
Targeting HIF-1α Regulatory Pathways as a Strategy to Hamper Tumor-Microenvironment Interactions in CLL

 , ,
, ,  , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients’ Samples
2.2. Cell Lines and Cell Culture
2.3. Western Blot (WB) Analysis
2.4. Akt and HIF-1α Activity
2.5. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
2.6. CXCL12 Quantification
2.7. Immunohistochemistry
2.8. Statistical Analysis
3. Results
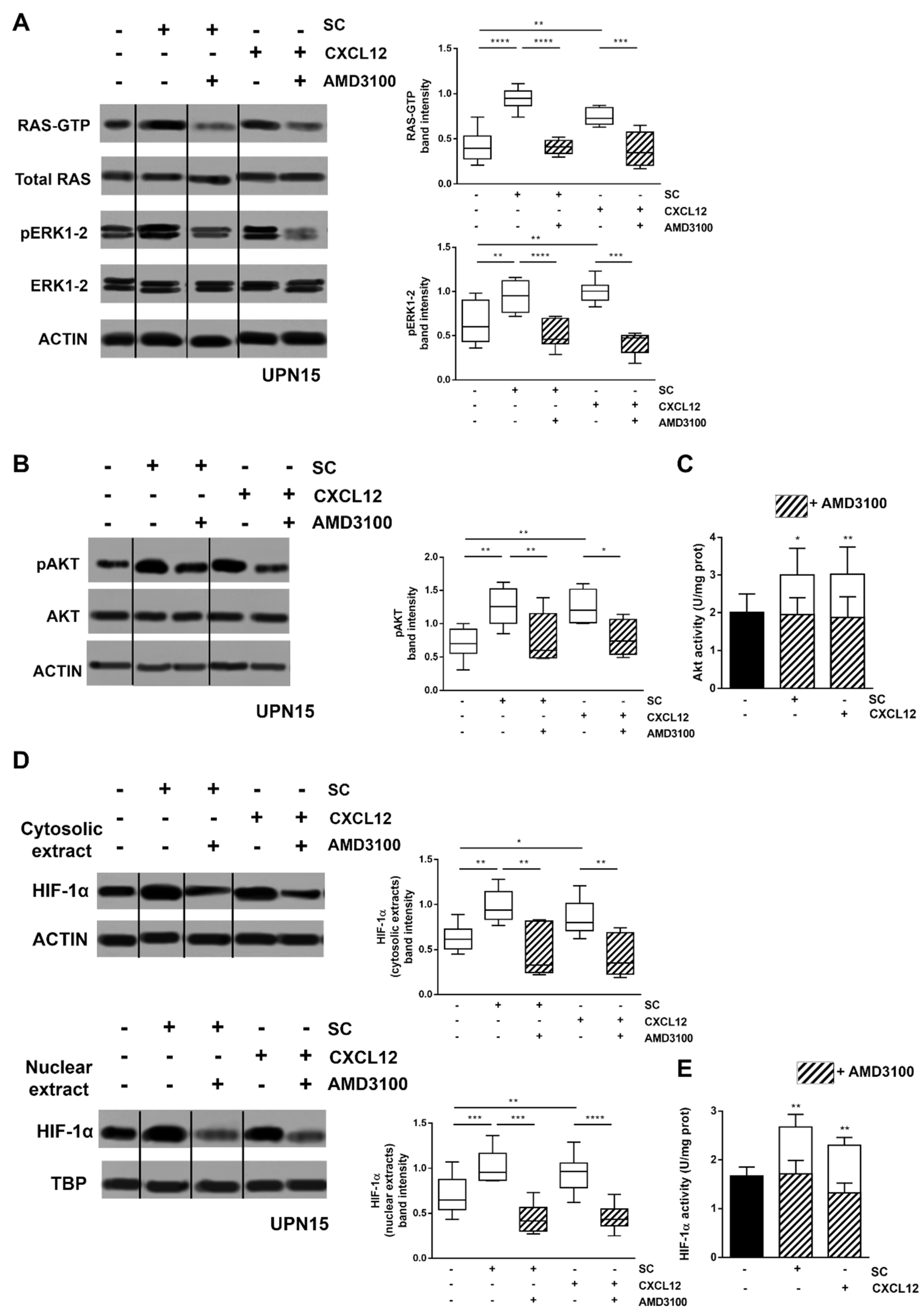
3.1. CXCL12/CXCR4 Axis Is a Main Regulator of SC-Induced HIF-1α Activation in CLL Cells
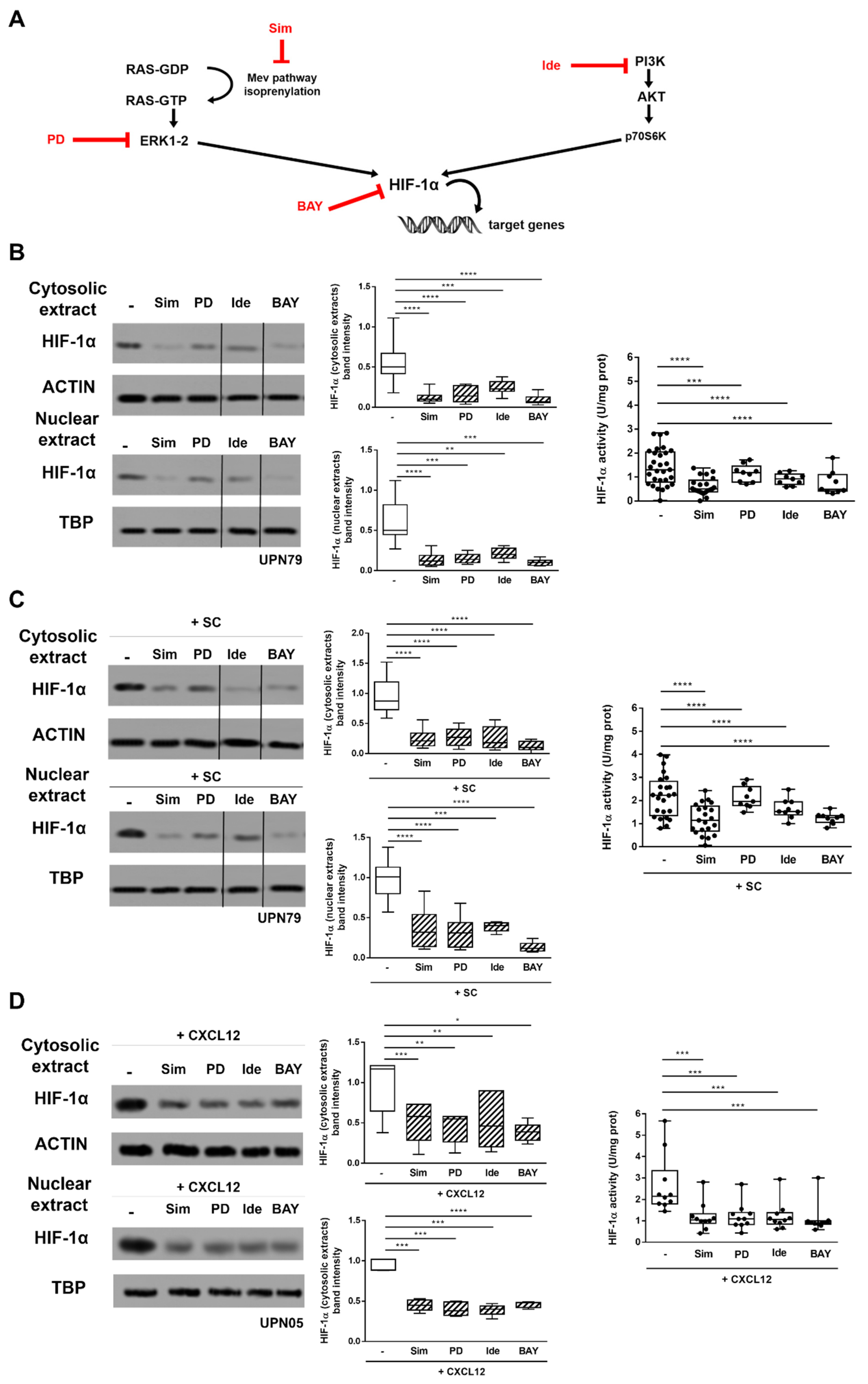
3.2. Inhibitors of CXCR4 Downstream Signalling Effectively Counteract SC- and CXCL12-Induced HIF-1α Upregulation in CLL Cells
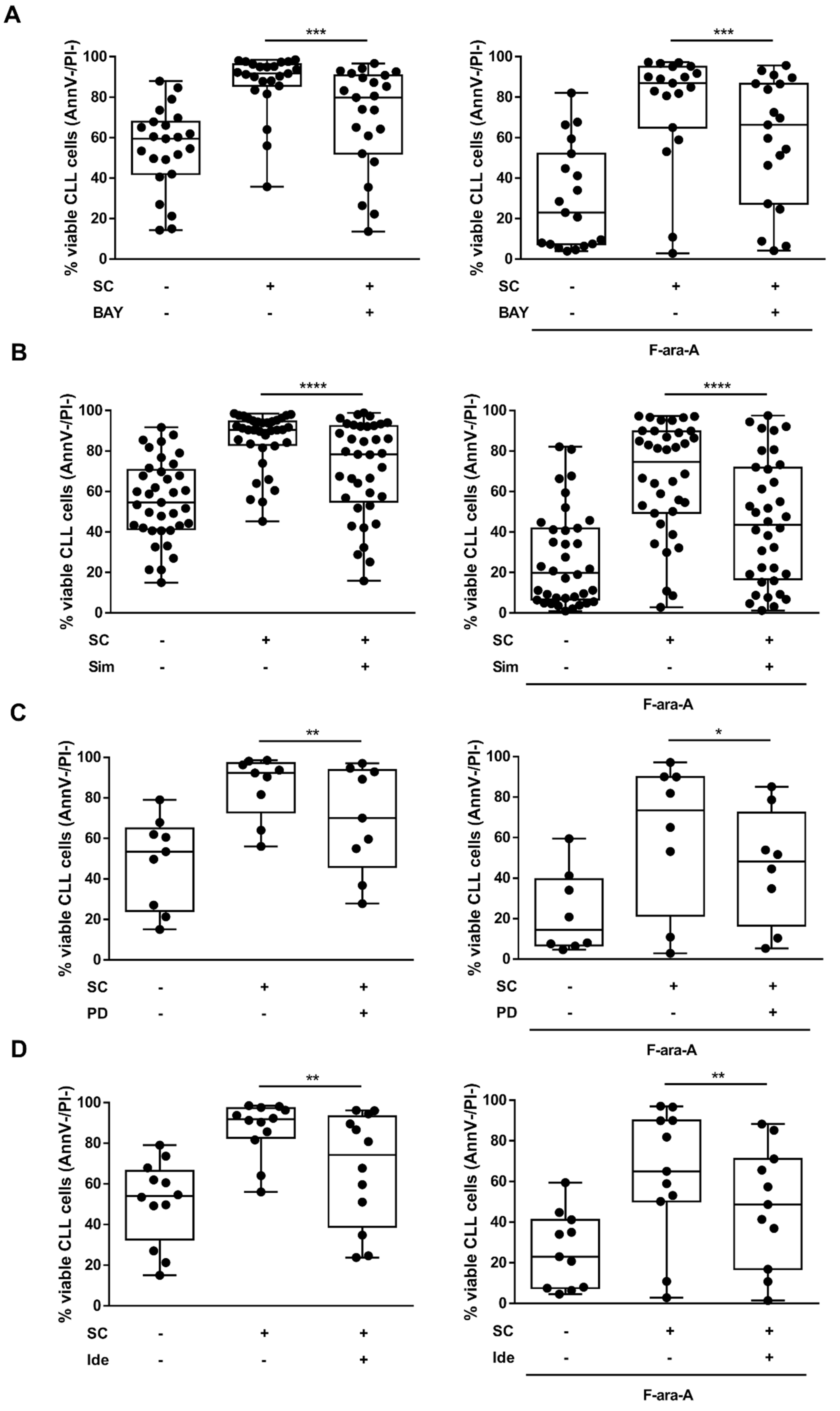
3.3. The Targeted Inhibition of HIF-1α Regulatory Pathways Hinders the SC-Mediated Protection from Spontaneous and Fludarabine-Induced Cell Death
3.4. The Inhibition of PI3K/AKT Pathway and Downstream HIF-1α Impairs CXCL12 Production in SC
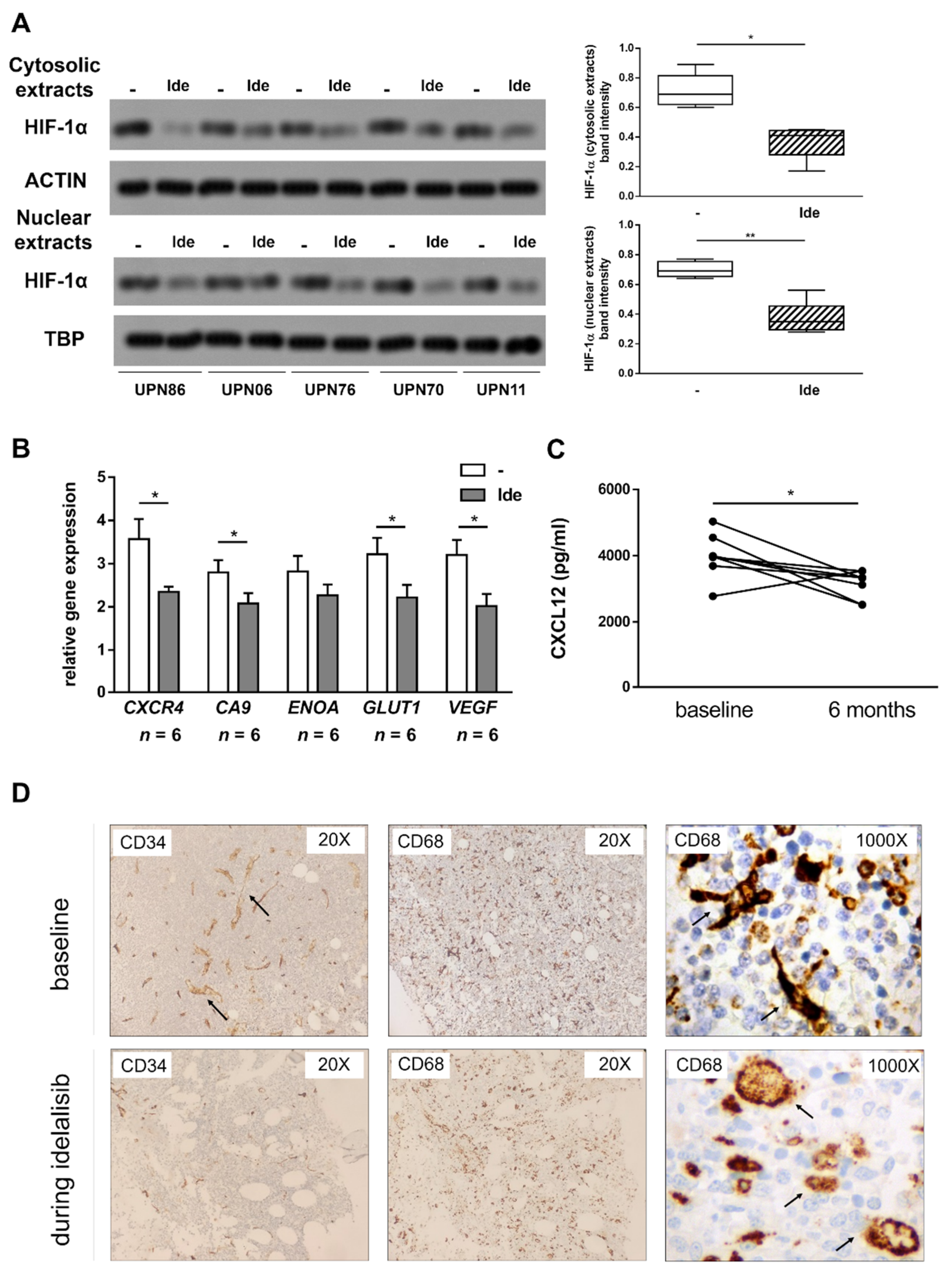
3.5. Idelalisib Hampers Stroma-Derived Survival Signals by Targeting HIF-1α at the SC- and CLL Cell-Level
3.6. Treatment with Idelalisib Affects HIF-1α Expression and Activity in CLL Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of hypoxia-inducible factor and metabolic pathways: Possible targets of cancer. Cell Biosci. 2017, 7, 62. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G. Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol. 2002, 64, 993–998. [Google Scholar] [CrossRef]
- Dimova, E.Y.; Michiels, C.; Kietzmann, T. Kinases as upstream regulators of the HIF system: Their emerging potential as anti-cancer drug targets. Curr. Pharm. Des. 2009, 15, 3867–3877. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Shanafelt, T.D.; Cimmino, A.; Taccioli, C.; Volinia, S.; Liu, C.G.; Calin, G.A.; Croce, C.M.; Chan, D.A.; Giaccia, A.J.; et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood 2009, 113, 5568–5574. [Google Scholar] [CrossRef]
- Rigoni, M.; Riganti, C.; Vitale, C.; Griggio, V.; Campia, I.; Robino, M.; Foglietta, M.; Castella, B.; Sciancalepore, P.; Buondonno, I.; et al. Simvastatin and downstream inhibitors circumvent constitutive and stromal cell-induced resistance to doxorubicin in IGHV unmutated CLL cells. Oncotarget 2015, 6, 29833–29846. [Google Scholar] [CrossRef]
- Valsecchi, R.; Coltella, N.; Belloni, D.; Ponente, M.; Hacken, E.T.; Scielzo, C.; Scarfo, L.; Bertilaccio, M.T.; Brambilla, P.; Lenti, E.; et al. HIF-1alpha regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood 2016, 127, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Griggio, V.; Vitale, C.; Todaro, M.; Riganti, C.; Kopecka, J.; Salvetti, C.; Bomben, R.; Bo, M.D.; Magliulo, D.; Rossi, D.; et al. HIF-1alpha is over-expressed in leukemic cells from TP53-disrupted patients and is a promising therapeutic target in chronic lymphocytic leukemia. Haematologica 2020, 105, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. The CLL cell microenvironment. Adv. Exp. Med. Biol. 2013, 792, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Hartmann, T.; Krome, M.; Rawluk, J.; Tamamura, H.; Fujii, N.; Kipps, T.J.; Burger, J.A. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood 2005, 106, 1824–1830. [Google Scholar] [CrossRef]
- Burger, J.A.; Burger, M.; Kipps, T.J. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood 1999, 94, 3658–3667. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Endo, T.; Tsukada, N.; Ohata, J.; Kitada, S.; Reed, J.C.; Zvaifler, N.J.; Kipps, T.J. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood 2005, 106, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Niedermeier, M.; Hennessy, B.T.; Knight, Z.A.; Henneberg, M.; Hu, J.; Kurtova, A.V.; Wierda, W.G.; Keating, M.J.; Shokat, K.M.; Burger, J.A. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: A novel therapeutic approach. Blood 2009, 113, 5549–5557. [Google Scholar] [CrossRef]
- Serra, S.; Vaisitti, T.; Audrito, V.; Bologna, C.; Buonincontri, R.; Chen, S.S.; Arruga, F.; Brusa, D.; Coscia, M.; Jaksic, O.; et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. 2016, 1, 47–61. [Google Scholar] [CrossRef]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, S.K.; Jung, B.J.; Choi, S.B.; Choi, E.Y.; Kim, C.S. Enhancing proliferation and optimizing the culture condition for human bone marrow stromal cells using hypoxia and fibroblast growth factor-2. Stem Cell Res. 2018, 28, 87–95. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Dohner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. Guidelines for diagnosis, indications for treatment, response assessment and supportive management of chronic lymphocytic leukemia. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef]
- Coscia, M.; Pantaleoni, F.; Riganti, C.; Vitale, C.; Rigoni, M.; Peola, S.; Castella, B.; Foglietta, M.; Griggio, V.; Drandi, D.; et al. IGHV unmutated CLL B cells are more prone to spontaneous apoptosis and subject to environmental prosurvival signals than mutated CLL B cells. Leukemia 2011, 25, 828–837. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Kurtova, A.V.; Balakrishnan, K.; Chen, R.; Ding, W.; Schnabl, S.; Quiroga, M.P.; Sivina, M.; Wierda, W.G.; Estrov, Z.; Keating, M.J.; et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: Development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009, 114, 4441–4450. [Google Scholar] [CrossRef]
- Riganti, C.; Orecchia, S.; Pescarmona, G.; Betta, P.G.; Ghigo, D.; Bosia, A. Statins revert doxorubicin resistance via nitric oxide in malignant mesothelioma. Int. J. Cancer 2006, 119, 17–27. [Google Scholar] [CrossRef]
- Ali, A.Y.; Guan, Q.; Wu, X.; Hou, S.; Banerji, V.; Johnston, J.B.; Wall, D.; Szwajcer, D.; Gibson, S.B.; Marshall, A.J. Expression and function of phosphoinositide 3-kinase delta in mesenchymal stromal cells from normal and leukaemic bone marrow. Br. J. Haematol. 2019, 185, 883–887. [Google Scholar] [CrossRef]
- Koczula, K.M.; Ludwig, C.; Hayden, R.; Cronin, L.; Pratt, G.; Parry, H.; Tennant, D.; Drayson, M.; Bunce, C.M.; Khanim, F.L.; et al. Metabolic plasticity in CLL: Adaptation to the hypoxic niche. Leukemia 2016, 30, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Butterworth, M.; Majid, A.; Walewska, R.J.; Sun, X.M.; Dyer, M.J.; Cohen, G.M. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009, 113, 4403–4413. [Google Scholar] [CrossRef] [PubMed]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef]
- Stamatopoulos, B.; Meuleman, N.; De Bruyn, C.; Pieters, K.; Mineur, P.; Le Roy, C.; Saint-Georges, S.; Varin-Blank, N.; Cymbalista, F.; Bron, D.; et al. AMD3100 disrupts the cross-talk between chronic lymphocytic leukemia cells and a mesenchymal stromal or nurse-like cell-based microenvironment: Pre-clinical evidence for its association with chronic lymphocytic leukemia treatments. Haematologica 2012, 97, 608–615. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Salanga, C.L.; Kipps, T.J.; Messmer, D.; Dorrestein, P.C.; Handel, T.M. Elucidating the CXCL12/CXCR4 signaling network in chronic lymphocytic leukemia through phosphoproteomics analysis. PLoS ONE 2010, 5, e11716. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Camnitz, W.; Burdick, M.D.; Strieter, R.M.; Mehrad, B.; Keeley, E.C. Dose-dependent Effect of Statin Therapy on Circulating CXCL12 Levels in Patients with Hyperlipidemia. Clin. Transl. Med. 2012, 1, 23. [Google Scholar] [CrossRef]
- Manso, B.A.; Zhang, H.; Mikkelson, M.G.; Gwin, K.A.; Secreto, C.R.; Ding, W.; Parikh, S.A.; Kay, N.E.; Medina, K.L. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia 2019, 33, 638–652. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitale, C.; Griggio, V.; Riganti, C.; Todaro, M.; Kopecka, J.; Jones, R.; Salvetti, C.; Boccellato, E.; Perutelli, F.; Voena, C.; et al. Targeting HIF-1α Regulatory Pathways as a Strategy to Hamper Tumor-Microenvironment Interactions in CLL. Cancers 2021, 13, 2883. https://doi.org/10.3390/cancers13122883
Vitale C, Griggio V, Riganti C, Todaro M, Kopecka J, Jones R, Salvetti C, Boccellato E, Perutelli F, Voena C, et al. Targeting HIF-1α Regulatory Pathways as a Strategy to Hamper Tumor-Microenvironment Interactions in CLL. Cancers. 2021; 13(12):2883. https://doi.org/10.3390/cancers13122883
Chicago/Turabian StyleVitale, Candida, Valentina Griggio, Chiara Riganti, Maria Todaro, Joanna Kopecka, Rebecca Jones, Chiara Salvetti, Elia Boccellato, Francesca Perutelli, Claudia Voena, and et al. 2021. "Targeting HIF-1α Regulatory Pathways as a Strategy to Hamper Tumor-Microenvironment Interactions in CLL" Cancers 13, no. 12: 2883. https://doi.org/10.3390/cancers13122883
APA StyleVitale, C., Griggio, V., Riganti, C., Todaro, M., Kopecka, J., Jones, R., Salvetti, C., Boccellato, E., Perutelli, F., Voena, C., Godio, L., Boccadoro, M., & Coscia, M. (2021). Targeting HIF-1α Regulatory Pathways as a Strategy to Hamper Tumor-Microenvironment Interactions in CLL. Cancers, 13(12), 2883. https://doi.org/10.3390/cancers13122883