
Inhibition of the Human Hsc70 System by Small Ligands as a Potential Anticancer Approach

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. High-Throughput Screening (HTS) and Validation of Modulators of Apg2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Name | IUPAC Name | Molecular Weight (Da) | Therapeutic Class |
|---|---|---|---|---|
| 1 | Chlorhexidine [42] | (1E)-2-[6-[[amino-[(E)-[amino-(4-chloroanilino)methylidene]amino]methylidene]amino]hexyl]-1-[amino-(4-chloroanilino)methylidene]guanidine | 505.46 | Infectiology |
| 2 | Pinaverium bromide [43] | 4-[(2-bromo-4,5-dimethoxyphenyl)methyl]-4-[2-[2-(6,6-dimethyl-2-bicyclo [3.1.1]heptanyl)ethoxy]ethyl]morpholin-4-ium bromide | 591.43 | Neuromuscular |
| 3 | Benzbromarone [44] | (3,5-dibromo-4-hydroxyphenyl)-(2-ethyl-1-benzofuran-3-yl)methanone | 424.1 | Cardiovascular |
| 4 | Beta-escin [45] | (beta-D-Xylopyrannosyl)-3(beta-D-glucopyrannosyl)-4(methyl-3acetoxybutyryl)-28tetrahydroxy-16alpha,21alpha,22beta,24oleanone-12 | 1131.28 | Metabolism Oncology |
| 5 | Mefloquine hydrochloride [46] | (S)-[2,8-bis(trifluoromethyl)quinolin-4-yl]-[(2R)-piperidin-2-yl]methanol hydrochloride | 414.78 | Infectiology |
| 6 | Tiratricol, 3,3′,5-triiodothyroacetic acid [47] | 2-[4-(4-hydroxy-3-iodophenoxy)-3,5-diiodophenyl]acetic acid | 621.94 | Endocrinology |
2.2. Apg2 Binders Also Interact with Hsc70
2.3. Effect of the Compounds on the Chaperone Activity of Hsc70 and Apg2
2.4. Toxicity of the Inhibitors in Human Melanoma Cell Lines
2.5. Melanoma Cell-Specific Inhibition by the Selected Compounds
2.6. Pinaverium Bromide (PB) Causes Melanoma Cell Death by Inducing Apoptosis
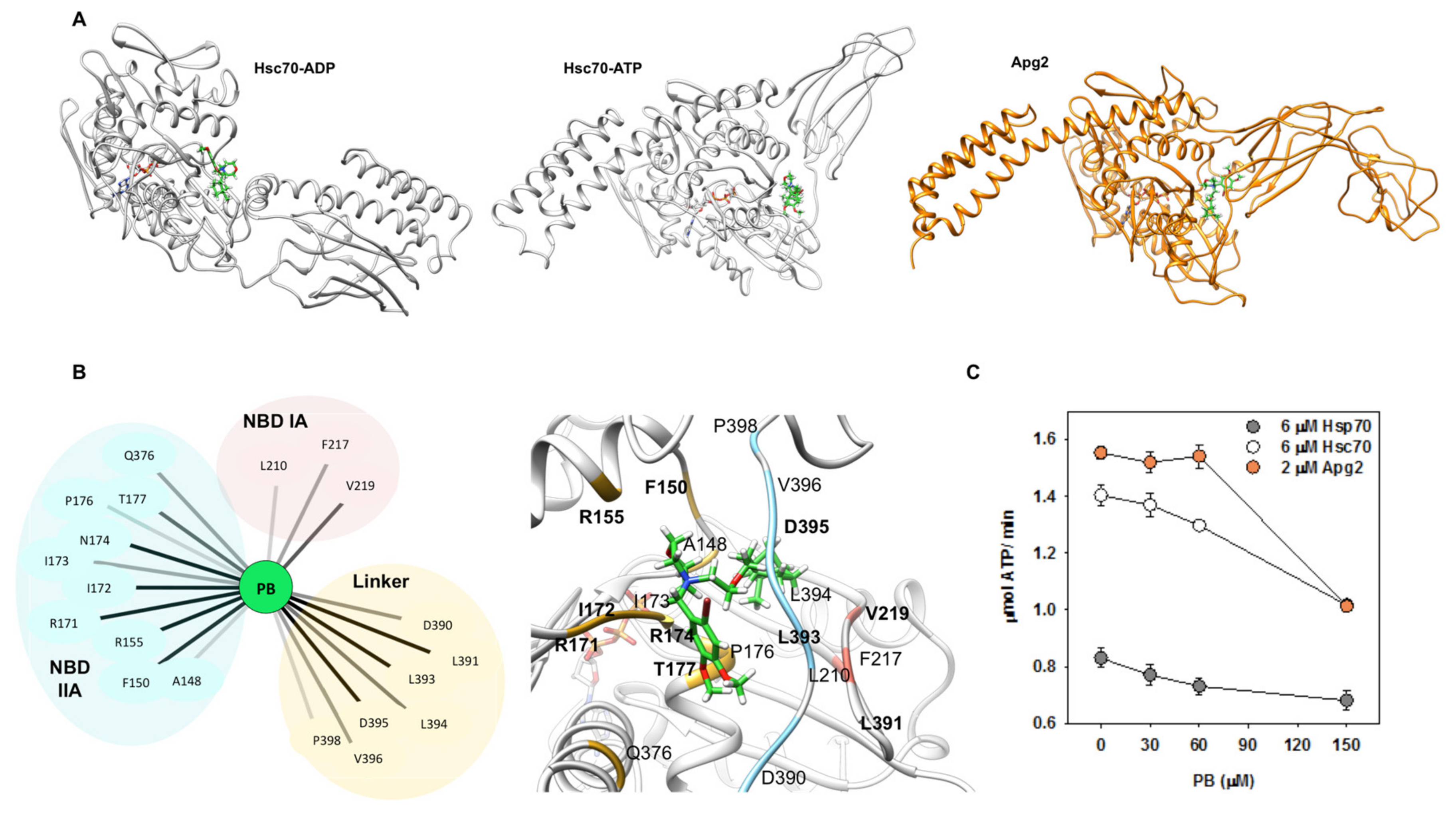
2.7. Identification of PB Binding Site by Docking and Molecular Dynamics
2.8. PB Inhibits Luciferase Protein Refolding in a Cellular Context
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression and Purification of Proteins
4.2. Differential Scanning Fluorimetry (DSF)
4.3. G6PDH Aggregate Reactivation
4.4. ATPase Measurements
4.5. Cell Culture
4.6. Recovery of Intracellular Luciferase Activity
4.7. Immunocytochemical Staining
4.8. Western Blot Analysis
4.9. Viability Assays
4.10. Time-Lapse Imaging
4.11. Homology Modeling
4.12. Protein–Ligand Docking
4.13. Molecular Dynamics Simulations
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, G.G.; Hipp, M.S.; Hartl, F.U. Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 2020, 12, a033951. [Google Scholar] [CrossRef]
- Jäättelä, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- English, C.A.; Sherman, W.; Meng, W.; Gierasch, L.M. The Hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J. Biol. Chem. 2017, 292, 14765–14774. [Google Scholar] [CrossRef]
- Easton, D.P.; Kaneko, Y.; Subjeck, J.R. The Hsp110 and Grp170 stress proteins: Newly recognized relatives of the Hsp70s. Cell Stress Chaperones 2000, 5, 276–290. [Google Scholar] [CrossRef]
- Oh, H.J.; Chen, X.; Subjeck, J.R. hsp110 Protects Heat-denatured Proteins and Confers Cellular Thermoresistance. J. Biol. Chem. 1997, 272, 31636–31640. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Easton, D.; Murawski, M.; Kaneko, Y.; Subjeck, J.R. The Chaperoning Activity of hsp. J. Biol. Chem. 1999, 274, 15712–15718. [Google Scholar] [CrossRef]
- Mattoo, R.U.H.; Sharma, S.K.; Priya, S.; Finka, A.; Goloubinoff, P. Hsp110 Is a Bona Fide Chaperone Using ATP to Unfold Stable Misfolded Polypeptides and Reciprocally Collaborate with Hsp70 to Solubilize Protein Aggregates. J. Biol. Chem. 2013, 288, 21399–21411. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The Hsp70 chaperone machinery: J-proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237.e4. [Google Scholar] [CrossRef] [PubMed]
- Cyr, D.M. Swapping Nucleotides, Tuning Hsp. Cell 2008, 133, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, S.; Socinski, M.A.; Burns, T.F. HSP90 inhibitors in lung cancer: Promise still unfulfilled. Clin. Adv. Hematol. Oncol. 2016, 14, 346–356. [Google Scholar] [PubMed]
- Nylandsted, J.; Brand, K.; Jäättelä, M. Heat Shock Protein 70 Is Required for the Survival of Cancer Cells. Ann. N. Y. Acad. Sci. 2006, 926, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jäättelä, M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Targeting heat shock proteins in cancer. Heat shock proteins in obesity: Links to cardiovascular disease. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef]
- Hatayama, T.; Yamagishi, N.; Minobe, E.; Sakai, K. Role of hsp105 in Protection against Stress-Induced Apoptosis in Neuronal PC12 Cells. Biochem. Biophys. Res. Commun. 2001, 288, 528–534. [Google Scholar] [CrossRef]
- Yamagishi, N.; Ishihara, K.; Saito, Y.; Hatayama, T. Hsp105 family proteins suppress staurosporine-induced apoptosis by inhibiting the translocation of Bax to mitochondria in HeLa cells. Exp. Cell Res. 2006, 312, 3215–3223. [Google Scholar] [CrossRef]
- Hosaka, S.; Nakatsura, T.; Tsukamoto, H.; Hatayama, T.; Baba, H.; Nishimura, Y. Synthetic small interfering RNA targeting heat shock protein 105 induces apoptosis of various cancer cells both in vitro and in vivo. Cancer Sci. 2006, 97, 623–632. [Google Scholar] [CrossRef]
- Duval, A.; Collura, A.; Berthenet, K.; Lagrange, A.; Garrido, C. Microsatellite Instability in Colorectal Cancer: Time to Stop Hiding! Oncotarget 2011, 2, 826–827. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, K.-J.; Rhee, Y.-Y.; Oh, S.; Cho, N.-Y.; Lee, H.S.; Kang, G.H. Expression status of wild-type HSP110 correlates with HSP110 T17 deletion size and patient prognosis in microsatellite-unstable colorectal cancer. Mod. Pathol. 2014, 27, 443–453. [Google Scholar] [CrossRef]
- Kimura, A.; Ogata, K.; Altan, B.; Yokobori, T.; Ide, M.; Mochiki, E.; Toyomasu, Y.; Kogure, N.; Yanoma, T.; Suzuki, M.; et al. Nuclear heat shock protein 110 expression is associated with poor prognosis and chemotherapy resistance in gastric cancer. Oncotarget 2016, 7, 18415–18423. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.E. The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.S.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.-H.D.B. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef]
- Sleire, L.; Førde-Tislevoll, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, G.J.; Gonzalez, D.; Boudesco, C.; Dias, A.M.M.; Gotthard, G.; Uyanik, B.; Dondaine, L.; Marcion, G.; Hermetet, F.; Denis, C.; et al. Selecting the first chemical molecule inhibitor of HSP110 for colorectal cancer therapy. Cell Death Differ. 2020, 27, 117–129. [Google Scholar] [CrossRef]
- Boudesco, C.; Verhoeyen, E.; Martin, L.; Chassagne-Clement, C.; Salmi, L.; Mhaidly, R.; Pangault, C.; Fest, T.; Ramla, S.; Jardin, F.; et al. HSP110 sustains chronic NF-κB signaling in activated B-cell diffuse large B-cell lymphoma through MyD88 stabilization. Blood 2018, 132, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Lee, T.H.; Kim, J.H.; Cho, N.-Y.; Kim, W.H.; Kang, G.H. Deletion in HSP110 T 17: Correlation with wild-type HSP110 expression and prognostic significance in microsatellite-unstable advanced gastric cancers. Hum. Pathol. 2017, 67, 109–118. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Cimmperman, P.; Baranauskienė, L.; Jachimovičiūtė, S.; Jachno, J.; Torresan, J.; Michailovienė, V.; Matulienė, J.; Sereikaitė, J.; Bumelis, V.; Matulis, D. A Quantitative Model of Thermal Stabilization and Destabilization of Proteins by Ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 Chaperone Ligands Control Domain Association via an Allosteric Mechanism Mediated by the Interdomain Linker. Mol. Cell 2007, 26, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Mcauley-Hecht, K.E. Microcalorimetry and the molecular recognition of peptides and proteins. Philos. Trans. R. Soc. London. Ser. A Phys. Eng. Sci. 1993, 345, 23–35. [Google Scholar] [CrossRef]
- Silvestri, D.L.; McEnery-Stonelake, M. Chlorhexidine: Uses and adverse reactions. Dermatitis 2013, 24, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Christen, M.O. Action of pinaverium bromide, a calcium-antagonist, on gastrointestinal motility disorders. Gen. Pharmacol. 1990, 21, 821–825. [Google Scholar] [CrossRef]
- Lee, M.-H.H.; Graham, G.G.; Williams, K.M.; Day, R.O. A Benefit-Risk Assessment of Benzbromarone in the Treatment of Gout: Was its withdrawal from the market in the best interest of patients? Drug Saf. 2008, 31, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Su, P. Effect of β-aescin extract from Chinese Buckeye Seed on chronic venous insufficiency. Pharmazie 2013, 68, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Schlagenhauf, P.; Adamcova, M.; Regep, L.; Schaerer, M.T.; Rhein, H.-G. The position of mefloquine as a 21st century malaria chemoprophylaxis. Malar. J. 2010, 9, 357. [Google Scholar] [CrossRef]
- Cohen-Lehman, J.; Charitou, M.M.; Klein, I. Tiratricol-Induced Periodic Paralysis: A review of nutraceuticals affecting thyroid function. Endocr. Pract. 2011, 17, 610–615. [Google Scholar] [CrossRef]
- Polier, S.; Dragovic, Z.; Hartl, F.U.; Bracher, A. Structural Basis for the Cooperation of Hsp70 and Hsp110 Chaperones in Protein Folding. Cell 2008, 133, 1068–1079. [Google Scholar] [CrossRef]
- Liu, Q.; Hendrickson, W.A. Insights into Hsp70 Chaperone Activity from a Crystal Structure of the Yeast Hsp110 Sse. Cell 2007, 131, 106–120. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef]
- Shipp, C.; Weide, B.; Derhovanessian, E.; Pawelec, G. Hsps are up-regulated in melanoma tissue and correlate with patient clinical parameters. Cell Stress Chaperones 2012, 18, 145–154. [Google Scholar] [CrossRef]
- Faria, G.; Cardoso, C.R.B.; Larson, R.E.; Silva, J.S.; Rossi, M.A. Chlorhexidine-induced apoptosis or necrosis in L929 fibroblasts: A role for endoplasmic reticulum stress. Toxicol. Appl. Pharmacol. 2009, 234, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, M.; Chellini, F.; Margheri, M.; Tonelli, P.; Tani, A. Effect of chlorhexidine digluconate on different cell types: A molecular and ultrastructural investigation. Toxicol. Vitr. 2008, 22, 308–317. [Google Scholar] [CrossRef]
- Weinstein, R.A.; Milstone, A.M.; Passaretti, C.L.; Perl, T.M.; Skowronski, D.M.; De Serres, G.; Scheifele, D.; Russell, M.L.; Warrington, R.; Dele Davies, H.; et al. Chlorhexidine: Expanding the Armamentarium for Infection Control and Prevention. Clin. Infect. Dis. 2008, 46, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner Caspase-3, -6, and -7 Perform Distinct, Non-redundant Roles during the Demolition Phase of Apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [PubMed]
- Pedregal, J.R.-G.; Sciortino, G.; Guasp, J.; Municoy, M.; Maréchal, J.-D. GaudiMM: A modular multi-objective platform for molecular modeling. J. Comput. Chem. 2017, 38, 2118–2126. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Hageman, J.; Van Waarde-Verhagen, M.; Zylicz, A.; Walerych, D.; Kampinga, H.H. The diverse members of the mammalian HSP70 machine show distinct chaperone-like activities. Biochem. J. 2011, 435, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.H.S.; Lopes, F.C.; John, E.B.O.; Carlini, C.R.; Ligabue-Braun, R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. Int. J. Mol. Sci. 2019, 20, 1322. [Google Scholar] [CrossRef] [PubMed]
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, M.R.; Valpuesta, J.M. Hsp70 chaperone: A master player in protein homeostasis [version 1; peer review: 3 approved]. F1000Research 2018, 7, 1497. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat Shock Proteins 27 and 70: Anti-Apoptotic Proteins with Tumorigenic Properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef]
- Perera, E.; Gnaneswaran, N.; Jennens, R.; Sinclair, R. Malignant melanoma. Healthcare 2014, 2, 1–19. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Gibney, G.T.; Sondak, V.K.; Smalley, K.S.M. Beyond BRAF: Where next for melanoma therapy? Br. J. Cancer 2015, 112, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Lang, B.J.; Guerrero-Giménez, M.E.; Prince, T.L.; Ackerman, A.; Bonorino, C.; Calderwood, S.K. Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond. Int. J. Mol. Sci. 2019, 20, 4507. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-S.; Park, C.-H.; Choi, B.-R.; Lim, M.-S.; Heo, S.-H.; Kim, C.-H.; Kang, S.-G.; Whang, K.U.; Cho, M.K. Expression of heat shock protein 105 and 70 in malignant melanoma and benign melanocytic nevi. J. Cutan. Pathol. 2009, 36, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Budina-kolomets, A.; Webster, M.R.; Leu, J.I.; Jennis, M.; Guerrini, A.; Kossenkov, A.V.; Xu, W.; Karakousis, G.; Amaravadi, R.K.; Wu, H.; et al. The response to melanoma treatment with BRAF inhibitors. Cancer Res. 2016, 76, 2720–2730. [Google Scholar] [CrossRef]
- Roufayel, R.; Kadry, S. Molecular Chaperone HSP70 and Key Regulators of Apoptosis—A Review. Curr. Mol. Med. 2019, 19, 315–325. [Google Scholar] [CrossRef]
- Komarova, E.Y.; Afanasyeva, E.A.; Bulatova, M.M.; Cheetham, M.E.; Margulis, B.A.; Guzhova, I.V. Downstream caspases are novel targets for the antiapoptotic activity of the molecular chaperone Hsp70. Cell Stress Chaperones 2004, 9, 265–275. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef]
- Mielczarek-Lewandowska, A.; Hartman, M.L.; Czyz, M. Inhibitors of HSP90 in melanoma. Apoptosis 2020, 25, 12–28. [Google Scholar] [CrossRef]
- Goloudina, A.R.; Demidov, O.N.; Garrido, C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012, 325, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Mereddy, G.R.; Ronayne, C.T. Repurposing Antimalarial Drug Mefloquine for Cancer Treatment. Transl. Med. 2018, 8. [Google Scholar] [CrossRef]
- Camilleri, M.; Ford, A.C. Pharmacotherapy for Irritable Bowel Syndrome. J. Clin. Med. 2017, 6, 101. [Google Scholar] [CrossRef]
- Feron, O.; Wibo, M.; Christen, M.; Godfraind, T. Interaction of pinaverium (a quaternary ammonium compound) with 1,4-dihydropyridine binding sites in rat ileum smooth muscle. Br. J. Pharmacol. 1992, 105, 480–484. [Google Scholar] [CrossRef]
- Macia, A.; Herreros, J.; Martí, R.M.; Cantí, C. Calcium Channel Expression and Applicability as Targeted Therapies in Melanoma. BioMed Res. Int. 2015, 2015, 587135. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Shevtsov, M.; Multhoff, G.; Mikhaylova, E.; Shibata, A.; Guzhova, I.; Margulis, B. Combination of Anti-Cancer Drugs with Molecular Chaperone Inhibitors. Int. J. Mol. Sci. 2019, 20, 5284. [Google Scholar] [CrossRef]
- Spiegelberg, D.; Abramenkovs, A.; Mortensen, A.C.L.; Lundsten, S.; Nestor, M.; Stenerlöw, B. The HSP90 inhibitor Onalespib exerts synergistic anti-cancer effects when combined with radiotherapy: An in vitro and in vivo approach. Sci. Rep. 2020, 10, 5923. [Google Scholar] [CrossRef]
- Cabrera, Y.; Dublang, L.; Fernández-Higuero, J.A.; Albesa-Jové, D.; Lucas, M.; Viguera, A.R.; Guerin, M.E.; Vilar, J.M.G.; Muga, A.; Moro, F. Regulation of Human Hsc70 ATPase and Chaperone Activities by Apg2: Role of the Acidic Subdomain. J. Mol. Biol. 2019, 431, 444–461. [Google Scholar] [CrossRef]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef] [PubMed]
- Støve, S.I.; Flydal, M.I.; Hausvik, E.; Underhaug, J.; Martinez, A. Chapter 15—Differential Scanning Fluorimetry in the Screening and Validation of pharmacological chaperones for Soluble and Membrane Proteins. In Protein Homeostasis Diseases; Pey, A., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 329–341. ISBN 978-0-12-819132-3. [Google Scholar]
- Fernández-Higuero, J.A.; Aguado, A.; Perales-Calvo, J.; Moro, F.; Muga, A. Activation of the DnaK-ClpB Complex is Regulated by the Properties of the Bound Substrate. Sci. Rep. 2018, 8, 5796. [Google Scholar] [CrossRef] [PubMed]
- Nørby, J.G. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 1988, 156, 116–119. [Google Scholar] [CrossRef]
- De Wet, J.R.; Wood, K.V.; DeLuca, M.; Helinski, D.R.; Subramani, S. Firefly luciferase gene: Structure and expression in mammalian cells. Mol. Cell. Biol. 1987, 7, 725–737. [Google Scholar] [CrossRef]
- Hageman, J.; Kampinga, H.H. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones 2009, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef]
- Yang, J.; Zong, Y.; Su, J.; Li, H.; Zhu, H.; Columbus, L.; Zhou, L.; Liu, Q. Conformation transitions of the polypeptide-binding pocket support an active substrate release from Hsp70s. Nat. Commun. 2017, 8, 1201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Prasad, K.; Lafer, E.M.; Sousa, R. Structural Basis of Interdomain Communication in the Hsc70 Chaperone. Mol. Cell 2005, 20, 513–524. [Google Scholar] [CrossRef]
- Yang, J.; Nune, M.; Zong, Y.; Zhou, L.; Liu, Q. Close and Allosteric Opening of the Polypeptide-Binding Site in a Human Hsp70 Chaperone BiP. Structure 2015, 23, 2191–2203. [Google Scholar] [CrossRef]
- O’Brien, M.C.; Flaherty, K.M.; McKay, D.B. Lysine 71 of the Chaperone Protein Hsc70 Is Essential for ATP Hydrolysis. J. Biol. Chem. 1996, 271, 15874–15878. [Google Scholar] [CrossRef] [PubMed]
- Schuermann, J.P.; Jiang, J.; Cuéllar, J.; Llorca, O.; Wang, L.; Gimenez, L.E.; Jin, S.; Taylor, A.B.; Demeler, B.; Morano, K.A.; et al. Structure of the Hsp110:Hsc70 Nucleotide Exchange Machine. Mol. Cell 2008, 31, 232–243. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Rodríguez-Guerra Pedregal, J.; Alonso-Cotchico, L.; Velasco-Carneros, L.; Maréchal, J.-D. OMMProtocol: A Command Line Application to Launch Molecular Dynamics Simulations with OpenMM. ChemRxiv 2018. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Dunbrack, R.L.J. Rotamer Libraries in the 21st Century. Curr. Opin. Struct. Biol. 2002, 12, 431–440. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M., Jr.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Meagher, K.L.; Redman, L.T.; Carlson, H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003, 24, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Allnér, O.; Nilsson, L.; Villa, A. Magnesium Ion–Water Coordination and Exchange in Biomolecular Simulations. J. Chem. Theory Comput. 2012, 8, 1493–1502. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Pande, V.S. OpenMM: A Hardware-Independent Framework for Molecular Simulations. Comput. Sci. Eng. 2010, 12, 34–39. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Brünger, A.; Brooks, C.L.; Karplus, M. Stochastic boundary conditions for molecular dynamics simulations of ST2 water. Chem. Phys. Lett. 1984, 105, 495–500. [Google Scholar] [CrossRef]
- Duane, S.; Kennedy, A.D.; Pendleton, B.J.; Roweth, D. Hybrid Monte Carlo. Phys. Lett. B 1987, 195, 216–222. [Google Scholar] [CrossRef]








| Compound | IC50 (µM) | |
|---|---|---|
| G6PDH Reactivation | ||
| Hsc70 + DnaJB1 | Hsc70 + DnaJB1 + Apg2 | |
| 1 | 11.9 ± 0.2 | 14.5 ± 1.8 |
| 2 | 58 ± 0.9 | 70.9 ± 0.1 |
| 3 | 10.1 ± 0.4 | 17.6 ± 0.1 |
| 4 | 37.3 ± 1.8 | 47 ± 5.6 |
| 5 | 106.8 ± 7.5 | 97.3 ± 21.1 |
| 6 | 18.7 ± 3.6 | 23.3 ± 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dublang, L.; Underhaug, J.; Flydal, M.I.; Velasco-Carneros, L.; Maréchal, J.-D.; Moro, F.; Boyano, M.D.; Martinez, A.; Muga, A. Inhibition of the Human Hsc70 System by Small Ligands as a Potential Anticancer Approach. Cancers 2021, 13, 2936. https://doi.org/10.3390/cancers13122936
Dublang L, Underhaug J, Flydal MI, Velasco-Carneros L, Maréchal J-D, Moro F, Boyano MD, Martinez A, Muga A. Inhibition of the Human Hsc70 System by Small Ligands as a Potential Anticancer Approach. Cancers. 2021; 13(12):2936. https://doi.org/10.3390/cancers13122936
Chicago/Turabian StyleDublang, Leire, Jarl Underhaug, Marte I. Flydal, Lorea Velasco-Carneros, Jean-Didier Maréchal, Fernando Moro, Maria Dolores Boyano, Aurora Martinez, and Arturo Muga. 2021. "Inhibition of the Human Hsc70 System by Small Ligands as a Potential Anticancer Approach" Cancers 13, no. 12: 2936. https://doi.org/10.3390/cancers13122936
APA StyleDublang, L., Underhaug, J., Flydal, M. I., Velasco-Carneros, L., Maréchal, J.-D., Moro, F., Boyano, M. D., Martinez, A., & Muga, A. (2021). Inhibition of the Human Hsc70 System by Small Ligands as a Potential Anticancer Approach. Cancers, 13(12), 2936. https://doi.org/10.3390/cancers13122936