
Predicting Colorectal Cancer Occurrence in IBD


Abstract
Simple Summary
Abstract
1. Introduction
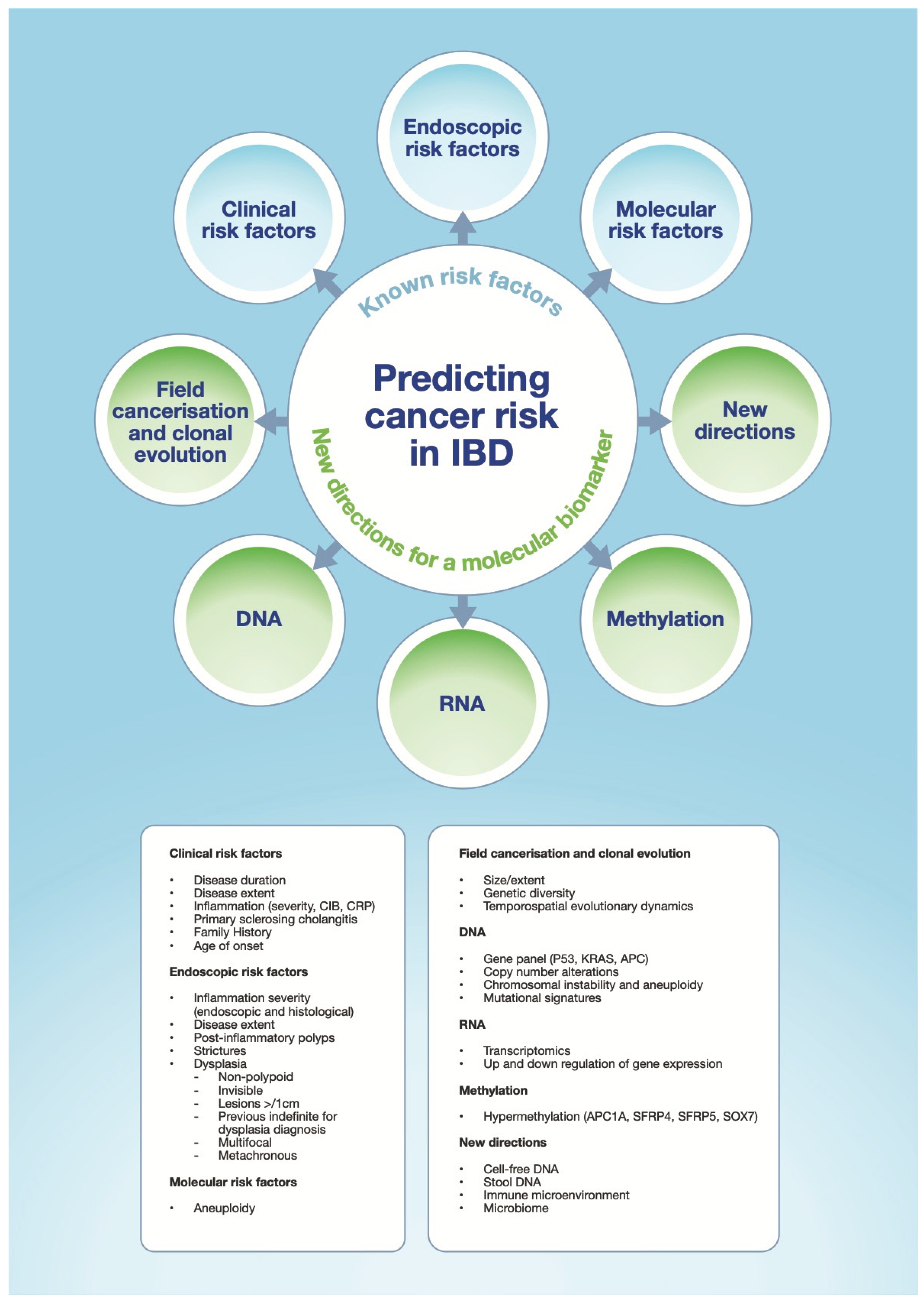
2. Risk Factors for Neoplasia Development and Progression to CA-CRC
2.1. Disease Duration
2.2. Disease Extent
2.3. Inflammation and Disease Activity
2.4. Primary Sclerosing Cholangitis, Stricturing Disease and Family History
2.5. Dysplasia
2.6. Other Associated Risk Factors

{kind=link}
| Risk Factor | Odds Ratio | Reference |
|---|---|---|
| Disease duration | ||
| Risk increases with duration of disease, most apparent after 6–8 years, with a cumulative stacked effect. | 4.74 | [6,7,39] |
| Disease extent | ||
| UC: Increasing risk respectively from pancolitis > Left sided colitis > proctitis | 2.43 | [7,39] |
| CD: Increased risk has been demonstrated with more extensive disease | N/A | [39,40,41] |
| Inflammation and severity | ||
| Risk increases with increasing disease severity (endoscopic). Even more apparent with cumulative inflammatory burden. | 2.62 | [39,47,49] |
| Risk increases with increasing disease severity (histological). | 1.98 | [39,47] |
| Primary Sclerosing Cholangitis | ||
| Associated with increased risk requiring annual surveillance from diagnosis. | 4.14 | [39,54] |
| Family history | ||
| Associated with increased risk depending on age of diagnosis and degree of relative. | 2.62 | [39,67,68] |
| Stricturing disease and post inflammatory polyps | ||
| Surrogates for previous severe inflammation and associated with higher risk | 7.78 and 3.29 | [39,62,63,101] |
| Dysplasia | ||
| Associated with increased and variable risk, with pooled CRC rate after LGD of 0.8 per 100 patient-years follow-up. | 10.7 | [14,39,60] |
| Age of onset and sex | ||
| Earlier age of onset (<16 yoa) is associated with increased risk. Males have a slightly higher risk. | 1.27 | [6,7,8,17,39,82] |
3. Current Practice in IBD Assessment and Surveillance
3.1. Clinical and Biochemical Assessment
3.2. Endoscopic Assessment
3.3. Histopathological Assessment
4. Molecular Understanding of Progression to CA-CRC
4.1. Field Cancerisation
4.2. DNA: Gene Panels and Copy Number Alterations
4.3. RNA: Transcriptomics
4.4. Methylation
4.5. Clonal Evolution and Evolutionary Dynamics
5. New Directions
5.1. Non-Invasive Tests
5.1.1. Circulating Tumour DNA (ctDNA) and Cell-Free DNA (cfDNA)
5.1.2. Stool DNA (sDNA)
5.2. Immune Micro-Environment
5.3. Microbiome
5.4. Artificial Intelligence
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Listrom, C.; Holt, K. Inflammatory bowel disease. Emerg. Med. 2004, 36, 24–28. [Google Scholar]
- Kaser, A.; Zeissig, S.B.R. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study. Lancet Gastroenterol. Hepatol. 2017, 5, 17–30. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- National Institute for Health and Clinical Excellence, Centre For Clinical Practice Scope, Ulcerative Colitis: The Management of Ulcerative Colitis. 2011, pp. 1–10. Available online: https://www.nice.org.uk/guidance/cg166/documents/ulcerative-colitis-final-scope2 (accessed on 12 May 2021).
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef]
- Ekbom, A.; Helmick, C.; Matthew Zack, H.-O.A. Ulcerative Colitis and colorectal Cancer. A population-Based study. N. Eng. J. Med. 1990, 323, 1120–1123. [Google Scholar] [CrossRef]
- Olén, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Colorectal cancer in ulcerative colitis: A Scandinavian population-based cohort study. Lancet 2020, 395, 123–131. [Google Scholar] [CrossRef]
- Castaño-Milla, C.; Chaparro, M.; Gisbert, J.P. Systematic review with meta-Analysis: The declining risk of colorectal cancer in ulcerative colitis. Aliment. Pharmacol. Ther. 2014, 39, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Simonsen, J.; Jorgensen, K.T.; Pedersen, B.V.; Nielsen, N.M.; Frisch, M. Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology 2012, 143, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kappelman, M.D.; Farkas, D.K.; Long, M.D.; Erichsen, R.; Sandler, R.S.; Sørensen, H.T.; Baron, J.A. Risk of Cancer in Patients with Inflammatory Bowel Diseases: A Nationwide Population-Based Cohort Study with 30 Years of Follow Up Michael. Clin. Gastroenterol. Hepatol. 2014, 12, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Lutgens, M.W.; van Oijen, M.G.; van der Heijden, G.J.; Vleggaar, F.P.; Siersema, P.D.; Oldenburg, B. Declining risk of colorectal cancer in inflammatory bowel disease: An updated meta-analysis of population-based cohort studies. Inflamm. Bowel Dis. 2013, 19, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Söderlund, S.; Brandt, L.; Lapidus, A.; Karlén, P.; Broström, O.; Löfberg, R.; Ekbom, A.; Askling, J. Decreasing Time-Trends of Colorectal Cancer in a Large Cohort of Patients with Inflammatory Bowel Disease. Gastroenterology 2009, 136, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.R.; Rutter, M.D.; Askari, A.; Lee, G.H.; Warusavitarne, J.; Moorghen, M.; Thomas-Gibson, S.; Saunders, B.P.; Graham, T.A.; Hart, A.L. Forty-year analysis of colonoscopic surveillance program for neoplasia in ulcerative colitis: An updated overview. Am. J. Gastroenterol. 2015, 110, 1022–1034. [Google Scholar] [CrossRef]
- Bye, W.A.; Ma, C.; Nguyen, T.M.; Parker, C.E.; Jairath, V.; East, J.E. Strategies for Detecting Colorectal Cancer in Patients with Inflammatory Bowel Disease: A Cochrane Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2018, 113, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Adami, H.O.; Bretthauer, M.; Emilsson, L.; Hernán, M.A.; Kalager, M.; Ludvigsson, J.F.; Ekbom, A. The continuing uncertainty about cancer risk in inflammatory bowel disease. Gut 2016, 65, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of Colorectal Cancer in Patients With Ulcerative Colitis: A Meta-analysis of Population-Based Cohort Studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef]
- Rutter, M.D.; Saunders, B.P.; Wilkinson, K.H.; Rumbles, S.; Schofield, G.; Kamm, M.A.; Williams, C.B.; Price, A.B.; Talbot, I.C.; Forbes, A. Thirty-Year Analysis of a Colonoscopic Surveillance Program for Neoplasia in Ulcerative Colitis. Gastroenterology 2006, 130, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68, 1–106. [Google Scholar] [CrossRef]
- Harbord, M.; Eliakim, R.; Bettenworth, D.; Karmiris, K.; Katsanos, K.; Kopylov, U.; Kucharzik, T.; Molnár, T.; Raine, T.; Sebastian, S.; et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 2: Current management. J. Crohns Colitis 2017, 11, 769–784. [Google Scholar] [CrossRef]
- Rubin, D.T.; Ananthakrishnan, A.N.; Siegel, C.A.; Sauer, B.G.; Long, M.D. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am. J. Gastroenterol. 2019, 114, 384–413. [Google Scholar] [CrossRef]
- Sebastian, S.; Hernández, V.; Myrelid, P.; Kariv, R.; Tsianos, E.; Toruner, M.; Marti-Gallostra, M.; Spinelli, A.; van der Meulen-de Jong, A.E.; Yuksel, E.S.; et al. Colorectal cancer in inflammatory bowel disease: Results of the 3rd ECCO pathogenesis scientific workshop (I). J. Crohns Colitis 2014, 8, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Cross, W.; Curtius, K.; Al Bakir, I.; Choi, C.H.R.; Davis, H.L.; Temko, D.; Biswas, S.; Martinez, P.; Williams, M.J.; et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 2019, 68, 985–995. [Google Scholar] [CrossRef]
- Ullman, T.; Odze, R.; Farraye, F.A. Diagnosis and Management of Dysplasia in Patients with Ulcerative Colitis and Crohn’s Disease of the Colon Thomas. Inflamm. Bowel Dis. 2009, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.; Abrams, K.; McKay, H.; Denley, H.; Mayberry, J. Inter-observer variation between general and specialist gastrointestinal pathologists when grading dysplasia in ulcerative colitis. J. Pathol. 2001, 194, 152–157. [Google Scholar] [CrossRef] [PubMed]
- DeRoche, T.C.; Xiao, S.Y.; Liu, X. Histological evaluation in ulcerative colitis. Gastroenterol. Rep. 2014, 2, 178–192. [Google Scholar] [CrossRef] [PubMed]
- van Schaik, F.D.; Offerhaus, G.J.; Schipper, M.E.; Siersema, P.D.; Vleggaar, F.P.; Oldenburg, B. Endoscopic and pathological aspects of colitis-associated dysplasia. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 671–678. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Health and Clinical Excellence. Colonoscopic Surveillance for Prevention of Colorectal Cancer in People with Ulcerative Colitis, Crohn’s Disease or Adenomas; Centre for Clinical Practice at NICE: London, UK, 2011.
- Ghosh, N.; Premchand, P. A UK cost of care model for inflammatory bowel disease. Frontline Gastroenterol. 2015, 6, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Shenbagaraj, L.; Thomas-Gibson, S.; Stebbing, J.; Broughton, R.; Dron, M.; Johnston, D.; Shaw, T.; Haboubi, H.N.; Green, J.T. Endoscopy in 2017: A national survey of practice in the UK. Frontline Gastroenterol. 2019, 10, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Mooiweer, E.; van der Meulen, A.E.; Ponsioen, C.Y.; van der Woude, C.J. Incidence of Interval Colorectal Cancer Among Inflammatory Bowel Disease Patients Undergoing Regular Colonoscopic Surveillance. Clin. Gastroenterol. Hepatol. 2015, 13, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.R.; Bakir, I.A.; Hart, A.L.; Graham, T.A. Clonal evolution of colorectal cancer in IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Zisman, T.L.; Bronner, M.P.; Rulyak, S.; Kowdley, K.V.; Saunders, M.; Lee, S.D.; Ko, C.; Kimmey, M.B.; Stevens, A.; Maurer, J.; et al. Prospective study of the progression of low-grade dysplasia in ulcerative colitis using current cancer surveillance guidelines. Inflamm. Bowel Dis. 2012, 18, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, P.L.; Lakatos, L. Risk for colorectal cancer in ulcerative colitis: Changes, causes and management strategies. World J. Gastroenterol. 2008, 14, 3937–3947. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Shen, Z.F.; Wu, B.S.; Xu, C.B.; He, Z.Q.; Chen, T.; Shang, H.T.; Xie, C.F.; Huang, S.Y.; Chen, Y.G.; et al. Risk of Colorectal Cancer in Ulcerative Colitis Patients: A Systematic Review and Meta-Analysis. Gastroenterol. Res. Pract. 2019. [Google Scholar] [CrossRef]
- Yoshino, T.; Nakase, H.; Takagi, T.; Bamba, S.; Okuyama, Y.; Kawamura, T.; Oki, T.; Obata, H.; Kawanami, C.; Katsushima, S.; et al. Risk factors for developing colorectal cancer in Japanese patients with ulcerative colitis: A retrospective observational study-CAPITAL (Cohort and Practice for IBD total management in Kyoto-Shiga Links) study I. BMJ Open Gastroenterol. 2016, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bopanna, S.; Ananthakrishnan, A.N.; Kedia, S.; Yajnik, V.; Ahuja, V. Risk of colorectal cancer in Asian patients with ulcerative colitis: A systematic review and meta-analysis Sawan. Lancet Gastroenterol. Hepatol. 2017, 2, 269–276. [Google Scholar] [CrossRef]
- Lutgens, M.; Vermeire, S.; Van Oijen, M.; Vleggaar, F.; Siersema, P.; van Assche, G.; Rutgeerts, P.; Oldenburg, B.; Dutch Initiative on Crohn and Colitis. A rule for determining risk of colorectal cancer in patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2015, 13, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Wijnands, A.M.; de Jong, M.E.; Lutgens, M.W.M.D.; Hoentjen, F.; Elias, S.G.; Oldenburg, B. Prognostic factors for advanced colorectal neoplasia in inflammatory bowel disease: Systematic review and meta-analysis. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Olén, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Colorectal cancer in Crohn’s disease: A Scandinavian population-based cohort study. Lancet Gastroenterol. Hepatol. 2020, 5, 475–484. [Google Scholar] [CrossRef]
- Dulai, P.S.; Sandborn, W.J.; Gupta, S. Colorectal cancer and dysplasia in inflammatory bowel disease: A review of disease epidemiology, pathophysiology, and management. Cancer Prev. Res. 2016, 9, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Gillen, D.G.C.; Walmsley, R.S.; Prior, P.; Andrews, H.A.; Allan, R.N. Ulcerative colitis and Crohn’s disease: A comparison of the colorectal cancer risk in extensive colitis. Gut 1994, 35, 1590–1592. [Google Scholar] [CrossRef]
- Choi, P.M.; Zelig, M.P. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: Implications for carcinogenesis and prevention. Gut 1994, 35, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Averboukh, F.; Ziv, Y.; Kariv, Y.; Zmora, O.; Dotan, I.; Klausner, J.M.; Rabau, M.; Tulchinsky, H. Colorectal carcinoma in inflammatory bowel disease: A comparison between Crohn’s and ulcerative colitis. Color. Dis. 2011, 13, 1230–1235. [Google Scholar] [CrossRef]
- Kiran, R.P.; Khoury, W.; Church, J.M.; Lavery, I.C.; Fazio, V.W.R.F. Colorectal cancer complicating inflammatory bowel disease: Similarities and differences between Crohn’s and ulcerative colitis based on three decades of experience. Ann. Surg. 2010, 252, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Löfberg, R.; Broström, O.; Karlén, P.; Ost, A.; Tribukait, B. Carcinoma and DNA aneuploidy in Crohn’s colitis—A histological and flow cytometric study. Gut 1991, 32, 900–904. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of Inflammation Is a Risk Factor for Colorectal Neoplasia in Ulcerative Colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Kirchgesner, J.; Svrcek, M.; Le Gall, G.; Landman, C.; Dray, X.; Bourrier, A.; Nion-Larmurier, I.; Hoyeau, N.; Sokol, H.; Seksik, P.; et al. Nancy Index Scores of Chronic Inflammatory Bowel Disease Activity Associate With Development of Colorectal Neoplasia. Clin. Gastroenterol. Hepatol. 2020, 18, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.R.; Al Bakir, I.; Ding, N.S.J.; Lee, G.H.; Askari, A.; Warusavitarne, J.; Moorghen, M.; Humphries, A.; Ignjatovic-Wilson, A.; Thomas-Gibson, S.; et al. Cumulative burden of inflammation predicts colorectal neoplasia risk in ulcerative colitis: A large single-centre study. Gut 2019, 68, 414–422. [Google Scholar] [CrossRef]
- Gupta, R.B.; Harpaz, N.; Itzkowitz, S.; Hossain, S.; Matula, S.; Kornbluth, A.; Bodian, C.; Ullman, T. Histologic Inflammation Is a Risk Factor for Progression to Colorectal Neoplasia in Ulcerative Colitis: A Cohort Study. Gastroenterology 2007, 133, 1099–1341. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.T.; Huo, D.; Kinnucan, J.A.; Sedrak, M.S.; McCullom, N.E.; Bunnag, A.P.; Raun-Royer, E.P.; Cohen, R.D.; Hanauer, S.B.; Hart, J.; et al. Inflammation is an independent risk factor for colonic neoplasia in patients with ulcerative colitis: A case-control study. Clin. Gastroenterol. Hepatol. 2013, 11, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Flores, B.M.; O’Connor, A.M.A. Impact of mucosal inflammation on risk of colorectal neoplasia in patients with ulcerative colitis: A systematic review and meta-analysis. Gastrointest Endosc. 2017, 86, 1006–1011. [Google Scholar] [CrossRef]
- Colman, R.J.; Rubin, D.T. Histological inflammation increases the risk of colorectal neoplasia in ulcerative colitis: A systematic review. Intest. Res. 2016, 14, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J. Crohns Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Soetikno, R.M.; Lin, O.S.; Heidenreich, P.A.; Young, H.S.B.M. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: A meta-analysis. Gastrointest. Endosc. 2002, 56, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, D.; Ekbom, A.; Ihre, T. Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut 1997, 41, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V.; Harewood, G.C.; Loftus, C.G.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeister, A.R.; Jewell, D.A.; Sandborn, W.J. PSC-IBD: A unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005, 54, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Guerra, I.; Bujanda, L.; Castro, J.; Merino, O.; Tosca, J.; Camps, B.; Gutiérrez, A.; Gordillo Ábalos, J.; De Castro, L.; Iborra, M.; et al. Clinical Characteristics, Associated Malignancies and Management of Primary Sclerosing Cholangitis in Inflammatory Bowel Disease Patients: A Multicentre Retrospective Cohort Study. J. Crohns Colitis 2019, 13, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, L.; Mester, G.; Erdelyi, Z.; David, G.; Pandur, T.; Balogh, M.; Fischer, S.; Vargha, P.L.P. Risk factors for ulcerative colitis-associated colorectal cancer in a Hungarian cohort of patients with ulcerative colitis: Results of a population-based study. Inflamm. Bowel Dis. 2006, 12, 205–211. [Google Scholar] [CrossRef]
- Fumery, M.; Dulai, P.S.; Gupta, S.; Prokop, L.J.; Ramamoorthy, S.; Sandborn, W.J.; Singh, S. Incidence, Risk Factors, and Outcomes of Colorectal Cancer in Patients with Ulcerative Colitis with Low-Grade Dysplasia: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2017, 15, 665–674. [Google Scholar] [CrossRef]
- Zheng, H.H.J.X. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: A meta-analysis of 16 observational studies. Eur. J. Gastroenterol. Hepatol. 2016, 28, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.D.; Saunders, B.P.; Wilkinson, K.H.; Rumbles, S.; Schofield, G.; Kamm, M.A.; Williams, C.B.; Price, A.B.; Talbot, I.C.; Forbes, A. Cancer surveillance in longstandinq ulcerative colitis: Endoscopic appearances help predict cancer risk. Gut 2004, 53, 1813–1816. [Google Scholar] [CrossRef]
- Sonnenberg, A.; Genta, R.M. Epithelial Dysplasia and Cancer in IBD Strictures. J. Crohns Colitis 2015, 9, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Lashner, B.A.; Turner, B.C.; Bostwick, D.G.; Frank, P.H.H.S. Dysplasia and cancer complicating strictures in ulcerative colitis. Dig. Dis. Sci. 1990, 35, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Fumery, M.; Pineton de Chambrun, G.; Stefanescu, C.; Buisson, A.; Bressenot, A.; Beaugerie, L.; Amiot, A.; Altwegg, R.; Savoye, G.; Abitbol, V.; et al. Detection of Dysplasia or Cancer in 3.5% of Patients With Inflammatory Bowel Disease and Colonic Strictures. Clin. Gastroenterol. Hepatol. 2015, 13, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Cremer, A.; Demetter, P.; De Vos, M.; Rahier, J.F.; Baert, F.; Moreels, T.; Macken, E.; Louis, E.; Ferdinande, L.; Fervaille, C.; et al. Risk of Development of More-advanced Lesions in Patients With Inflammatory Bowel Diseases and Dysplasia. Clin. Gastroenterol. Hepatol. 2020, 18, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Dickman, P.W.; Karlén, P.; Broström, O.; Lapidus, A.; Löfberg, R.; Ekbomz, A. Family history as a risk factor for colorectal cancer in inflammatory bowel disease. Gastroenterology 2001, 120, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Samadder, N.J.; Valentine, J.F.; Guthery, S.; Singh, H.; Bernstein, C.N.; Leighton, J.A.; Wan, Y.; Wong, J.; Boucher, K.; Pappas, L.; et al. Family History Associates With Increased Risk of Colorectal Cancer in Patients With Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2019, 17, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Nuako, K.W.; Ahlquist, D.A.; Mahoney, D.W.; Schaid, D.J.; Siems, D.M.L.N. Familial predisposition for colorectal cancer in chronic ulcerative colitis: A case-control study. Gastroenterology. Gastroenterology 1998, 115, 1079–1083. [Google Scholar] [CrossRef]
- De Jong, M.E.; Van Tilburg, S.B.; Nissen, L.H.; Kievit, W.; Nagtegaal, I.D.; Horjus, C.S.; Römkens, T.E.; Drenth, J.P.; Hoentjen, F.; Derikx, L.A. Long-term Risk of Advanced Neoplasia after Colonic Low-grade Dysplasia in Patients with Inflammatory Bowel Disease: A Nationwide Cohort Study. J. Crohns Colitis 2019, 13, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-h.R.; Ignjatovic-Wilson, A.; Askari, A.; Lee, G.H.; Warusavitarne, J.; Moorghen, M.; Thomas-Gibson, S.; Saunders, B.P.; Rutter, M.D.; Graham, T.A. Low-Grade Dysplasia in Ulcerative Colitis: Risk Factors for Developing High-Grade Dysplasia or Colorectal Cancer. Am. J. Gastroenterol. 2015, 110, 1461–1471. [Google Scholar] [CrossRef]
- Connell, W.R.; Lennard-Jones, J.E.; Williams, C.B.; Talbot, I.C.; Price, A.B.; Wilkinson, K.H. Factors affecting the outcome of endoscopic surveillance for cancer in ulcerative colitis. Gastroenterology 1994, 107, 934–944. [Google Scholar] [CrossRef]
- Lim, C.H.; Dixon, M.F.; Vail, A.; Forman, D.; Lynch, D.A.F.; Axon, A.T.R. Ten year follow up of ulcerative colitis patients with and without low grade dysplasia. Gut 2003, 52, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N.; Shanahan, F.W.W. Are we telling patients the truth about surveillance colonoscopy in ulcerative colitis? Lancet 1994, 8, 71–74. [Google Scholar]
- Ullman, T.; Croog, V.; Harpaz, N.; Sachar, D.I.S. Progression of flat low-grade dysplasia to advanced neoplasia in patients with ulcerative colitis. Gastroenterology 2003, 125, 1211–1219. [Google Scholar] [CrossRef]
- Befrits, R.; Ljung, T.; Jaramillo, E.R.C. Low-grade dysplasia in extensive, long-standing inflammatory bowel disease: A follow-up study. Dis. Colon. Rectum. 2002, 45, 615–620. [Google Scholar] [CrossRef]
- Kabir, M.; Fofaria, R.; Arebi, N.; Bassett, P.; Tozer, P.J.; Hart, A.L.; Thomas-gibson, S.; Humphries, A.; Suzuki, N.; Saunders, B.; et al. Systematic review with meta-analysis: IBD-associated colonic dysplasia prognosis in the videoendoscopic era (1990 to present ). Aliment. Pharmacol. Ther. 2020, 52, 5–19. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.E.; Kanne, H.; Nissen, L.H.C.; Drenth, J.P.H.; Derikx, L.A.A.P. Increased risk of high-grade dysplasia and colorectal cancer in inflammatory bowel disease patients with recurrent low-grade dysplasia. Gastrointest. Endosc. 2020, 91, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Ten Hove, J.R.; Shah, S.C.; Shaffer, S.R.; Bernstein, C.N.; Castaneda, D.; Palmela, C.; Mooiweer, E.; Elman, J.; Kumar, A.; Glass, J.; et al. Consecutive negative findings on colonoscopy during surveillance predict a low risk of advanced neoplasia in patients with inflammatory bowel disease with long-standing colitis: Results of a 15-year multicentre, multinational cohort study. Gut 2019, 68, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, R.; Shah, S.C.; Torres, J.; Castaneda, D.; Glass, J.; Elman, J.; Kumar, A.; Axelrad, J.; Harpaz, N.; Ullman, T.; et al. Association Between Indefinite Dysplasia and Advanced Neoplasia in Patients With Inflammatory Bowel Diseases Undergoing Surveillance. Clin. Gastroenterol. Hepatol. 2020, 18, 1518–1527. [Google Scholar] [CrossRef]
- Curtius, K.; Kabir, M.; Al Bakir, I.; Choi, C.-H.R.; Hartono, J.L.; Johnson, M.; East, J.E.; Oxford IBD Cohort Study Investigators; Lindsay, J.O.; Vega, R.; et al. Multi-centre derivation and validation of a colitis-associated colorectal cancer risk prediction web-tool. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Olén, O.; Askling, J.; Sachs, M.C.; Frumento, P.; Neovius, M.; Smedby, K.E.; Ekbom, A.; Malmborg, P.; Ludvigsson, J.F. Childhood onset inflammatory bowel disease and risk of cancer: A Swedish nationwide cohort study. BMJ 2017, 358, 3951. [Google Scholar] [CrossRef] [PubMed]
- Pinczowski, D.; Ekbom, A.; Baron, J.; Yuen, J.; Adami, H.O. Risk factors for colorectal cancer in patients with ulcerative colitis: A case-control study. Gastroenterology 1994, 107, 117–120. [Google Scholar] [CrossRef]
- Cheng, Y.; Desreumaux, P. 5-Aminosalicylic acid is an attractive candidate agent for chemoprevention of colon cancer in patients with inflammatory bowel disease. World J. Gastroenterol. 2005, 11, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Van Staa, T.P.; Card, T.; Logan, R.F.; Leufkens, H.G.M. 5-Aminosalicylate use and colorectal cancer risk in inflammatory bowel disease: A large epidemiological study. Gut 2005, 54, 1573–1578. [Google Scholar] [CrossRef]
- Ullman, T.; Croog, V.; Harpaz, N.; Hossain, S.; Kornbluth, A.; Bodian, C.; Itzkowitz, S. Progression to Colorectal Neoplasia in Ulcerative Colitis: Effect of Mesalamine. Clin. Gastroenterol. Hepatol. 2008, 6, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Ma, J.; Wang, K.; Zhang, H. Chemopreventive effects of 5-aminosalicylic acid on inflammatory bowel disease-associated colorectal cancer and dysplasia: A systematic review with meta-analysis. Oncotarget 2017, 8, 1031–1045. [Google Scholar] [CrossRef]
- Zhao, L.N.; Li, J.Y.; Yu, T.; Chen, G.C.; Yuan, Y.H.; Chen, Q.K. 5-Aminosalicylates reduce the risk of colorectal neoplasia in patients with ulcerative colitis: An updated meta-analysis. PLoS ONE 2014, 9, e94208. [Google Scholar] [CrossRef] [PubMed]
- Bonovas, S.; Fiorino, G.; Lytras, T.; Nikolopoulos, G.; Peyrin-Biroulet, L.D.S. Systematic review with meta-analysis: Use of 5-aminosalicylates and risk of colorectal neoplasia in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2017, 45, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Velayos, F.S.; Terdiman, J.P.W.J. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: A systematic review and metaanalysis of observational studies. Am. J. Gastroenterol. 2005, 100, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Matula, S.; Croog, V.; Itzkowitz, S.; Harpaz, N.; Bodian, C.; Hossain, S.; Ullman, T. Chemoprevention of colorectal neoplasia in ulcerative colitis: The effect of 6-mercaptopurine. Clin. Gastroenterol. Hepatol. 2005, 3, 1015–1021. [Google Scholar] [CrossRef]
- Carrat, F.; Seksik, P.; Colombel, J.F.; Peyrin-Biroulet, L.; Beaugerie, L.; CESAME Study Group. The effects of aminosalicylates or thiopurines on the risk of colorectal cancer in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2017, 45, 533–541. [Google Scholar] [CrossRef]
- Jess, T.; Lopez, A.; Andersson, M.; Beaugerie, L.; Peyrin-Biroulet, L. Thiopurines and Risk of Colorectal Neoplasia in Patients With Inflammatory Bowel Disease: A Meta-analysis. Clin. Gastroenterol. Hepatol. 2014, 12, 1793–1800.e1. [Google Scholar] [CrossRef] [PubMed]
- Actis, G.C.; Fadda, M.; Pellicano, R.; David, E.; Rizzetto, M.S.A. The 17-year single-center experience with the use of azathioprine to maintain remission in ulcerative colitis. Biomed. Pharmacother. 2009, 63, 362–365. [Google Scholar] [CrossRef] [PubMed]
- van Schaik, F.D.M.; van Oijen, M.G.H.; Smeets, H.M.; van der Heijden, G.J.M.G.; Siersema, P.D.; Oldenburg, B. Thiopurines prevent advanced colorectal neoplasia in patients with inflammatory bowel disease. Gut 2012, 61, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Mei, Z.; Guo, Y.; Wang, G.; Wu, T.; Cui, X.; Huang, Z.; Zhu, Y.; Wen, D.; Song, J.; et al. Reduced risk of inflammatory bowel disease-associated colorectal neoplasia with use of thiopurines: A systematic review and meta-analysis. J. Crohns Colitis 2018, 12, 546–558. [Google Scholar] [CrossRef]
- Lu, M.J.; Qiu, X.Y.; Mao, X.Q.; Li, X.T.; Zhang, H.J. Systematic review with meta-analysis: Thiopurines decrease the risk of colorectal neoplasia in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2018, 47, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Bezzio, C.; Festa, S.; Saibeni, S.P.C. Chemoprevention of colorectal cancer in ulcerative colitis: Digging deep in current evidence. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Pouillon, L.; Beaugerie, L.; Danese, S.P.-B.L. Colorectal cancer prevention in patients with ulcerative colitis. Best Pr. Res. Clin. Gastroenterol. 2018, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.K.; Kramer, J.R.; Richardson, P.; Mei, M.; El-Serag, H.B. Risk of colorectal cancer among Caucasian and African American veterans with ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.D.; Riddell, R.H. Colorectal dysplasia in inflammatory bowel disease: A clinicopathologic perspective. Clin. Gastroenterol. Hepatol. 2014, 12, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Koutroubakis, I.E.; Regueiro, M.; Schoen, R.E.; Ramos-Rivers, C.; Hashash, J.G.; Schwartz, M.; Swoger, J.; Barrie, A.; Baidoo, L.; Dunn, M.A.; et al. Multiyear Patterns of Serum Inflammatory Biomarkers and Risk of Colorectal Neoplasia in Patients with Ulcerative Colitis. Inflamm. Bowel Dis. 2016, 22, 100–105. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ananthakrishnan, A.N.; Cheng, S.-C.; Cai, T.; Cagan, A.; Gainer, V.S.; Szolovits, P.; Shaw, S.Y.; Churchill, S.; Karlson, E.W.; Murphy, S.N.; et al. Serum Inflammatory Markers and Risk of Colorectal Cancer in Patients with Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2014, 12, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Rasic, I.; Radovic, S.; Aksamija, G. Relationship Between Chronic Inflammation and the Stage and Histopathological Size of Colorectal Carcinoma. Med. Arch. 2016, 70, 104–107. [Google Scholar] [CrossRef]
- Walsh, A.; Kormilitzin, A.; Hinds, C.; Sexton, V.; Brain, O.; Keshav, S.; Uhlig, H.; Geddes, J.; Goodwin, G.; Peters, M.; et al. Defining Faecal Calprotectin Thresholds as a Surrogate for Endoscopic and Histological Disease Activity in Ulcerative Colitis-a Prospective Analysis. J. Crohns Colitis 2019, 13, 424–430. [Google Scholar] [CrossRef]
- Hart, L.; Chavannes, M.; Kherad, O.; Maedler, C.; Mourad, N.; Marcus, V.; Afif, W.; Bitton, A.; Lakatos, P.L.; Brassard, P.B.T. Faecal Calprotectin Predicts Endoscopic and Histological Activity in Clinically Quiescent Ulcerative Colitis. J. Crohns Colitis 2020, 14, 46–52. [Google Scholar] [CrossRef]
- Schoepfer, A.M.; Beglinger, C.; Straumann, A.; Trummler, M.; Renzulli, P.S.F. Ulcerative colitis: Correlation of the Rachmilewitz endoscopic activity index with fecal calprotectin, clinical activity, C-reactive protein, and blood leukocytes. Inflamm. Bowel Dis. 2009, 15, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Mosli, M.H.; Zou, G.; Garg, S.K.; Feagan, S.G.; MacDonald, J.K.; Chande, N.; Sandborn, W.J.F.B. C-Reactive Protein, Fecal Calprotectin, and Stool Lactoferrin for Detection of Endoscopic Activity in Symptomatic Inflammatory Bowel Disease Patients: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2015, 110, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Mooiweer, E.; Fidder, H.H.; Siersema, P.D.; Laheij, R.J.F.; Oldenburg, B. Fecal calprotectin testing can identify ineffective colorectal cancer surveillance procedures in patients with longstanding colitis. Inflamm. Bowel Dis. 2014, 20, 1079–1084. [Google Scholar] [CrossRef]
- von Roon, A.C.; Karamountzos, L.; Purkayastha, S.; Reese, G.E.; Darzi, A.W.; Teare, J.P.; Paraskeva, P.T.P. Diagnostic precision of fecal calprotectin for inflammatory bowel disease and colorectal malignancy. Am. J. Gastroenterol. 2007, 102, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, R.; Fritsch, J.; Holtmann, M.; Koehler, H.H.; Stolte, M.; Kanzler, S.; Nafe, B.; Jung, M.; Galle, P.R.; Neurath, M.F. Methylene blue-aided chromoendoscopy for the detection of intraepithelial neoplasia and colon cancer in ulcerative colitis. Gastroenterology 2003, 124, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Hurlstone, D.P.; Sanders, D.S.; Lobo, A.J.; McAlindon, M.E.C.S. Indigo carmine-assisted high-magnification chromoscopic colonoscopy for the detection and characterisation of intraepithelial neoplasia in ulcerative colitis: A prospective evaluation. Endoscopy 2005, 37, 1186–1192. [Google Scholar] [CrossRef] [PubMed]
- Marion, J.F.; Waye, J.D.; Present, D.H.; Israel, Y.; Bodian, C.; Harpaz, N.; Chapman, M.; Itzkowitz, S.; Steinlauf, A.F.; Abreu, M.T.; et al. Chromoendoscopy-targeted biopsies are superior to standard colonoscopic surveillance for detecting dysplasia in inflammatory bowel disease patients: A prospective endoscopic trial. Am. J. Gastroenterol. 2008, 103, 2342–2349. [Google Scholar] [CrossRef]
- Subramanian, V.; Mannath, J.; Ragunath, K.; Hawkey, C.J. Meta-analysis: The diagnostic yield of chromoendoscopy for detecting dysplasia in patients with colonic inflammatory bowel disease. Aliment. Pharmacol. Ther. 2011, 33, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Soetikno, R.; Subramanian, V.; Kaltenbach, T.; Rouse, R.V.; Sanduleanu, S.; Suzuki, N.; Tanaka, S.; McQuaid, K. The detection of nonpolypoid (flat and depressed) colorectal neoplasms in patients with inflammatory bowel disease. Gastroenterology 2013, 144, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Mooiweer, E.; van der Meulen-de Jong, A.E.; Ponsioen, C.Y.; Fidder, H.H.; Siersema, P.D.; Dekker, E.O.B. Chromoendoscopy for Surveillance in Inflammatory Bowel Disease Does Not Increase Neoplasia Detection Compared With Conventional Colonoscopy With Random Biopsies: Results From a Large Retrospective Study. Am. J. Gastroenterol. 2015, 110, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Bressenot, A.; Salleron, J.; Bastien, C.; Danese, S.; Boulagnon-Rombi, C.P.-B.L. Comparing histological activity indexes in, U.C. Gut 2015, 64, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Osterman, M.T.; Scott, F.I.; Fogt, F.F.; Gilroy, E.D.; Parrott, S.; Galanko, J.; Cross, R.; Moss, A.; Herfarth, H.H.; Higgins, P.D.R.; et al. Endoscopic and Histological Assessment, Correlation, and Relapse in Clinically Quiescent Ulcerative Colitis (MARQUEE). Inflamm. Bowel Dis. 2020, 27, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Jauregui-Amezaga, A.; Geerits, A.; Das, Y.; Lemmens, B.; Sagaert, X.; Bessissow, T.; Lobatón, T.; Ferrante, M.; Van Assche, G.; Bisschops, R.; et al. A Simplified Geboes Score for Ulcerative Colitis. J. Crohns. Colitis 2017, 11, 305–313. [Google Scholar] [CrossRef]
- Marchal-Bressenot, A.; Salleron, J.; Boulagnon-Rombi, C.; Bastien, C.; Cahn, V.; Cadiot, G.; Diebold, M.D.; Danese, S.; Reinisch, W.; Schreiber, S.; et al. Development and validation of the Nancy histological index for UC. Gut 2017, 66, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Marchal-Bressenot, A.; Scherl, A.; Salleron, J.; Peyrin-Biroulet, L. A practical guide to assess the Nancy histological index for, U.C. Gut. 2016, 65, 1919–1920. [Google Scholar] [CrossRef] [PubMed]
- Popp, C.; Nichita, L.; Voiosu, T.; Bastian, A.; Cioplea, M.; Micu, G.; Pop, G.; Sticlaru, L.; Bengus, A.; Voiosu, A.; et al. Expression Profile of p53 and p21 in Large Bowel Mucosa as Biomarkers of Inflammatory-Related Carcinogenesis in Ulcerative Colitis. Dis. Markers 2016, 2016, 3625279. [Google Scholar] [CrossRef] [PubMed]
- Noffsinger, A.E.; Belli, J.M.; Miller, M.A.; Fenoglio-Preiser, C.M. A unique basal pattern of p53 expression in ulcerative colitis is associated with mutation in the p53 gene. Histopathology 2001, 39, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Machinami, R. Immunohistochemistry of ulcerative colitis-associated with dysplasia and carcinoma. Pathol. Int. 1999, 49, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Fujimori, T.; Mitomi, H.; Tomita, S.; Ichikawa, K.; Imura, J.; Fujii, S.; Itabashi, M.; Kameoka, S.; Igarashi, Y. Immunohistochemical assessment of a unique basal pattern of p53 expression in ulcerative-colitis-associated neoplasia using computer-assisted cytometry. Diagn. Pathol. 2014, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, Y.; Tan, S. p53 expression in patients with ulcerative colitis - associated with dysplasia and carcinoma: A systematic meta-analysis. BMC Gastroenterol. 2017, 17, 1–8. [Google Scholar] [CrossRef]
- Brahim, E.B.; Mrabet, A.; Jouini, R.; Koubaa, W.; Sidhom, R.B.; Elloumi, H.C.A. Immunohistochemistry in the diagnosis of dysplasia in chronic inflammatory bowel disease colorectal polyps. Arab J. Gastroenterol. 2016, 17, 121–126. [Google Scholar] [CrossRef] [PubMed]
- van Schaik, F.D.; Oldenburg, B.; Offerhaus, G.J.; Schipper, M.E.; Vleggaar, F.P.; Siersema, P.D.; van Oijen, M.G.H.; ten Kate, F.J.W. Role of immunohistochemical markers in predicting progression of dysplasia to advanced neoplasia in patients with ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 480–488. [Google Scholar] [CrossRef]
- Kaserer, K.; Schmaus, J.; Bethge, U.; Migschitz, B.; Fasching, S.; Walch, A.; Herbst, F.; Teleky, B.; Wrba, F. Staining patterns of p53 immunohistochemistry and their biological significance in colorectal cancer. J. Pathol. 2000, 190, 450–456. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tomita, H.; Shimizu, M.; Tanaka, T.; Suzui, N.; Miyazaki, T.; Hara, A. P53 expression as a diagnostic biomarker in ulcerative colitis-associated cancer. Int. J. Mol. Sci. 2017, 18, 1284. [Google Scholar] [CrossRef]
- Toritani, K.; Kimura, H.; Kunisaki, R.; Watanabe, J.; Kunisaki, C.; Ishibe, A.; Chiba, S.; Inayama, Y.; Endo, I. Uselessness of serum p53 antibody for detecting colitis-associated cancer in the era of immunosuppressive therapy. In Vivo 2020, 34, 723–728. [Google Scholar] [CrossRef]
- Pratt, P.K.; David, N.; Weber, H.C.; Little, F.F.; Kourkoumpetis, T.; Patts, G.J.; Weinberg, J.; Farraye, F.A. Antibody Response to Hepatitis B Virus Vaccine is Impaired in Patients with Inflammatory Bowel Disease on Infliximab Therapy. Inflamm. Bowel Dis. 2018, 24, 380–386. [Google Scholar] [CrossRef]
- Ford, A.C.; Sandborn, W.J.; Khan, K.J.; Hanauer, S.B.; Talley, N.J.; Moayyedi, P. Efficacy of biological therapies in inflammatory bowel disease: Systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 644–659. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, G.; Peyrin-Biroulet, L.; Naccarato, P.; Szabò, H.; Sociale, O.R.; Vetrano, S.; Fries, W.; Montanelli, A.; Repici, A.; Malesci, A.D.S. Effects of immunosuppression on immune response to pneumococcal vaccine in inflammatory bowel disease: A prospective study. Inflamm. Bowel Dis. 2012, 18, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Loustaunau, C.; Facista, A.; Ramsey, L.; Hassounah, N.; Taylor, H.; Krouse, R.; Payne, C.M.; Tsikitis, V.L.; Goldschmid, S.; et al. Deficient Pms2, ERCC1, Ku86, CcOI in field defects during progression to colon cancer. J. Vis. Exp. 2010, 28, 1931. [Google Scholar] [CrossRef] [PubMed]
- Al Bakir, I.; Curtius, K.; Graham, T.A. From colitis to cancer: An evolutionary trajectory that merges maths and biology. Front. Immunol. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Huels, D.J.; Ridgway, R.A.; Sansom, O.J.; Kholodenko, B.N.; Kolch, W.; Cho, K.H. The APC network regulates the removal of mutated cells from colonic crypts. Cell Rep. 2014, 7, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Kozar, S.; Morrissey, E.; Nicholson, A.M.; van der Heijden, M.; Zecchini, H.I.; Kemp, R.; Tavaré, S.; Vermeulen, L.; Winton, D.J. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem. Cell 2013, 13, 626–633. [Google Scholar] [CrossRef]
- Baker, A.M.; Cereser, B.; Melton, S.; Fletcher, A.G.; Rodriguez-Justo, M.; Tadrous, P.J.; Humphries, A.; Elia, G.; McDonald, S.A.C.; Wright, N.A.; et al. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 2014, 8, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, D.P.; Southwick, H.W.S.W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2017, 18, 19–32. [Google Scholar] [CrossRef]
- Braakhuis, B.J.M.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar] [PubMed]
- Leedham, S.J.; Graham, T.A.; Oukrif, D.; McDonald, S.A.C.; Rodriguez-Justo, M.; Harrison, R.F.; Shepherd, N.A.; Novelli, M.R.; Jankowski, J.A.Z.; Wright, N.A. Clonality, Founder Mutations, and Field Cancerization in Human Ulcerative Colitis-Associated Neoplasia. Gastroenterology 2009, 136, 542–550.e6. [Google Scholar] [CrossRef] [PubMed]
- Galandiuk, S.; Rodriguez-Justo, M.; Jeffery, R.; Nicholson, A.M.; Cheng, Y.; Oukrif, D.; Elia, G.; Leedham, S.J.; McDonald, S.A.C.; Wright, N.A.; et al. Field Cancerization in the Intestinal Epithelium of Patients with Crohn’s Ileocolitis. Gastroenterology 2012, 142, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Salipante, S.J.; Risques, R.A.; Crispin, D.A.; Li, L.; Bronner, M.P.; Brentnall, T.A.; Rabinovitch, P.S.; Horwitz, M.S.; Loeb, L.A. Clonal expansions in ulcerative colitis identify patients with neoplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 20871–20876. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Bansal, A.; Lai, L.A.; Crispin, D.A.; Ussakli, C.H.; Horwitz, M.S.; Bronner, M.P.; Brentnall, T.A.; Loeb, L.A.; Rabinovitch, P.S.; et al. Clonal expansions and short telomeres are associated with neoplasia in early-onset, but not late-onset, ulcerative colitis. Inflamm. Bowel Dis. 2013, 19, 2593–2602. [Google Scholar] [CrossRef]
- Lai, L.A.; Risques, R.A.; Bronner, M.P.; Rabinovitch, P.S.; Crispin, D.; Chen, R.; Brentnall, T.A. Pan-colonic field defects are detected by CGH in the colons of, U.C. patients with dysplasia/cancer. Cancer Lett. 2012, 320, 180–188. [Google Scholar] [CrossRef]
- Rubin, C.E.; Haggitt, R.C.; Burmer, G.C.; Brentnall, T.A.; Stevens, A.C.; Levine, D.S.; Dean, P.J.; Kimmey, M.; Perera, D.R.; Rabinovitch, P.S. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 1992, 103, 1611–1620. [Google Scholar] [CrossRef]
- Meyer, R.; Freitag-Wolf, S.; Blindow, S.; Büning, J.; Habermann, J.K. Combining aneuploidy and dysplasia for colitis’ cancer risk assessment outperforms current surveillance efficiency: A meta-analysis. Int. J. Colorectal. Dis. 2017, 32, 171–182. [Google Scholar] [CrossRef]
- Söderlund, S.; Tribukait, B.; Öst, Å.; Broström, O.; Karlén, P.; Löfberg, R.; Askling, J.S.U. Colitis-associated DNA aneuploidy and dysplasia in Crohn’s disease and risk of colorectal cancer. Inflamm. Bowel Dis. 2011, 17, 1101–1107. [Google Scholar] [CrossRef]
- Gerling, M.; Nousiainen, K.; Hautaniemi, S.; Krüger, S.; Fritzsche, B.; Homann, N.; Bruch, H.P.; Auer, G.; Roblick, U.J.; Ried, T.H.J. Aneuploidy-associated gene expression signatures characterize malignant transformation in ulcerative colitis. Inflamm. Bowel Dis. 2013, 19, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.F.; Nause, S.L.; Freitag-Wolf, S.; Krüger, S.; Bruch, H.P.; Roblick, U.J.H.J. Aneuploidy characterizes adjacent non-malignant mucosa of ulcerative colitis-associated but not sporadic colorectal carcinomas: A matched-pair analysis. Scand. J. Gastroenterol. 2013, 48, 679–687. [Google Scholar] [CrossRef]
- Kakiuchi, N.; Yoshida, K.; Uchino, M.; Kihara, T.; Akaki, K.; Inoue, Y.; Kawada, K.; Nagayama, S.; Yokoyama, A.; Yamamoto, S.; et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 2020, 577, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Nanki, K.; Fujii, M.; Shimokawa, M.; Matano, M.; Nishikori, S.; Date, S.; Takano, A.; Toshimitsu, K.; Ohta, Y.; Takahashi, S.; et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 2020, 577, 254–259. [Google Scholar] [CrossRef]
- Esserman, L.J.; Thompson, I.M.; Reid, B.; Nelson, P.; Ransohoff, D.F.; Welch, H.G.; Hwang, S.; Berry, D.A.; Kinzler, K.W.; William, C.P.U. Addressing overdiagnosis and overtreatment in cancer: A prescription for change Prof. Lancet Oncol. 2014, 15, e234–e242. [Google Scholar] [CrossRef]
- Mayer, R.; Wong, W.D.; Rothenberger, D.A.; Goldberg, S.M.M.R. Colorectal cancer in inflammatory bowel disease: A continuing problem. Dis. Colon. Rectum. 1999, 42, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer—IV.; Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.V.B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Hao, X.P.; Frayling, I.M.; Sgouros, J.G.; Du, M.Q.; Willcocks, T.C.; Talbot, I.C.; Tomlinson, I.P.M. The spectrum of p53 mutations in colorectal adenomas differs from that in colorectal carcinomas. Gut 2002, 50, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Cross, W.; Kovac, M.; Mustonen, V.; Temko, D.; Davis, H.; Baker, M.; Biswas, S.; Arnold, R.; Chegwidden, L.; Gatenbee, C.; et al. Europe PMC Funders Group The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2018, 2, 1661–1672. [Google Scholar] [CrossRef]
- Cross, W.C.H.; Graham, T.A.; Wright, N.A. New paradigms in clonal evolution: Punctuated equilibrium in cancer. J. Pathol. 2016, 240, 126–136. [Google Scholar] [CrossRef]
- Yin, J.; Harpaz, N.; Tong, Y.; Huang, Y.; Laurin, J.; Greenwald, B.D.; Hontanosas, M.; Newkirk, C.; Meltzer, S.J. Point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993, 104, 1633–1639. [Google Scholar] [CrossRef]
- Wanders, L.K.; Cordes, M.; Voorham, Q.; Sie, D.; De Vries, S.D.; D’Haens, G.R.A.M.; De Boer, N.K.H.; Ylstra, B.; Van Grieken, N.C.T.; Meijer, G.A.; et al. IBD-Associated Dysplastic Lesions Show More Chromosomal Instability Than Sporadic Adenomas. Inflamm. Bowel Dis. 2020, 26, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Tarmin, L.; Yin, J.; Kozam, M.; Noordzij, J.; Jiang, H.Y.; Meltzer, S.J.; Harpaz, N.; Antonio, L.B.; Chan, O.; Cymes, K. Adenomatous Polyposis Coli Gene Mutations in Ulcerative Colitis-associated Dysplasias and Cancers versus Sporadic Colon Neoplasms. Cancer Res. 1995, 55, 2035–2038. [Google Scholar] [PubMed]
- Burmer, G.C.; Levine, D.S.; Kulander, B.G.; Haggitt, R.C.; Rubin, C.E.; Rabinovitch, P.S. C-Ki-ras mutations in chronic ulcerative colitis and sporadic colon carcinoma. Gastroenterology 1990, 99, 416–420. [Google Scholar] [CrossRef]
- Lgfberg, R.; Brostrom, O.; Karlen, P.; Ost, A.; Tribukait, B. DNA Aneuploidy in Ulcerative Colitis: Reproducibility, Topographic Distribution, and Relation to Dysplasia. Gastroenterology 1992, 102, 1149–1154. [Google Scholar] [CrossRef]
- Tsai, J.H.; Rabinovitch, P.S.; Huang, D.; Small, T.; Mattis, A.N.; Kakar, S.C.W. Association of Aneuploidy and Flat Dysplasia With Development of High-Grade Dysplasia or Colorectal Cancer in Patients With Inflammatory Bowel Disease. Gastroenterology 2017, 153, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jin, Z.; Li, X.; Wu, L.; Jin, J. Associations between single-nucleotide polymorphisms and inflammatory bowel disease-associated colorectal cancers in inflammatory bowel disease patients: A meta-analysis. Clin. Transl. Oncol. 2017, 19, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-exome Sequencing analyses of Inflammatory Bowel Disease-associated Colorectal Cancers Ana. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-associated Cancers are Distinct from Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2017, 151, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, S.; Varghese, V.K.; Sahu, P.; Jayaram, P.; Shivakumar, B.M.; Pai, C.G.; Satyamoorthy, K. Targeted sequencing-based analyses of candidate gene variants in ulcerative colitis-associated colorectal neoplasia. Br. J. Cancer 2017, 117, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, M.; Ota, H.; Tateishi, Y.; Nakajima, T.; Riddell, R.C.J. Colitis-associated colorectal adenocarcinomas are frequently associated with non-intestinal mucin profiles and loss of SATB2 expression. Mod. Pathol. 2019, 32, 884–892. [Google Scholar] [CrossRef]
- Chen, R.; Lai, L.A.; Brentnall, T.A.; Pan, S. Biomarkers for colitis-associated colorectal cancer. World J. Gastroenterol. 2016, 22, 7882–7891. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.N.; Finley, J.C.; Risques, R.A.; Shen, W.T.; Gollahon, K.A.R.P. Quantitative fluorescence in situ hybridization (QFISH) of telomere lengths in tissue and cells. Curr. Protoc. Cytom. 2005, 33. [Google Scholar] [CrossRef] [PubMed]
- Risques, R.A.; Lai, L.A.; Brentnall, T.A.; Li, L.; Feng, Z.; Gallaher, J.; Mandelson, M.T.; Potter, J.D.; Bronner, M.P.R.P. Ulcerative colitis is a disease of accelerated colon aging: Evidence from telomere attrition and DNA damage. Gastroenterology 2008, 135, 410–418. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.N.; Bronner, M.P.; Brentnall, T.A.; Finley, J.C.; Shen, W.T.; Emerson, S.; Emond, M.J.; Gollahon, K.A.; Moskovitz, A.H.; Crispin, D.A.; et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat. Genet. 2002, 32, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, G.; Goranova, T.E.; De, S.D.; Ennis, D.; Anna, M.; Eldridge, M.; Sie, D.; Lewsley, L.; Hanif, A.; Dowson, S.; et al. Europe PMC Funders Group Copy-number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2019, 50, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Scheinin, I.; Sie, D.; Bengtsson, H.; Van De Wiel, M.A.; Olshen, A.B.; Van Thuijl, H.F.; Van Essen, H.F.; Eijk, P.P.; Rustenburg, F.; Meijer, G.A.; et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome. Res. 2014, 24, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, B.M.; Rotti, H.; Vasudevan, T.G.; Balakrishnan, A.; Chakrabarty, S.; Bhat, G.; Rao, L.; Pai, C.G.; Atyamoorthy, K. Copy number variations are progressively associated with the pathogenesis of colorectal cancer in ulcerative colitis. World J. Gastroenterol. 2015, 21, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Colliver, D.W.; Crawford, N.P.; Eichenberger, M.R.; Zacharius, W.; Petras, R.E.; Stromberg, A.J.G.S. Molecular profiling of ulcerative colitis-associated neoplastic progression. Exp. Mol. Pathol. 2006, 80, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.; Dougherty, U.; Huang, Y.; Gometz, E.; Nathanson, J.; Cohen, G.; Levy, S.; Kocherginsky, M.; Venu, N.; Westerhoff, M.; et al. Gene Signature Distinguishes Patients with Chronic Ulcerative Colitis Harboring Remote Neoplastic Lesions. Inflamm. Bowel Dis. 2013, 19, 461–470. [Google Scholar] [CrossRef]
- Watanabe, T.; Kobunai, T.; Yamamoto, Y.; Ikeuchi, H.; Matsuda, K.; Ishihara, S.; Nozawa, K.; Iinuma, H.; Kanazawa, T.; Tanaka, T.; et al. Predicting ulcerative colitis-associated colorectal cancer using reverse-transcription polymerase chain reaction analysis. Clin. Color. Cancer 2011, 10, 134–141. [Google Scholar] [CrossRef]
- Huang, R.; Wang, K.; Gao, L.; Gao, W. TIMP1 is a potential key gene associated with the pathogenesis and prognosis of ulcerative colitis-associated colorectal cancer. Onco Targets Ther. 2019, 12, 8895–8904. [Google Scholar] [CrossRef]
- de Barrios, O.; Sanchez-Moral, L.; Cortés, M.; Ninfali, C.; Profitós-Pelejà, N.; Martínez-Campanario, M.C.; Siles, L.; Del Campo, R.; Fernández-Aceñero, M.J.; Darling, D.S.; et al. ZEB1 promotes inflammation and progression towards inflammation-driven carcinoma through repression of the DNA repair glycosylase MPG in epithelial cells. Gut 2019, 68, 2129–2141. [Google Scholar] [CrossRef]
- Sheng, Y.H.; Wong, K.Y.; Seim, I.; Wang, R.; He, Y.; Wu, A.; Patrick, M.; Lourie, R.; Schreiber, V.; Giri, R.; et al. MUC13 promotes the development of colitis-associated colorectal tumors via β-catenin activity. Oncogene 2019, 38, 7294–7310. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Okugawa, Y.; Kondo, S.; Okita, Y.; Araki, T.; Kusunoki, K.; Uchino, M.; Ikeuchi, H.; Hirota, S.; Mitsui, A.; et al. Comprehensive analysis identifying aberrant DNA methylation in rectal mucosa from ulcerative colitis patients with neoplasia. Oncotarget 2018, 9, 33149–33159. [Google Scholar] [CrossRef]
- Tominaga, K.; Fujii, S.; Mukawa, K.; Fujita, M.; Ichikawa, K.; Tomita, S.; Imai, Y.; Kanke, K.; Ono, Y.; Terano, A.; et al. Prediction of colorectal neoplasia by quantitative methylation analysis of estrogen receptor gene in nonneoplastic epithelium from patients with ulcerative colitis. Clin. Cancer Res. 2005, 11, 8880–8885. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luo, Y.; Yu, M.; Grady, W.M. Field cancerization in the colon: A role for aberrant DNA methylation? Gastroenterol. Rep. 2014, 2, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Kondo, Y.; Rosner, G.L.; Xiao, L.; Hernandez, N.S.; Vilaythong, J.; Houlihan, P.S.; Krouse, R.S.; Prasad, A.R.; Einspahr, J.G.; et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J. Natl. Cancer Inst. 2005, 97, 1330–1338. [Google Scholar] [CrossRef]
- Beggs, A.D.; Domingo, E.; Abulafi, M.; Hodgson, S.V.; Tomlinson, I.P.M. A study of genomic instability in early preneoplastic colonic lesions. Oncogene 2013, 32, 5333–5337. [Google Scholar] [CrossRef]
- Beggs, A.D.; Mehta, S.; Deeks, J.J.; James, J.D.; Caldwell, G.M.; Dilworth, M.P.; Stockton, J.D.; Blakeway, D.; Pestinger, V.; Vince, A.; et al. Validation of epigenetic markers to identify colitis associated cancer: Results of module 1 of the ENDCAP-C study. EBioMedicine 2019, 39, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.J.; Ahuja, N.; Toyota, M.; Bronner, M.P.; Brentnall, T.A. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001, 61, 3573–3577. [Google Scholar]
- Parang, B.; Kaz, A.M.; Barrett, C.W.; Short, S.P.; Ning, W.; Keating, C.E.; Mittal, M.K.; Naik, R.D.; Washington, M.K.; Frank, L.; et al. BVES regulates c-Myc stability via PP2A and suppresses colitis- induced tumorigenesis. Gut 2017, 66, 852–862. [Google Scholar] [CrossRef]
- Emmett, R.A.; Davidson, K.L.; Gould, N.J.A.R. DNA methylation patterns in ulcerative colitis-associated cancer: A systematic review. Epigenomics 2017, 9, 1029–1042. [Google Scholar] [CrossRef]
- Gerecke, C.; Scholtka, B.; Löwenstein, Y.; Fait, I.; Gottschalk, U.; Rogoll, D.; Melcher, R.K.B. Hypermethylation of ITGA4, TFPI2 and VIMENTIN promoters is increased in inflamed colon tissue: Putative risk markers for colitis-associated cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Scarpa, M.; Castagliuolo, I.; Erroi, F.; Kotsafti, A.; Basato, S.; Brun, P.; D’Incà, R.; Rugge, M.; Angriman, I.; et al. Aberrant gene methylation in non-neoplastic mucosa as a predictive marker of ulcerative colitis-associated CRC. Oncotarget 2016, 7, 10322–10331. [Google Scholar] [CrossRef]
- Azuara, D.; Rodriguez-Moranta, F.; de Oca, J.; Sanjuan, X.; Guardiola, J.; Lobaton, T.; Wang, A.; Boadas, J.; Piqueras, M.; Monfort, D.; et al. Novel methylation panel for the early detection of neoplasia in high-risk ulcerative colitis and Crohn’s colitis patients. Inflamm. Bowel Dis. 2013, 19, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Papadia, C.; Louwagie, J.; Del Rio, P.; Grooteclaes, M.; Coruzzi, A.; Montana, C.; Novelli, M.; Bordi, C.; de’ Angelis, G.L.; Bassett, P.; et al. FOXE1 and SYNE1 genes hypermethylation panel as promising biomarker in colitis-associated colorectal neoplasia. Inflamm. Bowel Dis. 2014, 20, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Okugawa, Y.; Tanaka, K.; Araki, T.; Uchida, K.; Hishida, A.; Uchino, M.; Ikeuchi, H.; Hirota, S.; Kusunoki, M.; et al. A Panel of Methylated MicroRNA Biomarkers for Identifying High-Risk Patients with Ulcerative Colitis-Associated Colorectal Cancer Yuji. Gastroenterology 2017, 153, 1634–1646. [Google Scholar] [CrossRef] [PubMed]
- Saraggi, D.; Fassan, M.; Mescoli, C.; Scarpa, M.; Valeri, N.; Michielan, A.; D’Incá, R.R.M. The molecular landscape of colitis-associated carcinogenesis. Dig. Liver. Dis. 2017, 49, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Carlo, C.; Maley, P.M. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Timmer, M.R.; Lau, C.T.; Calpe, S.; Sancho-Serra, M.D.C.; Straub, D.; Baker, A.M.; Meijer, S.L.; Kate, F.J.W.T.; Mallant-Hent, R.C.; et al. Dynamic clonal equilibrium and predetermined cancer risk in Barrett’s oesophagus. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Leedham, S.J.; Preston, S.L.; McDonald, S.A.C.; Elia, G.; Bhandari, P.; Poller, D.; Harrison, R.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.T.; Sanchez, C.A.; Prevo, L.J.; Wong, D.J.; Galipeau, P.C.; Paulson, T.G.; Rabinovitch, P.S.; Reid, B.J. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat. Genet. 1999, 22, 106–109. [Google Scholar] [CrossRef]
- Maley, C.C.; Galipeau, P.C.; Finley, J.C.; Wongsurawat, V.J.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Risques, R.A.; Rabinovitch, P.S. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006, 38, 468–473. [Google Scholar] [CrossRef]
- Willenbucher, R.F.; Zelman, S.J.; Ferrell, L.D.; Moore, D.H., 2nd; Waldman, W.F. Chromosomal alterations in ulcerative colitis-related neoplastic progression. Gastroenterology 1997, 113, 791–801. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D. A Big Bang model of human colorectal tumor growth HHS Public Access new model provides a quantitative framework to interpret tumor growth dynamics and the origins of ITH with significant clinical implications. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts CE, S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Uchida, K.; Tanaka, K.; Ide, S.; Otake, K.; Okita, Y.; Inoue, M.; Araki, T.; Mizoguchi, A.; Kusunoki, M. Dynamic pathology for circulating free DNA in a dextran sodium sulfate colitis mouse model. Pediatr. Surg. Int. 2014, 30, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Vymetalkova, V.; Cervena, K.; Bartu, L.; Vodicka, P. Circulating cell-free dna and colorectal cancer: A systematic review. Int. J. Mol. Sci. 2018, 19, 3356. [Google Scholar] [CrossRef]
- Xie, W.; Xie, L.; Song, X. The diagnostic accuracy of circulating free DNA for the detection of KRAS mutation status in colorectal cancer: A meta-analysis. Cancer Med. 2019, 8, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Deng, Z.; Li, B.; Peng, Y.; Song, M.; Liu, J. Circulating tumor DNA is effective for detection of KRAS mutation in colorectal cancer: A meta-analysis. Int. J. Biol. Markers 2017, 32, e421–e427. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.G.; Boysen, A.K.; Pallisgård, N.; Johansen, J.S.; Tabernero, J.; Sørensen, M.M.; Jensen, B.V.; Hansen, T.F.; Sefrioui, D.; Andersen, R.F.; et al. Cell-Free DNA in Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis. Oncologist 2017, 22, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.; Carroll, G.; Gould, T.; Pockney, P.; Dun, M.S.R. Cell-Free DNA as a Diagnostic Blood-Based Biomarker for Colorectal Cancer: A Systematic Review. J. Surg. Res. 2019, 236, 184–197. [Google Scholar] [CrossRef]
- Wang, X.; Shi, X.-Q.; Zeng, P.-W.; Mo, F.-M.; Chen, Z.-H. Circulating cell free DNA as the diagnostic marker for colorectal cancer: A systematic review and meta-analysis. Oncotarget 2018, 9, 24514–24524. [Google Scholar] [CrossRef]
- Cree, I.A.; Uttley, L.; Buckley Woods, H.; Kikuchi, H.; Reiman, A.; Harnan, S.; Whiteman, B.L.; Philips, S.T.; Messenger, M.; Cox, A.; et al. The evidence base for circulating tumour DNA blood-based biomarkers for the early detection of cancer: A systematic mapping review. BMC Cancer 2017, 17, 1–17. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal liquid biopsy and mathematical modeling of clonal evolution forecast time to treatment failure in the prospect-c phase ii colorectal cancer clinical trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Lauricella, C.; Lamba, S.; et al. Monitoring clonal evolution and resistance to EGFR blockade in the blood of metastatic colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Kisiel, J.B.; Yab, T.C.; Nazer Hussain, F.T.; Taylor, W.R.; Garrity-Park, M.M.; Sandborn, W.J.; Loftus, E.V.; Wolff, B.G.; Smyrk, T.C.; Itzkowitz, S.H.; et al. Stool DNA testing for the detection of colorectal neoplasia in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2013, 37, 546–554. [Google Scholar] [CrossRef]
- Kisiel, J.B.; Konijeti, G.G.; Piscitello, A.J.; Chandra, T.; Goss, T.F.; Ahlquist, D.A.; Farraye, F.A.; Ananthakrishnan, A.N. Analysis of DNA Methylation at Specific Loci in Stool Samples Detects Colorectal Cancer and High-Grade Dysplasia in Patients with Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 914–921. [Google Scholar] [CrossRef]
- Klepp, P.; Kisiel, J.B.; Småstuen, M.C.; Røseth, A.; Andersen, S.N.; Vatn, M.H.; Ahlquist, D.A.; Moum, B.A.B.S. Multi-target stool DNA test in the surveillance of inflammatory bowel disease: A cross-sectional cohort study. Scand. J. Gastroenterol. 2018, 53, 273–278. [Google Scholar] [CrossRef]
- Kisiel, J.B.; Konijeti, G.G.; Piscitello, A.J.; Chandra, T.; Goss, T.F.; Ahlquist, D.A.; Farraye, F.A.; Ananthakrishnan, A.N. Stool DNA Analysis is Cost Effective for Colorectal Cancer Surveillance in Patients with Ulcerative Colitis John. Clin. Gastroenterol Hepatol. 2016, 14, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef]
- Gatenbee, C.D.; Baker, A.; Schenck, R.O.; Neves, M.P.; Yakub, S.; Martinez, P.; Cross, W.C.H.; Jansen, M.; Rodriguez-justo, M.; Leedham, S.; et al. Niche engineering drives early passage through an immune bottleneck in progression to colorectal cancer Chandler. bioRxiv 2019. [Google Scholar] [CrossRef]
- Lakatos, E.; Williams, M.J.; Schenck, R.O.; Cross, W.C.H.; Househam, J.; Werner, B.; Gatenbee, C.; Robertson-Tessi, M.; Barnes, C.P.; Anderson, A.R.A.; et al. Evolutionary dynamics of neoantigens in growing tumours. bioRxiv 2019, 52, 1–41. [Google Scholar]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the “Immunoscore” in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Michael-Robinson, J.M.; Pandeya, N.; Walsh, M.D.; Biemer-Huttmann, A.E.; Eri, R.D.; Buttenshaw, R.L.; Lincoln, D.; Clouston, A.D.; Jaas, J.R.; Radford-Smith, G.L. Characterization of tumour-infiltrating lymphocytes and apoptosis in colitis-associated neoplasia: Comparison with sporadic colorectal cancer. J. Pathol. 2006, 208, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Hirata, I.; Nakano, N.; Tahara, S.; Horiguchi, N.; Kawamura, T.; Okubo, M.; Ishizuka, T.; Yamada, H.; Yoshida, D.; et al. Potential link between Fusobacterium enrichment and DNA methylation accumulation in the inflammatory colonic mucosa in ulcerative colitis. Oncotarget 2017, 8, 61917–61926. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naïve microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jobin, C. Novel insights into microbiome in colitis and colorectal cancer. Curr. Opin. Gastroenterol. 2017, 33, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Liguori, G.; Lamas, B.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes 2018, 9, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Lopez-Siles, M.; Martinez-Medina, M.; Surís-Valls, R.; Aldeguer, X.; Sabat-Mir, M.; Duncan, S.H.; Flint, H.J.; Garcia-Gil, L.J. Changes in the Abundance of Faecalibacterium prausnitzii Phylogroups i and, I.I.in the Intestinal Mucosa of Inflammatory Bowel Disease and Patients with Colorectal Cancer. Inflamm. Bowel Dis. 2016, 22, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Khilkin, M.; Kerjaschki, D.; Schreiber, S.; Ortner, M.; Weber, J.L.H. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology 1998, 115, 281–286. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.R.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014, 63, 761–770. [Google Scholar] [CrossRef]
- Coleman, O.I.; Haller, D. Bacterial signaling at the intestinal epithelial interface in inflammation and cancer. Front. Immunol. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Ahmad, O.F.; Soares, A.S.; Mazomenos, E.; Brandao, P.; Vega, R.; Seward, E.; Stoyanov, D.; Chand, M.L.L. Artificial intelligence and computer-aided diagnosis in colonoscopy: Current evidence and future directions. Lancet 2019, 4, 71–80. [Google Scholar] [CrossRef]
- Abadir, A.P.; Ali, M.F.; Karnes, W.; Samarasena, J.B. Artificial Intelligence in Gastrointestinal Endoscopy. Clin. Endosc. 2020, 53, 132–141. [Google Scholar] [CrossRef]
- Tajbakhsh, N.; Gurudu, S.R.L.J. Automated Polyp Detection in Colonoscopy Videos Using Shape and Context Information. IEEE Trans. Med. Imaging 2016, 35, 630–644. [Google Scholar] [CrossRef]
- Fernández-Esparrach, G.; Bernal, J.; López-Cerón, M.; Córdova, H.; Sánchez-Montes, C.; de Miguel, C.S.F.R. Exploring the clinical potential of an automatic colonic polyp detection method based on the creation of energy maps. Endoscopy 2019, 48, 837–842. [Google Scholar] [CrossRef]
- Le Berre, C.; Sandborn, W.J.; Aridhi, S.; Devignes, M.D.; Fournier, L.; Smaïl-Tabbone, M.; Danese, S.P.-B.L. Application of Artificial Intelligence to Gastroenterology and Hepatology. Gastroenterology 2020, 158, 76–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, J.; Zhou, W.; An, P.; Shen, L.; Liu, J.; Jiang, X.; Huang, X.; Mu, G.; Wan, X.; et al. Randomised controlled trial of WISENSE, a real-time quality improving system for monitoring blind spots during esophagogastroduodenoscopy. Gut 2019, 68, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Berzin, T.M.; Glissen Brown, J.R.; Bharadwaj, S.; Becq, A.; Xiao, X.; Liu, P.; Li, L.; Song, Y.; Zhang, D.; et al. Real-time automatic detection system increases colonoscopic polyp and adenoma detection rates: A prospective randomised controlled study. Gut 2019, 68, 1813–1819. [Google Scholar] [CrossRef]
- Ozawa, T.; Ishihara, S.; Fujishiro, M.; Saito, H.; Kumagai, Y.; Shichijo, S.; Aoyama, K.T. Novel computer-assisted diagnosis system for endoscopic disease activity in patients with ulcerative colitis. Gastrointest. Endosc. 2019, 89, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Stidham, R.W.; Liu, W.; Bishu, S.; Rice, M.D.; Higgins, P.D.R.; Zhu, J.; Nallamothu, B.K.; Waljee, A.K. Performance of a Deep Learning Model vs Human Reviewers in Grading Endoscopic Disease Severity of Patients with Ulcerative Colitis. JAMA Netw. Open 2019, 2, e193963. [Google Scholar] [CrossRef] [PubMed]
- Abadir, A.P.; Requa, J.; Ninh, A.; Karnes WM, M. Unambiguous real- time endoscopic scoring of ulcerative colitis using a convolutional neural network. Am. J. Gastroenterol 2018, 113, S349. [Google Scholar] [CrossRef]
- Maeda, Y.; Kudo, S.; Mori, Y.; Misawa, M.; Ogata, N.; Sasanuma, S.; Wakamura, K.; Oda, M.; Mori, K.; Ohtsuka, K. Fully automated diagnostic system with artificial intelligence using endocytoscopy to identify the presence of histologic inflammation associated with ulcerative colitis (with video). Gastrointest. Endosc. 2019, 89, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Matalka, I.I.; Al-Omari, F.A.; Salama, R.M.; Mohtaseb, A.H. A novel approach for quantitative assessment of mucosal damage in inflammatory bowel disease. Diagn. Pathol. 2013, 8, 1. [Google Scholar] [CrossRef] [PubMed]

| Risk factors | Pancolitis with no active inflammation | Pancolitis with mildly active inflammation | Pancolitis with moderate-severe inflammation (endoscopic or histological), |
| (endoscopic or histological), | (endoscopic or histological), | or dysplasia or strictures within past 5 years (±surgery) | |
| or left sided UC or CD of similar extent | or presence of post-inflammatory polyps, | or PSC, | |
| (i.e., <50% mucosa involved) | or family history of CRC in 1st degree relative > 50 yoa | or family history of CRC in 1st degree relative < 50 yoa | |
| Risk | Low | Intermediate | High |
| Surveillance | 5 year | 3 year | Annual |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yalchin, M.; Baker, A.-M.; Graham, T.A.; Hart, A. Predicting Colorectal Cancer Occurrence in IBD. Cancers 2021, 13, 2908. https://doi.org/10.3390/cancers13122908
Yalchin M, Baker A-M, Graham TA, Hart A. Predicting Colorectal Cancer Occurrence in IBD. Cancers. 2021; 13(12):2908. https://doi.org/10.3390/cancers13122908
Chicago/Turabian StyleYalchin, Mehmet, Ann-Marie Baker, Trevor A. Graham, and Ailsa Hart. 2021. "Predicting Colorectal Cancer Occurrence in IBD" Cancers 13, no. 12: 2908. https://doi.org/10.3390/cancers13122908
APA StyleYalchin, M., Baker, A.-M., Graham, T. A., & Hart, A. (2021). Predicting Colorectal Cancer Occurrence in IBD. Cancers, 13(12), 2908. https://doi.org/10.3390/cancers13122908