
Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer


Simple Summary
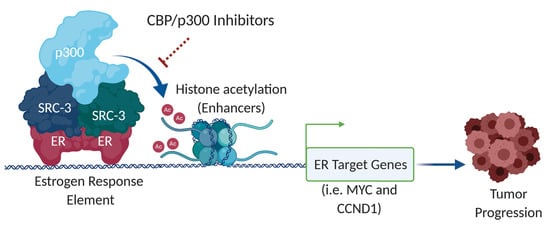
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Analysis of CREBBP/EP300 Dependency and Expression in Cell Lines and Tumors
2.2. TCGA CBP/p300 Interactome Analysis
2.3. Reagents and Cell Culture
2.4. Immunoblot Analysis
2.5. RT-qPCR of ER Target Genes
2.6. ERE Luciferase Assay
2.7. Microarray and RNA-Seq Analysis
2.8. Immunofluorescence Microscopy
2.9. Chromatin Immunoprecipitation (ChIP), Library Preparation, Sequencing (ChIP-Seq), and ChIP-qPCR
2.10. ChIP-Seq Data Analysis
2.11. ChromHMM Analysis
2.12. Association of ChIP-Seq Data and Microarray Gene Expression Data
2.13. Cell Viability, Growth, and Colony Formation Assays
2.14. Senescence Staining and Image Analysis
2.15. Statistical Analysis
3. Results
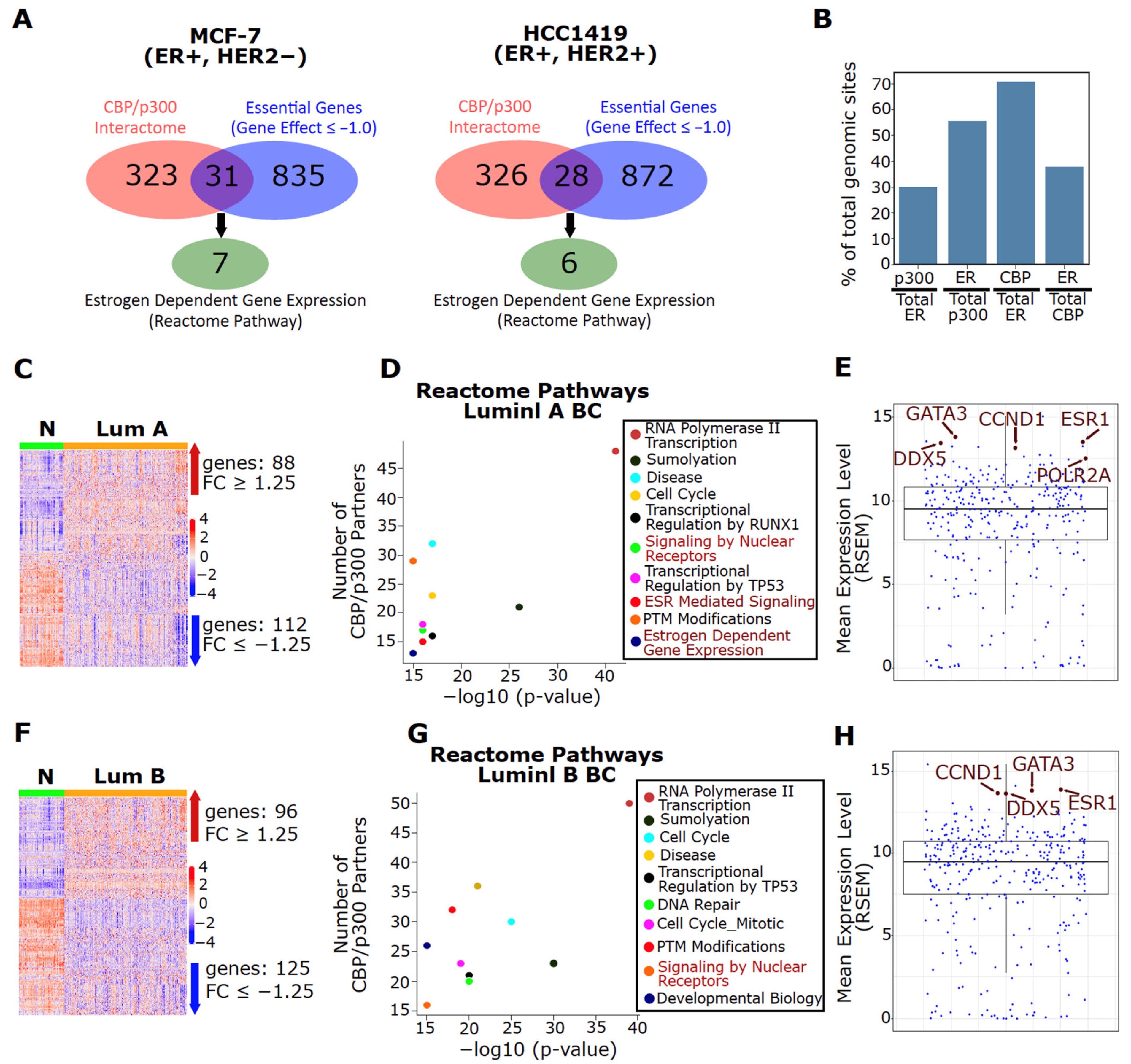
3.1. Upregulation of CBP/p300 in BC Cell Lines and Tumors
3.2. ER Is a Major Interaction Partner for Both CBP and p300 in Luminal BC
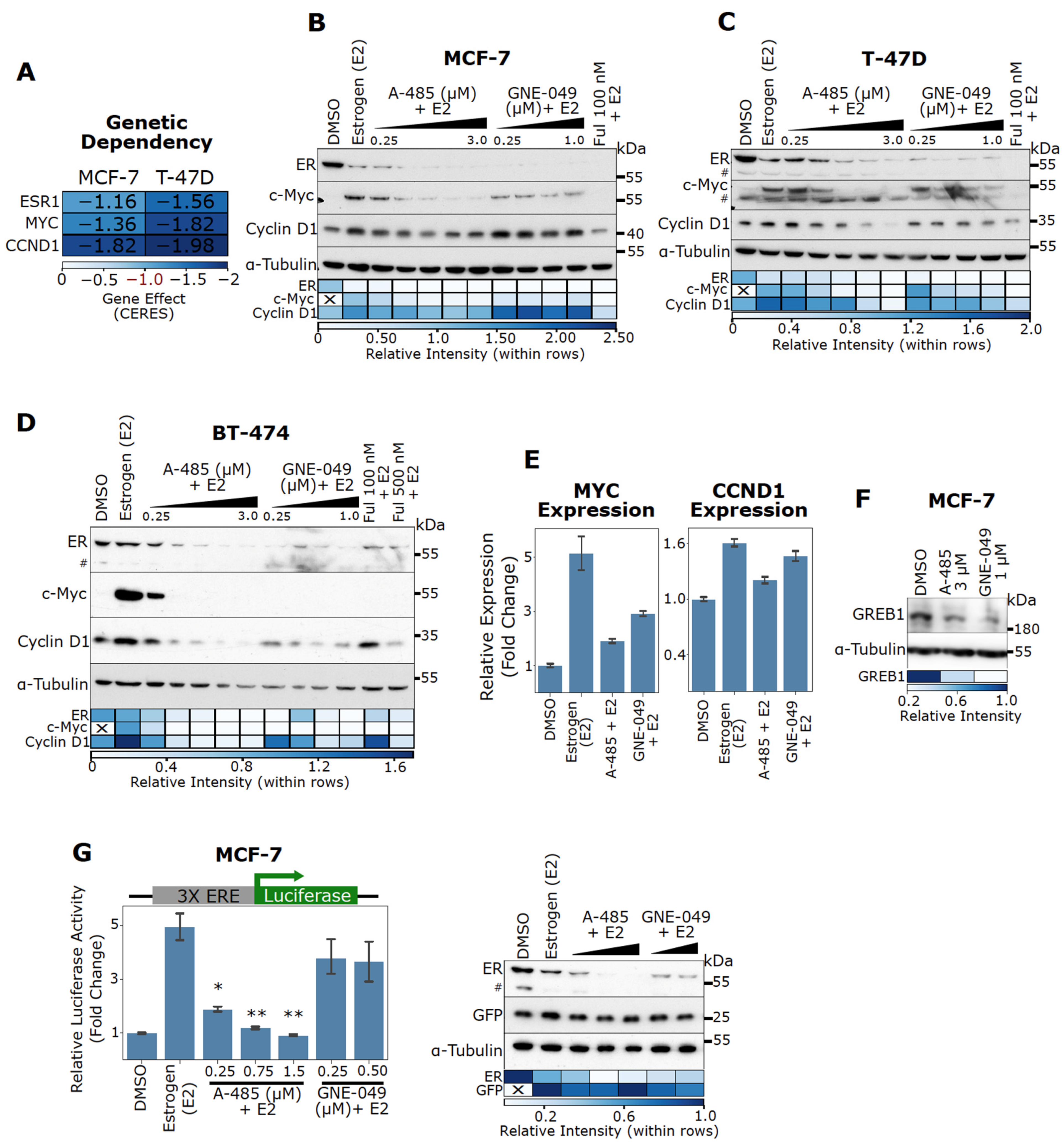
3.3. CBP/p300 KAT (A-485) and BD (GNE-049) Inhibitors Attenuate Estrogen-Induced Gene Expression and ER Activity in ER+ BC Cells
3.4. CBP/p300 Are Critical for ER Signaling and Expression of Estrogen-Regulated Genes
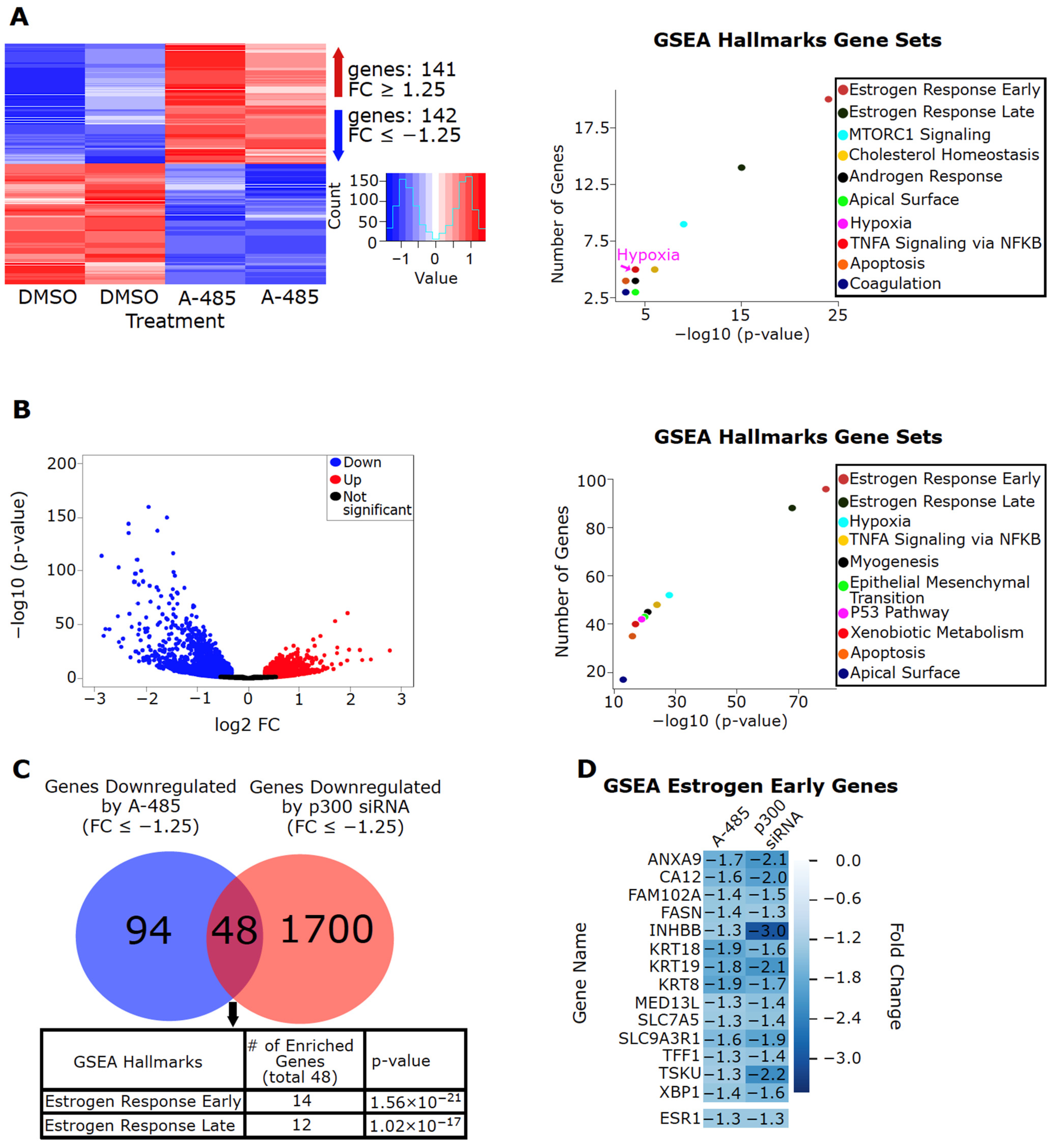
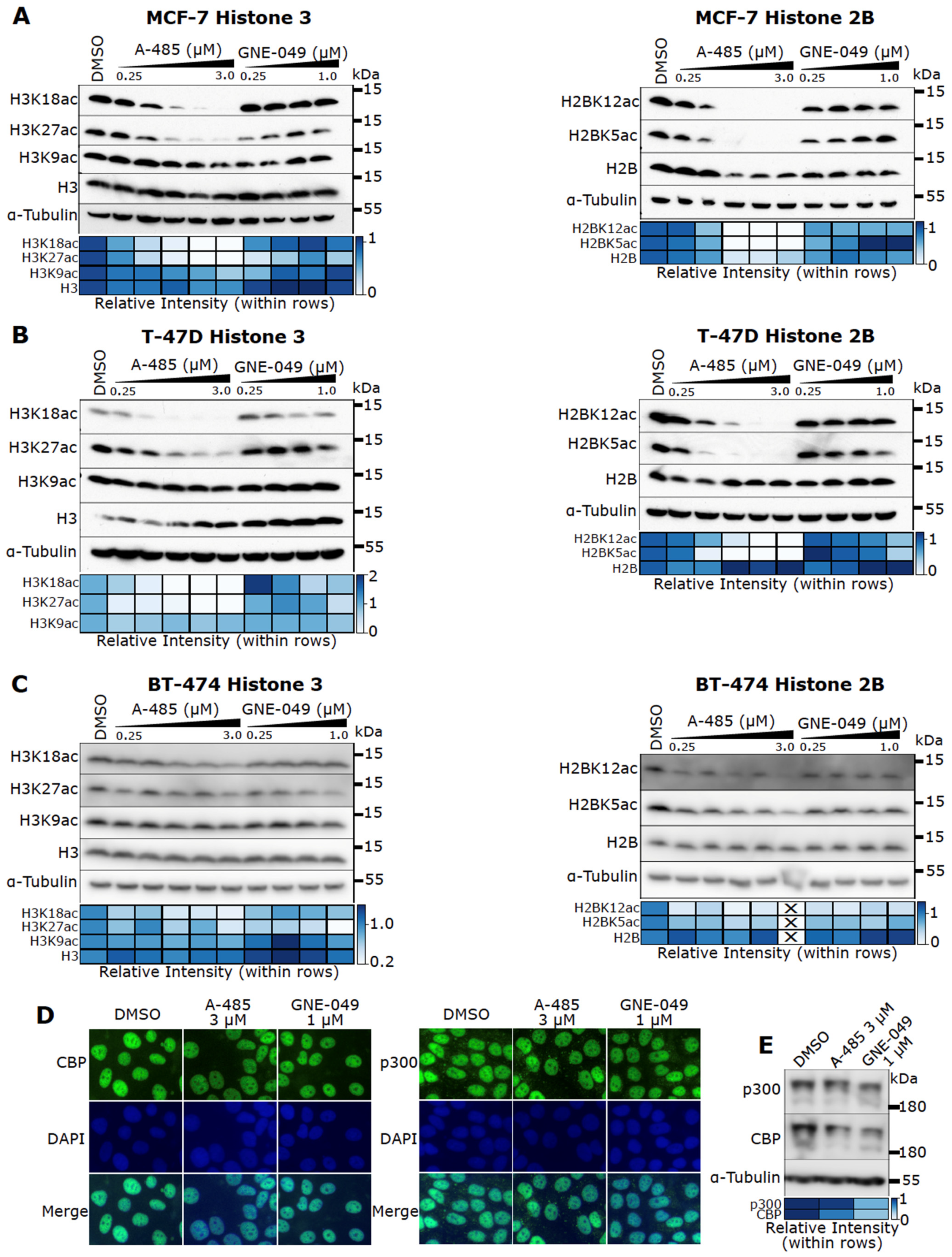
3.5. Effects of A-485 and GNE-049 on Histone Acetylation
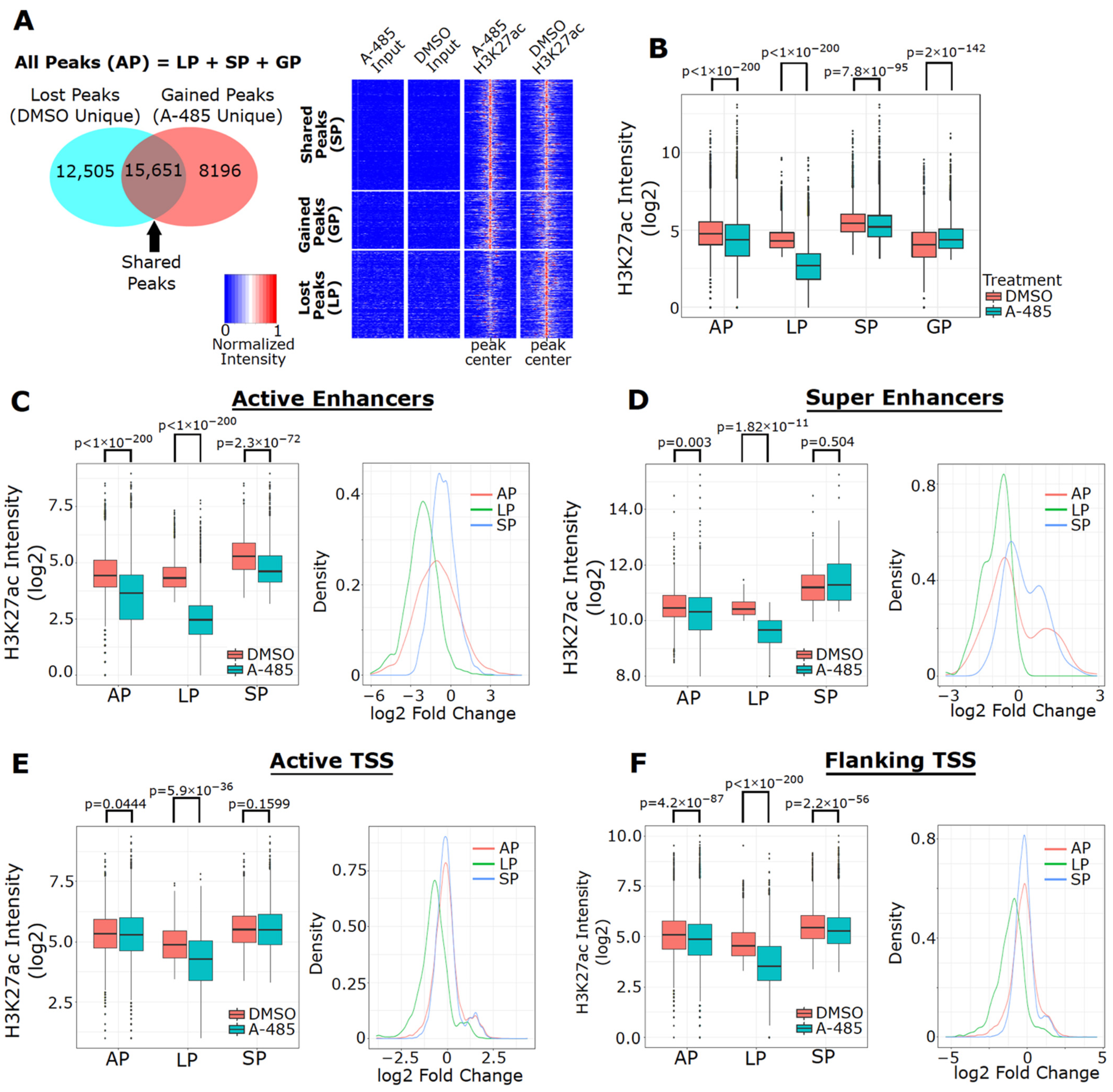
3.6. A-485 Preferentially Suppresses H3K27ac at Active Enhancers
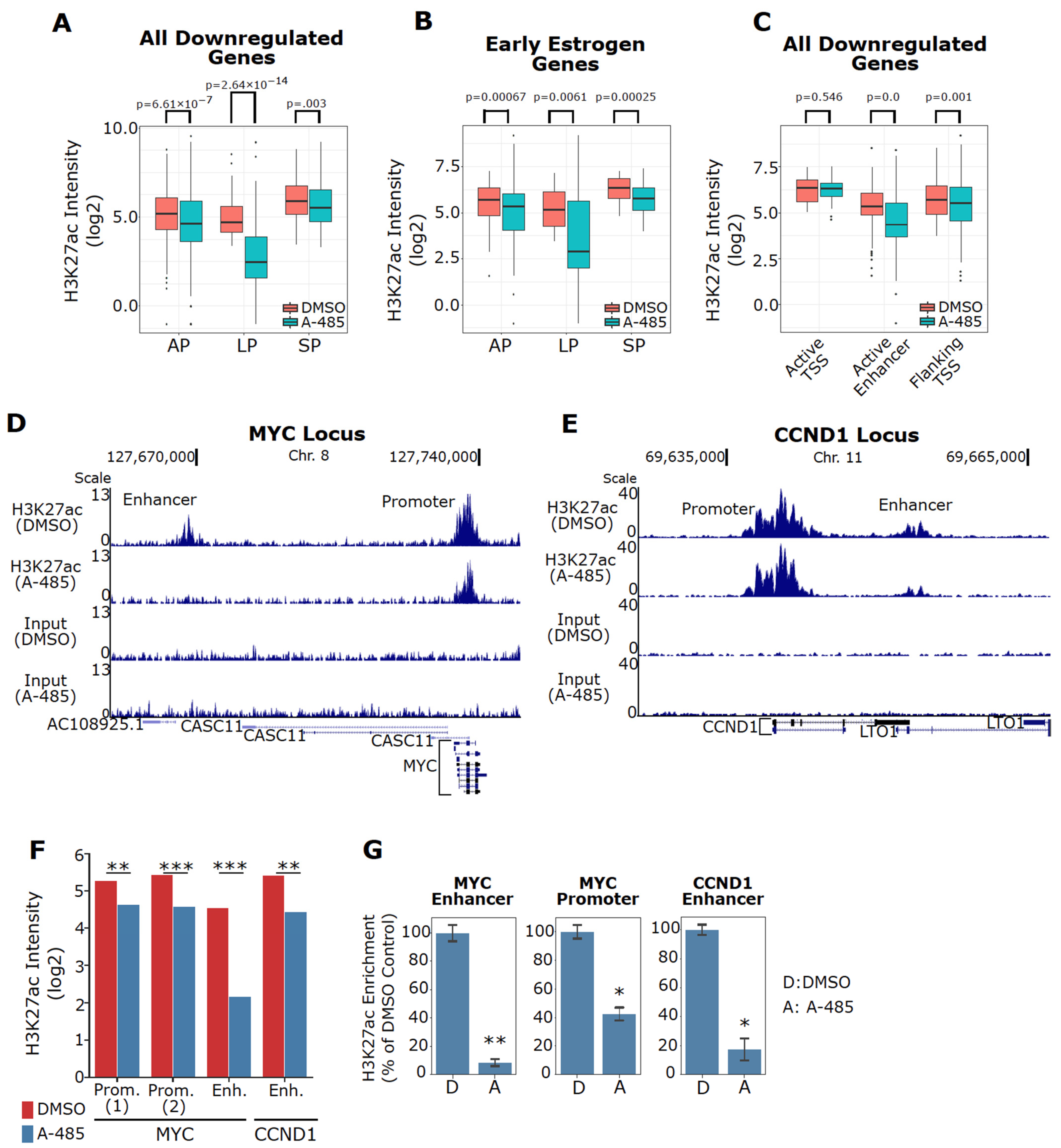
3.7. A-485 Inhibits H3K27ac at the Enhancers of Downregulated ER Target Genes
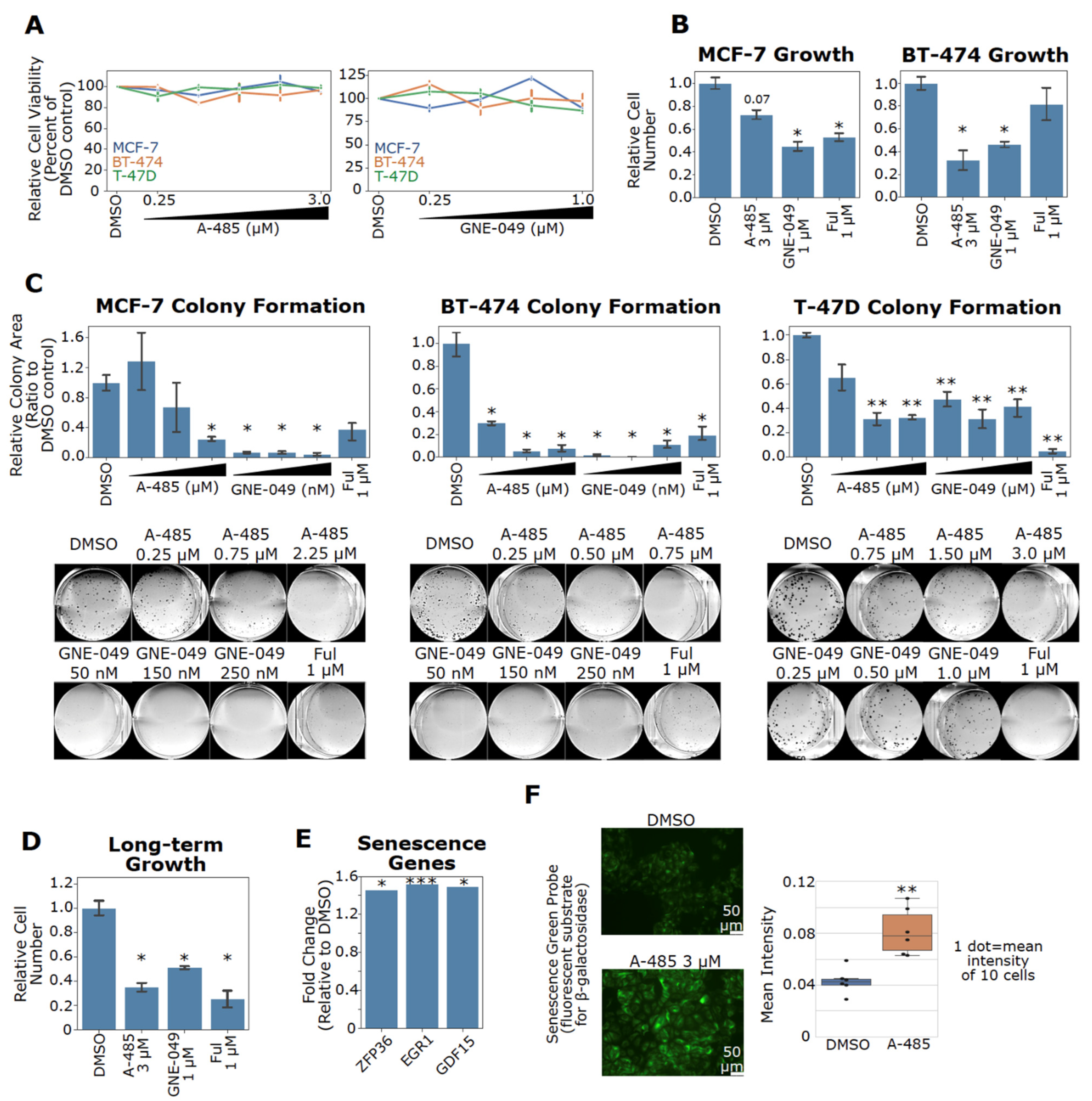
3.8. Pharmacological Inhibition of CBP/p300 Suppresses Cell Growth and Induces Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Stice, J.P.; Knowlton, A.A. Estrogen, NFkappaB, and the heat shock response. Mol. Med. 2008, 14, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Prall, O.W.; Rogan, E.M.; Musgrove, E.A.; Watts, C.K.; Sutherland, R.L. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol. Cell. Biol. 1998, 18, 4499–4508. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mayer, J.A.; Mazumdar, A.; Fertuck, K.; Kim, H.; Brown, M.; Brown, P.H. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol. Endocrinol. 2011, 25, 1527–1538. [Google Scholar] [CrossRef]
- Butt, A.J.; McNeil, C.M.; Musgrove, E.A.; Sutherland, R.L. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: The potential roles of c-Myc, cyclin D1 and cyclin E. Endocr. Relat. Cancer 2005, 12, S47–S59. [Google Scholar] [CrossRef]
- Dubik, D.; Dembinski, T.C.; Shiu, R.P. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987, 47, 6517–6521. [Google Scholar]
- Jordan, V.C. A century of deciphering the control mechanisms of sex steroid action in breast and prostate cancer: The origins of targeted therapy and chemoprevention. Cancer Res. 2009, 69, 1243–1254. [Google Scholar] [CrossRef]
- Ali, S.; Buluwela, L.; Coombes, R.C. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu. Rev. Med. 2011, 62, 217–232. [Google Scholar] [CrossRef]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, N.P.; Jha, P.; Hafiz, A.; Mahfooz, M.; Abdus Sami, G.; Azhar Kamal, M.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef]
- Giuliano, M.; Schifp, R.; Osborne, C.K.; Trivedi, M.V. Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast 2011, 20 (Suppl. 3), S42–S49. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sotiriou, C. Luminal breast cancer: From biology to treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506. [Google Scholar] [CrossRef]
- Alluri, P.G.; Speers, C.; Chinnaiyan, A.M. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Guo, S.; Yang, C.; Sun, L.; Zong, B.; Li, K.; Liu, L.; Tu, G.; Liu, M.; Liu, S. Although cMYC contributes to tamoxifen resistance, it improves cisplatin sensitivity in ERpositive breast cancer. Int. J. Oncol. 2020, 56, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Zwijsen, R.M.; Wientjens, E.; Klompmaker, R.; van der Sman, J.; Bernards, R.; Michalides, R.J. CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997, 88, 405–415. [Google Scholar] [CrossRef]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug. Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 Inhibitors: Game Changers in the Management of Hormone Receptor–Positive Advanced Breast Cancer? Oncology 2018, 32, 216–222. [Google Scholar]
- Yi, P.; Wang, Z.; Feng, Q.; Chou, C.K.; Pintilie, G.D.; Shen, H.; Foulds, C.E.; Fan, G.; Serysheva, I.; Ludtke, S.J.; et al. Structural and Functional Impacts of ER Coactivator Sequential Recruitment. Mol. Cell 2017, 67, 733–743.e4. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.E.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Hsiao, S.J.; Kraus, W.L. A role for coactivators and histone acetylation in estrogen receptor alpha-mediated transcription initiation. EMBO J. 2001, 20, 6084–6094. [Google Scholar] [CrossRef] [PubMed]
- Hanstein, B.; Eckner, R.; DiRenzo, J.; Halachmi, S.; Liu, H.; Searcy, B.; Kurokawa, R.; Brown, M. p300 is a component of an estrogen receptor coactivator complex. Proc. Natl. Acad. Sci. USA 1996, 93, 11540–11545. [Google Scholar] [CrossRef]
- Acevedo, M.L.; Kraus, W.L. Mediator and p300/CBP-steroid receptor coactivator complexes have distinct roles, but function synergistically, during estrogen receptor alpha-dependent transcription with chromatin templates. Mol. Cell. Biol. 2003, 23, 335–348. [Google Scholar] [CrossRef]
- Kraus, W.L.; Kadonaga, J.T. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev. 1998, 12, 331–342. [Google Scholar] [CrossRef]
- Song, X.; Chen, J.; Zhao, M.; Zhang, C.; Yu, Y.; Lonard, D.M.; Chow, D.C.; Palzkill, T.; Xu, J.; O’Malley, B.W.; et al. Development of potent small-molecule inhibitors to drug the undruggable steroid receptor coactivator-3. Proc. Natl. Acad. Sci. USA 2016, 113, 4970–4975. [Google Scholar] [CrossRef]
- Yang, H.; Pinello, C.E.; Luo, J.; Li, D.; Wang, Y.; Zhao, L.Y.; Jahn, S.C.; Saldanha, S.A.; Chase, P.; Planck, J.; et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol. Cancer Ther. 2013, 12, 610–620. [Google Scholar] [CrossRef]
- Pegg, N.; Brooks, N.; Worthington, J.; Young, B.; Prosser, A.; Lane, J.; Taddei, D.; Brown, R.; Harbottle, G.; Shannon, J.; et al. Characterisation of CCS1477: A novel small molecule inhibitor of p300/CBP for the treatment of castration resistant prostate cancer. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef]
- Zou, L.J.; Xiang, Q.P.; Xue, X.Q.; Zhang, C.; Li, C.C.; Wang, C.; Li, Q.; Wang, R.; Wu, S.; Zhou, Y.L.; et al. Y08197 is a novel and selective CBP/EP300 bromodomain inhibitor for the treatment of prostate cancer. Acta. Pharmacol. Sin. 2019, 40, 1436–1447. [Google Scholar] [CrossRef]
- Spriano, F.; Gaudio, E.; Cascione, L.; Tarantelli, C.; Melle, F.; Motta, G.; Priebe, V.; Rinaldi, A.; Golino, G.; Mensah, A.A.; et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020, 4, 4124–4135. [Google Scholar] [CrossRef]
- Xiang, Q.; Wang, C.; Zhang, Y.; Xue, X.; Song, M.; Zhang, C.; Li, C.; Wu, C.; Li, K.; Hui, X.; et al. Discovery and optimization of 1-(1H-indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain inhibitors for the treatment of castration-resistant prostate cancer. Eur. J. Med. Chem. 2018, 147, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.A.; Fedorov, O.; Martin, S.; Singleton, D.C.; Tallant, C.; Wells, C.; Picaud, S.; Philpott, M.; Monteiro, O.P.; Rogers, C.M.; et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319. [Google Scholar] [CrossRef] [PubMed]
- Romero, F.A.; Murray, J.; Lai, K.W.; Tsui, V.; Albrecht, B.K.; An, L.; Beresini, M.H.; de Leon Boenig, G.; Bronner, S.M.; Chan, E.W.; et al. GNE-781, A Highly Advanced Potent and Selective Bromodomain Inhibitor of Cyclic Adenosine Monophosphate Response Element Binding Protein, Binding Protein (CBP). J. Med. Chem. 2017, 60, 9162–9183. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, J.; Wang, D.; Lin, D.; Pang, X.; Wang, S.; Zhao, Y.; Shi, L.; Xue, H.; Pan, Y.; et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 2019, 11, e10659. [Google Scholar] [CrossRef]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Scholz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018, 174, 231–244.e12. [Google Scholar] [CrossRef]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Pradeepa, M.M. Causal role of histone acetylations in enhancer function. Transcription 2017, 8, 40–47. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, L.; Han, Y.; Wu, F.; Yang, W.S.; Zhang, Z.; Huo, T.; Zhu, Y.; Yu, C.; Kim, H.; et al. Acetylation of histone H3K27 signals the transcriptional elongation for estrogen receptor alpha. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef]
- Narita, T.; Ito, S.; Higashijima, Y.; Chu, W.K.; Neumann, K.; Walter, J.; Satpathy, S.; Liebner, T.; Hamilton, W.B.; Maskey, E.; et al. Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, Q.; Gerona-Navarro, G.; Moshkina, N.; Zhou, M.M. Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure 2008, 16, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims, R.J., 3rd. Bromodomains: A new target class for drug development. Nat. Rev. Drug. Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef]
- Michaelides, M.R.; Kluge, A.; Patane, M.; Van Drie, J.H.; Wang, C.; Hansen, T.M.; Risi, R.M.; Mantei, R.; Hertel, C.; Karukurichi, K.; et al. Discovery of Spiro Oxazolidinediones as Selective, Orally Bioavailable Inhibitors of p300/CBP Histone Acetyltransferases. ACS Med. Chem. Lett. 2018, 9, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, R.; Li, Z.; Mei, L.; Wan, S.; Ding, H.; Chen, Z.; Xing, J.; Feng, H.; Han, J.; et al. Discovery of Highly Potent, Selective, and Orally Efficacious p300/CBP Histone Acetyltransferases Inhibitors. J. Med. Chem. 2020, 63, 1337–1360. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.E.; Patel, G.; Patel, C.; Brucelle, F.; Huhn, A.; Gardberg, A.S.; Poy, F.; Cantone, N.; Bommi-Reddy, A.; Sims, R.J., 3rd; et al. Discovery of CPI-1612: A Potent, Selective, and Orally Bioavailable EP300/CBP Histone Acetyltransferase Inhibitor. ACS Med. Chem. Lett. 2020, 11, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Waddell, A.R.; Huang, H.; Liao, D. CBP/p300: Critical Co-activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers. Cancers 2021, 13. in press. [Google Scholar]
- Zwart, W.; Theodorou, V.; Kok, M.; Canisius, S.; Linn, S.; Carroll, J.S. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. EMBO J. 2011, 30, 4764–4776. [Google Scholar] [CrossRef]
- Shang, Y.; Hu, X.; DiRenzo, J.; Lazar, M.A.; Brown, M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 2000, 103, 843–852. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Network, T.C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Waddell, A.R.; Liao, D. Assays for Validating Histone Acetyltransferase Inhibitors. J. Vis. Exp. 2020. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman, M.; Constantinou, S.; Min, H.; Gallon, J.; Lin, M.-L.; Singh, P.; Raguz, S.; Ali, S.; Shousha, S.; Coombes, R.C.; et al. Tumour suppressor EP300, a modulator of paclitaxel resistance and stemness, is downregulated in metaplastic breast cancer. Breast Cancer Res. Treat. 2017, 163, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Siam, A.; Baker, M.; Amit, L.; Regev, G.; Rabner, A.; Najar, R.A.; Bentata, M.; Dahan, S.; Cohen, K.; Araten, S.; et al. Regulation of alternative splicing by p300-mediated acetylation of splicing factors. RNA 2019, 25, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Vannam, R.; Sayilgan, J.; Ojeda, S.; Karakyriakou, B.; Hu, E.; Kreuzer, J.; Morris, R.; Herrera Lopez, X.I.; Rai, S.; Haas, W.; et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem. Biol. 2020. [Google Scholar] [CrossRef]
- Nawaz, Z.; Lonard, D.M.; Dennis, A.P.; Smith, C.L.; O’Malley, B.W. Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1858–1862. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Nguyen, J.; Sundaresan, A.; Banaszynski, L.A. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol. Cell. Biol. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Eeckhoute, J.; Carroll, J.S.; Geistlinger, T.R.; Torres-Arzayus, M.I.; Brown, M. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev. 2006, 20, 2513–2526. [Google Scholar] [CrossRef]
- Kim, E.; Zucconi, B.E.; Wu, M.; Nocco, S.E.; Meyers, D.J.; McGee, J.S.; Venkatesh, S.; Cohen, D.L.; Gonzalez, E.C.; Ryu, B.; et al. MITF Expression Predicts Therapeutic Vulnerability to p300 Inhibition in Human Melanoma. Cancer Res. 2019, 79, 2649–2661. [Google Scholar] [CrossRef]
- Al-Mudares, F.; Reddick, S.; Ren, J.; Venkatesh, A.; Zhao, C.; Lingappan, K. Role of Growth Differentiation Factor 15 in Lung Disease and Senescence: Potential Role Across the Lifespan. Front. Med. 2020, 7, 919. [Google Scholar] [CrossRef]
- Krones-Herzig, A.; Adamson, E.; Mercola, D. Early growth response 1 protein, an upstream gatekeeper of the p53 tumor suppressor, controls replicative senescence. Proc. Natl. Acad. Sci. USA 2003, 100, 3233–3238. [Google Scholar] [CrossRef]
- Sanduja, S.; Blanco, F.F.; Young, L.E.; Kaza, V.; Dixon, D.A. The role of tristetraprolin in cancer and inflammation. Front. Biosci. 2012, 17, 174–188. [Google Scholar] [CrossRef]
- Caslini, C.; Hong, S.; Ban, Y.J.; Chen, X.S.; Ince, T.A. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene 2019, 38, 6599–6614. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waddell, A.; Mahmud, I.; Ding, H.; Huo, Z.; Liao, D. Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer. Cancers 2021, 13, 2799. https://doi.org/10.3390/cancers13112799
Waddell A, Mahmud I, Ding H, Huo Z, Liao D. Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer. Cancers. 2021; 13(11):2799. https://doi.org/10.3390/cancers13112799
Chicago/Turabian StyleWaddell, Aaron, Iqbal Mahmud, Haocheng Ding, Zhiguang Huo, and Daiqing Liao. 2021. "Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer" Cancers 13, no. 11: 2799. https://doi.org/10.3390/cancers13112799
APA StyleWaddell, A., Mahmud, I., Ding, H., Huo, Z., & Liao, D. (2021). Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer. Cancers, 13(11), 2799. https://doi.org/10.3390/cancers13112799