648/371) | 4663/15,169 (30.74%) | 2001/2662 | 3253/15,169 (21.45%) | 1378/1875 | 24.06 | 0.17 |
| SKCM | 5 | 486 (409/77) | 4158/11,651 (35.69%) | 2271/1887 | 2752/11,651 (23.62%) | 1453/1299 | 13.43 | 0.38 |
| STCA | 9 | 664 (393/271) | 7047/15,066 (46.77%) | 3766/3281 | 5243/15,066 (34.8%) | 2920/2323 | 44.96 | 0..42 |
| THCA | 5 | 245 (139/106) | 8662/20,536 (42.18%) | 4271/4391 | 6356/20,536 (30.95%) | 2975/3381 | 10.02 | 0.39 |
| Study | Source | N (Cases/Controls) | Tested SNPs after Merging with SNP List | Mean Chi2 | Lambda GC | Max Chi2 | Genome-Wide Significant SNPs | SNP Heritability |
|---|---|---|---|---|---|---|---|---|
| AD 1 | IGAP [61] | 54,162 (17,008/37,154) | 1,150,200 | 1.11 | 1.09 | 565.21 | 165 | 0.07 |
| AD 2 | GR@ACE project [62] | 21,235 (11,999/9236) | 1,204,123 | 1.09 | 1.068 | 1123.06 | 59 | 0.13 |
| AD 3 | GWAS catalog [63] | 455,258 (71,880AD */383,378) | 1,203,908 | 1.12 | 1.08 | 1009.11 | 320 | 0.01 |
| PD 1 | Nalls et al. [64] | 482,730 (33,674/449,056) | 1,137,530 | 1.14 | 1.09 | 180.42 | 276 | 0.02 |
| PD 2 | 23andMe [65] | 308,557 (6477/302,080) | 1,211,658 | 1.10 | 1.08 | 164.95 | 142 | 0.02 |
| PRCA 1 | GWAS catalog [66] | 140,254 (79,148/61,106) | 1,206,082 | 1.51 | 1.23 | 846.34 | 2733 | 0.16 |
| PRCA 2 | UK Biobank | 206,770 (6879/199,891) | 1,211,361 | 1.13 | 1.09 | 181.15 | 397 | 0.03 |
| BRCA 1 | BCAC [67] | 247,173 (133,384/113,789) | 519,352 | 1.72 | 1.38 | 481.43 | 1389 | 0.12 |
| BRCA 2 | GWAS catalog [68] | 139,274 (76,192/63,082) | 1,128,758 | 1.68 | 1.36 | 1424.99 | 2832 | 0.22 |
| BRCA 3 | UK Biobank | 245,494 (10,478/235,016) | 1,211,361 | 1.11 | 1.08 | 314.15 | 276 | 0.02 |
| CRCA | UK Biobank | 387,318 (4562/382,756) | 1,215,182 | 1.058 | 1.052 | 51.049 | 22 | 0.01 |
| OVCA | GWAS catalog [69] | 85,426 (16,924/68,502) | 1,149,515 | 1.09 | 1.06 | 169.17 | 209 | 0.04 |
| SKCM 1 | UK Biobank | 452,264 (2465/449,799) | 1,211,361 | 1.04 | 1.03 | 121.09 | 144 | 0.00 |
| SKCM 2 | Genomel [70] | 32,383 (11,523/20,860) | 1,100,284 | 1.12 | 1.08 | 372.76 | 561 | 0.17 |
| LGCA | UK Biobank | 452,264 (1655/450,609) | 1,211,361 | 1.02 | 1.01 | 26.03 | 0 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forés-Martos, J.; Boullosa, C.; Rodrigo-Domínguez, D.; Sánchez-Valle, J.; Suay-García, B.; Climent, J.; Falcó, A.; Valencia, A.; Puig-Butillé, J.A.; Puig, S.; et al. Transcriptomic and Genetic Associations between Alzheimer’s Disease, Parkinson’s Disease, and Cancer. Cancers 2021, 13, 2990. https://doi.org/10.3390/cancers13122990
Forés-Martos J, Boullosa C, Rodrigo-Domínguez D, Sánchez-Valle J, Suay-García B, Climent J, Falcó A, Valencia A, Puig-Butillé JA, Puig S, et al. Transcriptomic and Genetic Associations between Alzheimer’s Disease, Parkinson’s Disease, and Cancer. Cancers. 2021; 13(12):2990. https://doi.org/10.3390/cancers13122990
Chicago/Turabian StyleForés-Martos, Jaume, Cesar Boullosa, David Rodrigo-Domínguez, Jon Sánchez-Valle, Beatriz Suay-García, Joan Climent, Antonio Falcó, Alfonso Valencia, Joan Anton Puig-Butillé, Susana Puig, and et al. 2021. "Transcriptomic and Genetic Associations between Alzheimer’s Disease, Parkinson’s Disease, and Cancer" Cancers 13, no. 12: 2990. https://doi.org/10.3390/cancers13122990
APA StyleForés-Martos, J., Boullosa, C., Rodrigo-Domínguez, D., Sánchez-Valle, J., Suay-García, B., Climent, J., Falcó, A., Valencia, A., Puig-Butillé, J. A., Puig, S., & Tabarés-Seisdedos, R. (2021). Transcriptomic and Genetic Associations between Alzheimer’s Disease, Parkinson’s Disease, and Cancer. Cancers, 13(12), 2990. https://doi.org/10.3390/cancers13122990