
Identification of the Neurokinin-1 Receptor as Targetable Stratification Factor for Drug Repurposing in Pancreatic Cancer

 ,
,
Abstract
Simple Summary
Abstract
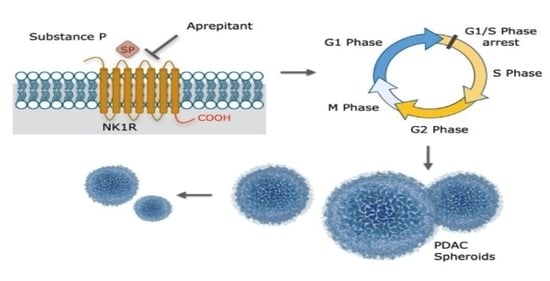
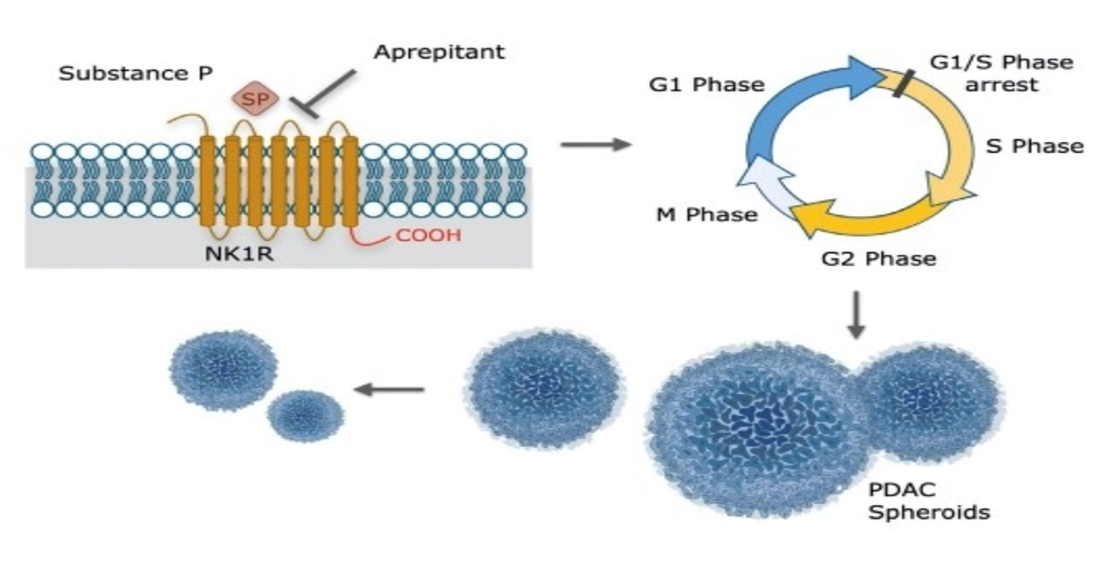
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of PSC Conditioned Media
2.3. Drugs
2.4. Viability Assay
2.5. Sphere and Colony Formation Culture
2.6. RNA Isolation and RT-qPCR
2.7. Apoptosis Detection Assay
2.8. Caspase Detection Assay
2.9. Cell Cycle Detection
2.10. ELISA
2.11. Statistical Analysis
3. Results
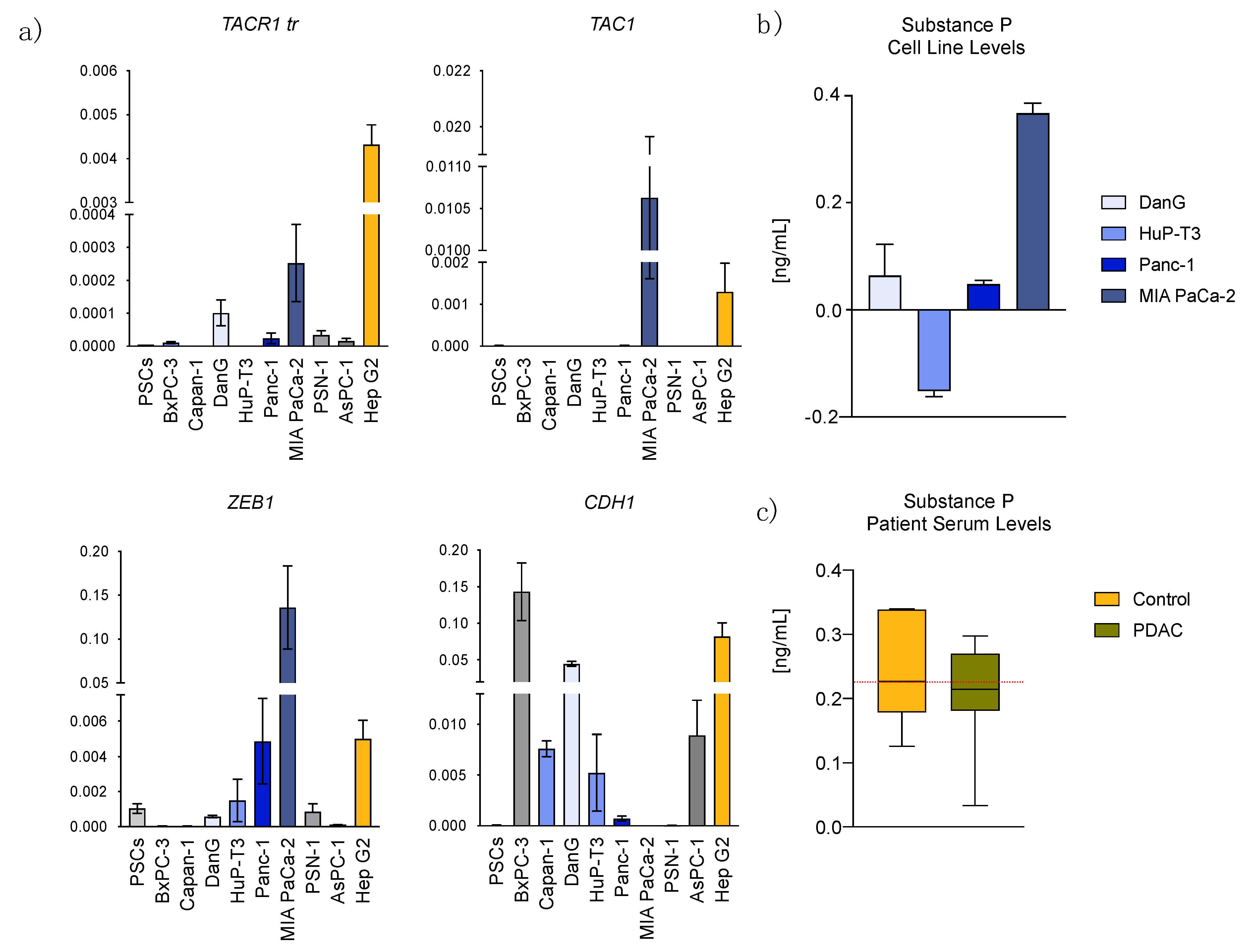
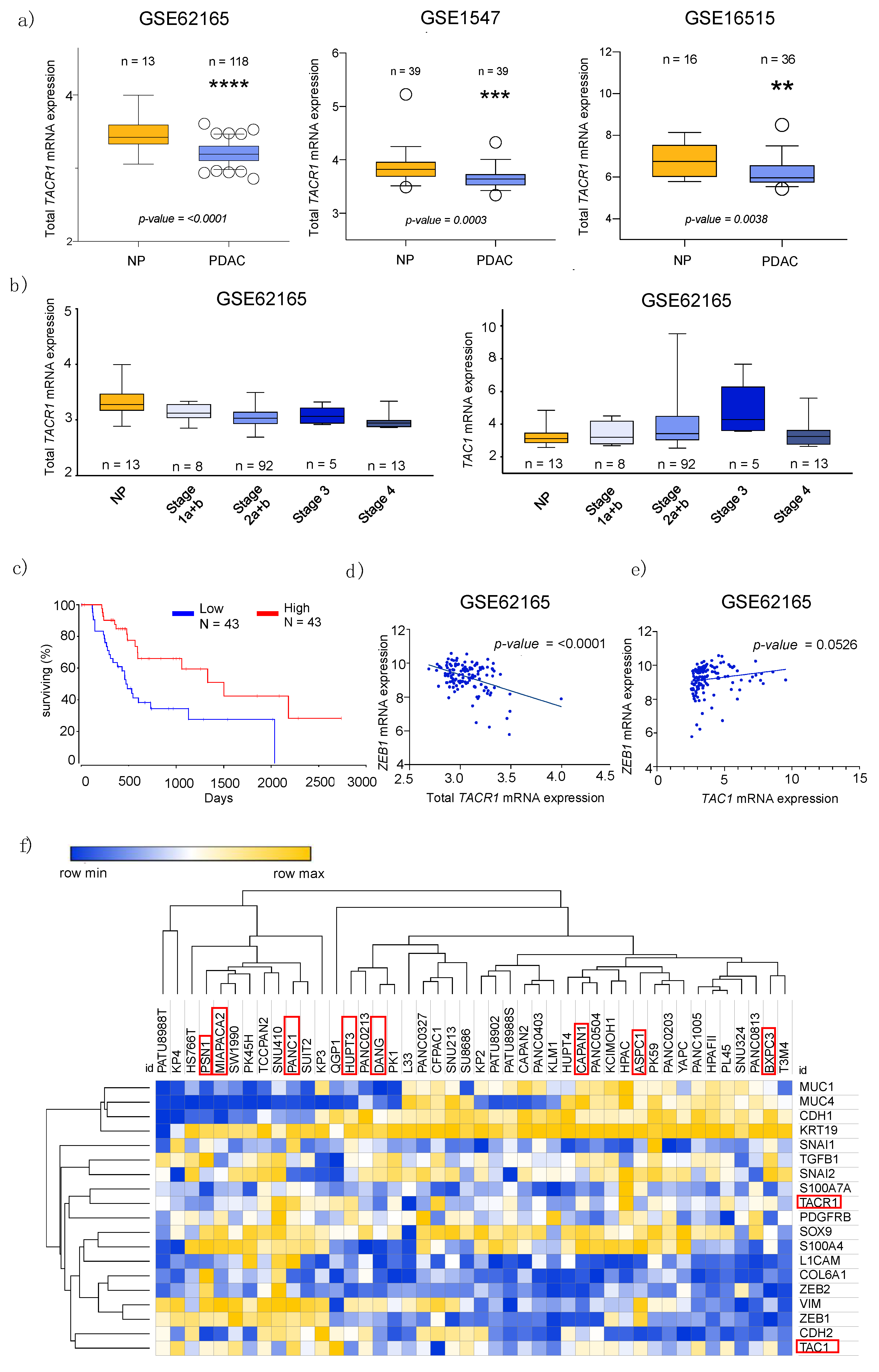
3.1. Transcriptome Data Analysis Reveals Diverging Expression of TACR1 and TAC1 in PDAC and Normal Pancreatic Tissue
3.2. Epithelial to Mesenchymal Transition State Correlates with Expression of TACR1 and TAC1
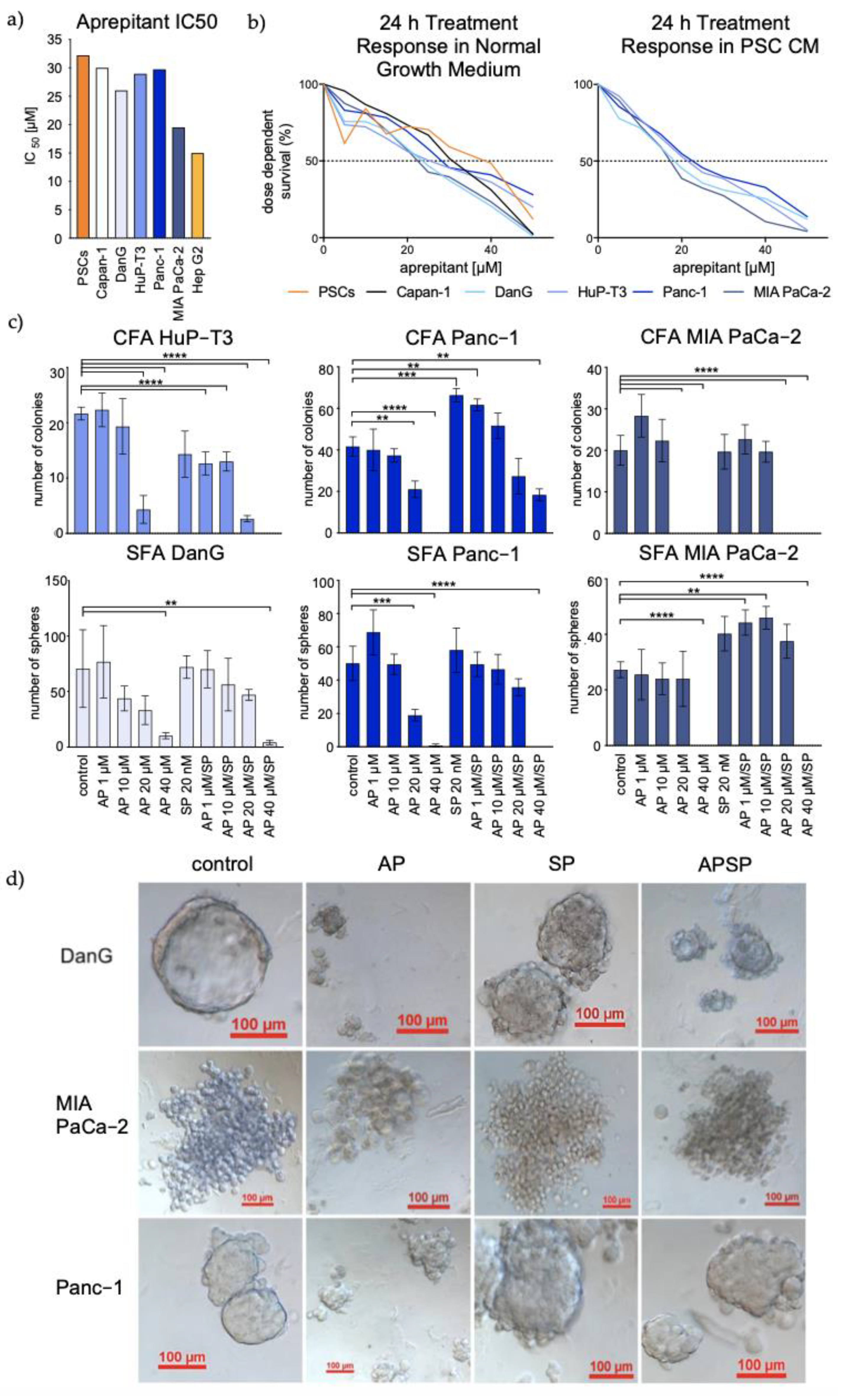
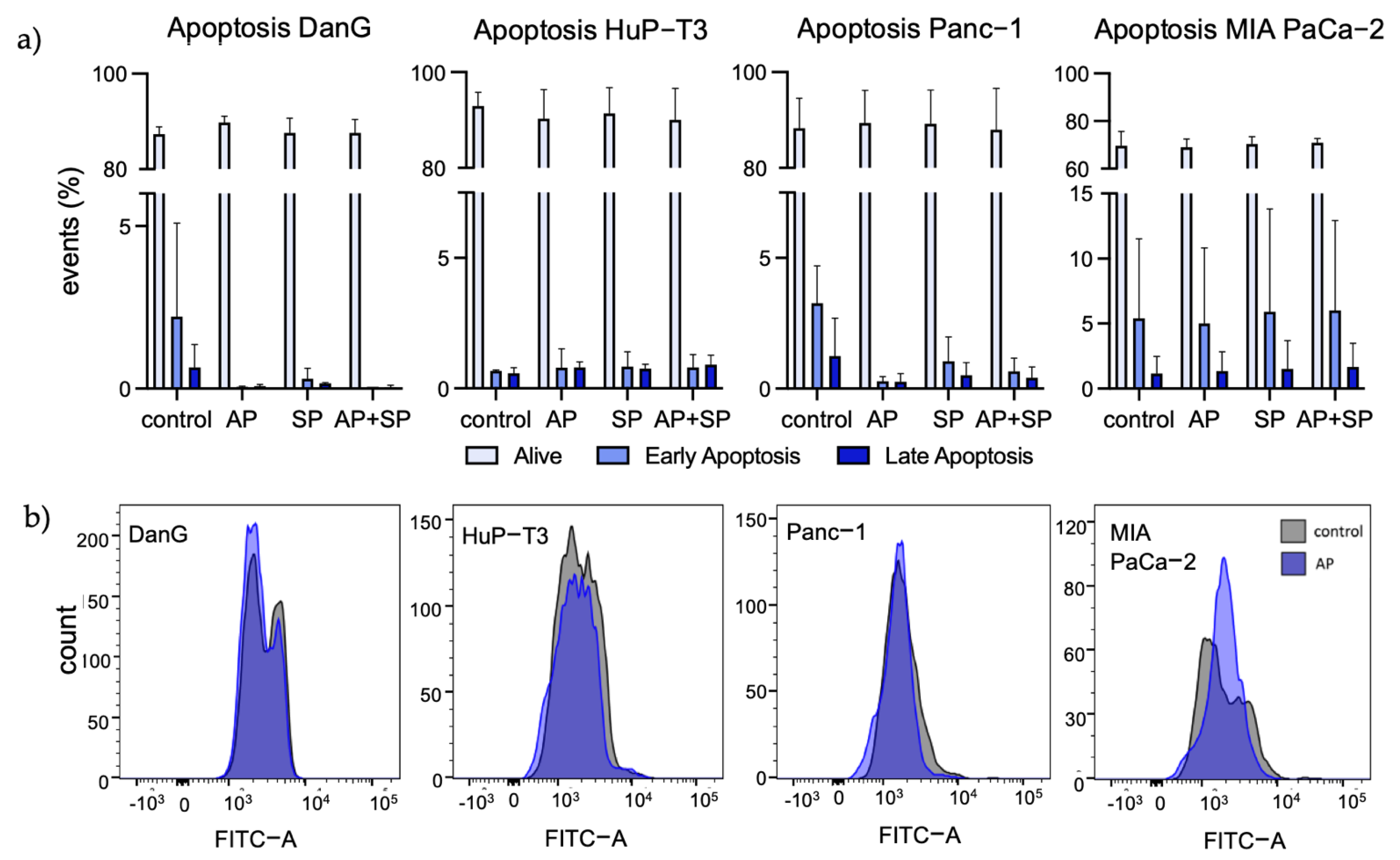
3.3. Aprepitant Significantly Reduces Growth in PDAC Cell Lines and Cancer Stem Cell-Like Cells
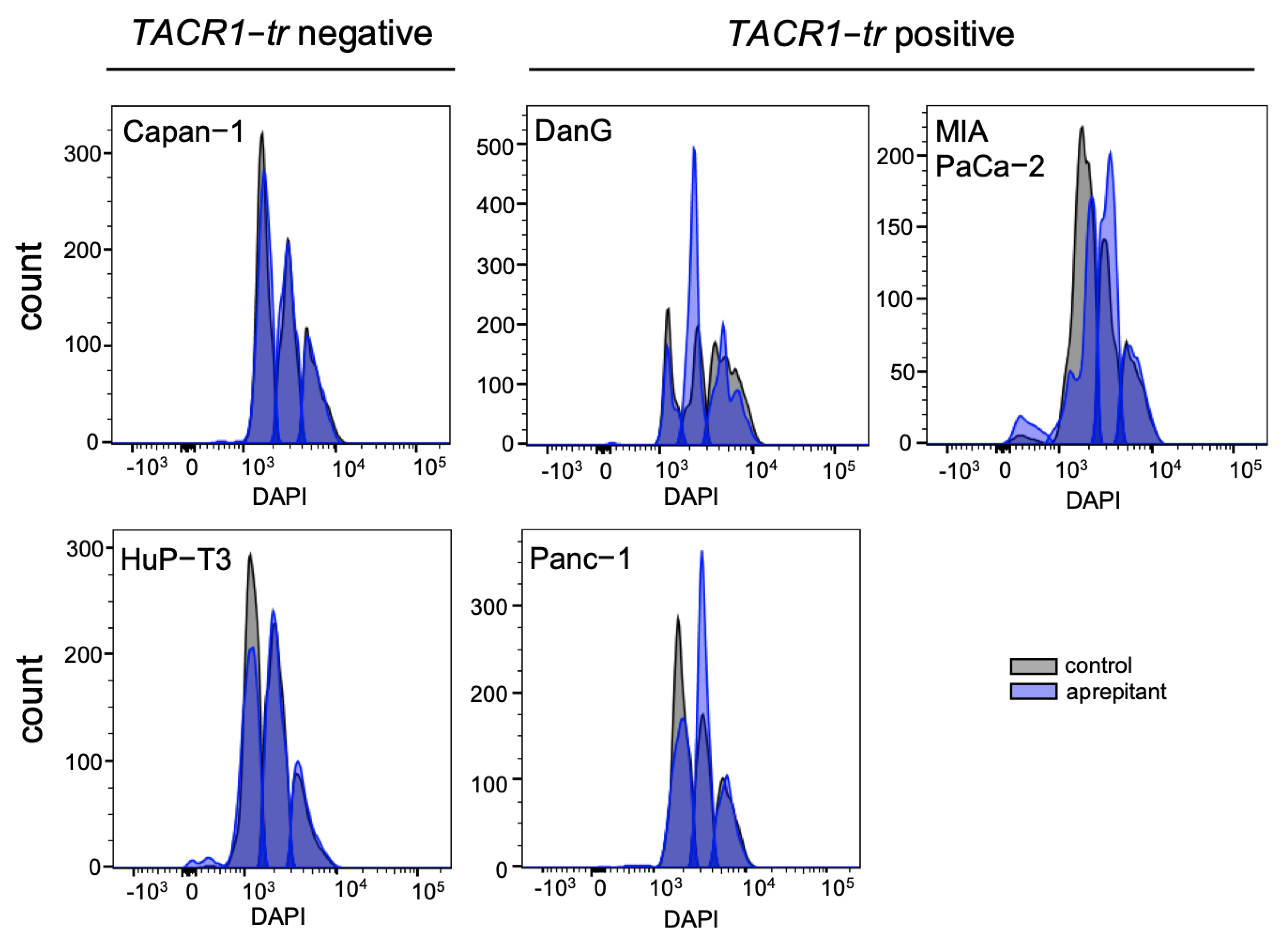
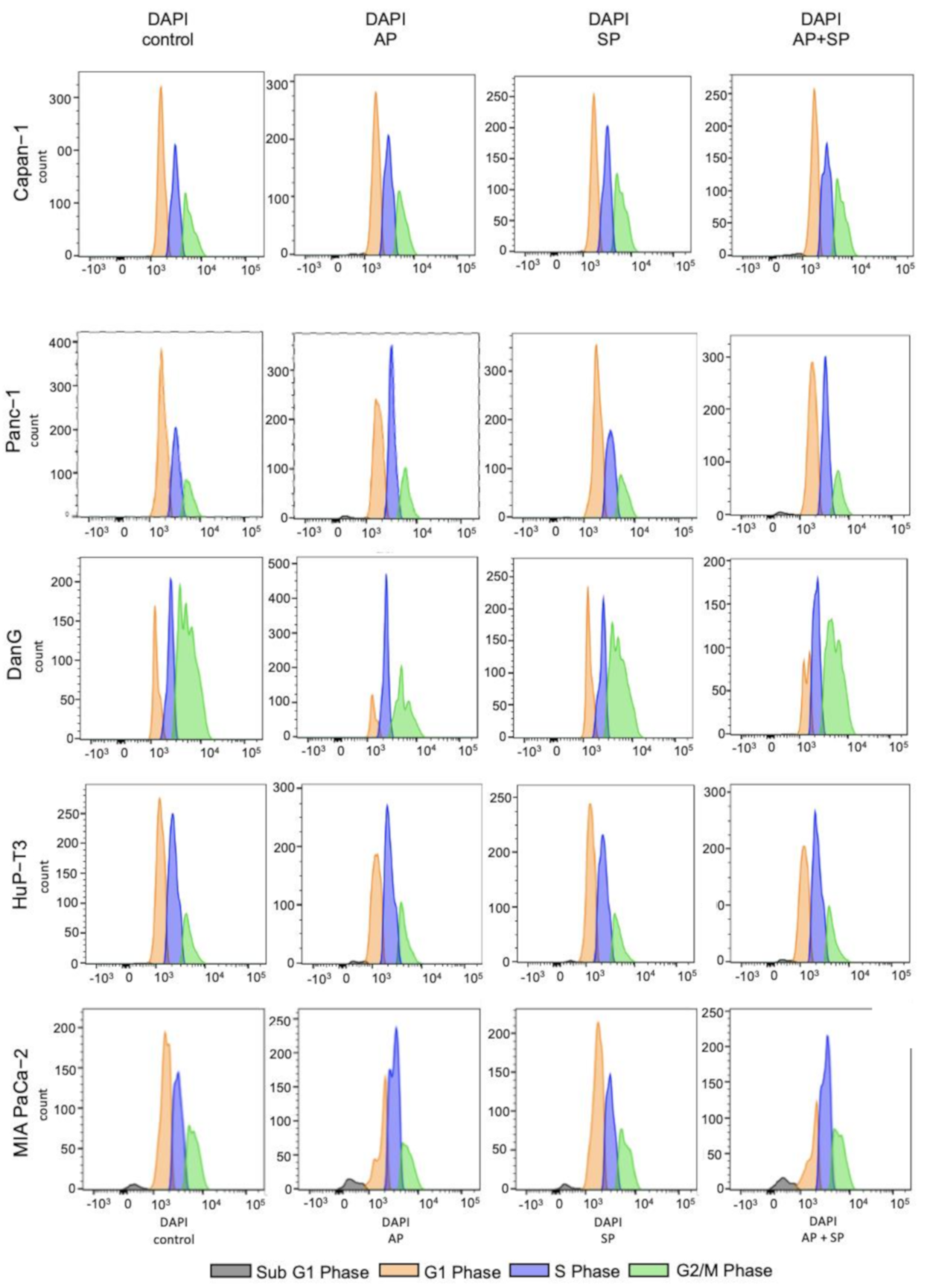
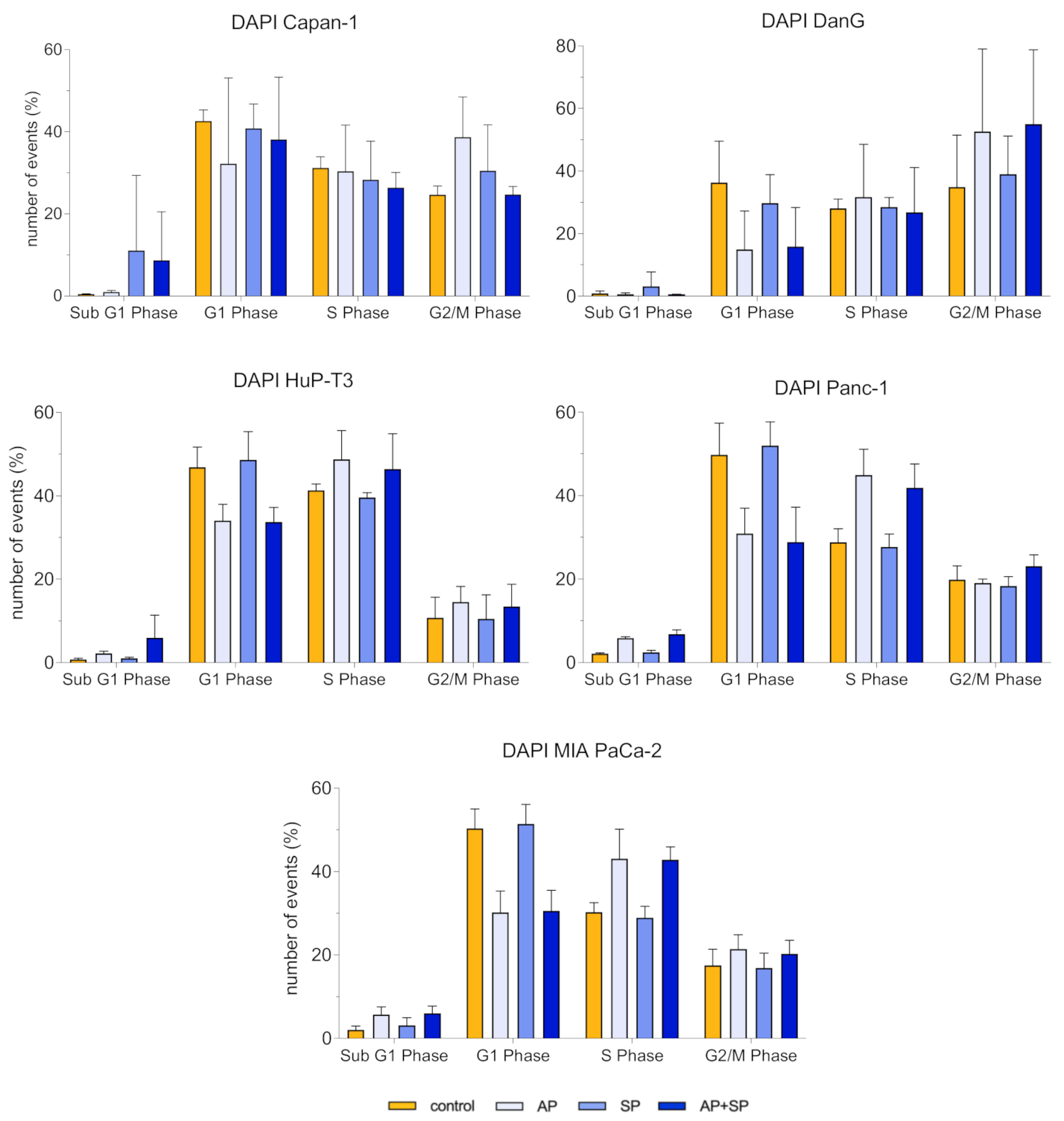
3.4. Aprepitant Affects Cell Cycle Progression in PDAC Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Ariston Gabriel, A.N.; Jiao, Q.; Yvette, U.; Yang, X.; Al-Ameri, S.A.; Du, L.; Wang, Y.-S.; Wang, C. Differences between KC and KPC pancreatic ductal adenocarcinoma mice models, in terms of their modelling biology and their clinical relevance. Pancreatology 2020, 20, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Quante, A.S.; Ming, C.; Rottmann, M.; Engel, J.; Boeck, S.; Heinemann, V.; Westphalen, C.B.; Strauch, K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016, 5, 2649–2656. [Google Scholar] [CrossRef]
- Gasparini, G.; Pellegatta, M.; Crippa, S.; Schiavo Lena, M.; Belfiori, G.; Doglioni, C.; Taveggia, C.; Falconi, M. Nerves and Pancreatic Cancer: New Insights into A Dangerous Relationship. Cancers 2019, 11, 893. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 adrenergic-neurotrophin feed-forward loop promotes pancreatic cancer. Cancer Cell. 2018, 33, 75–90. [Google Scholar] [CrossRef]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic Signaling via Muscarinic Receptors Directly and Indirectly Suppresses Pancreatic Tumorigenesis and Cancer Stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef]
- Saloman, J.L.; Albers, K.M.; Li, D.; Hartman, D.J.; Crawford, H.C.; Muha, E.A.; Rhim, A.D.; Davis, B.M. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 3078–3083. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Martinez, A.N.; Philipp, M.T. Substance P and Antagonists of the Neuokinin-1 Receptor in Neuroinflammation Associated with Infectious and Neurodegenerative Diseases of the Central Nervous System. J. Neurol. Neuromed. 2016, 1, 29–36. [Google Scholar]
- Li, X.; Ma, G.; Ma, Q.; Li, W.; Liu, J.; Han, L.; Duan, W.; Xu, Q.; Liu, H.; Wang, Z.; et al. Neurotransmitter substance P mediates pancreatic cancer perineural invasion via NK-1R in cancer cells. Mol. Cancer Res. 2013, 11, 294–302. [Google Scholar] [CrossRef]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and their receptors: Contributions to physiological control and the mechanisms of disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef]
- Krause, J.E.; Takeda, Y.; Hershey, A.D. Structure, functions, and mechanisms of substance P receptor action. J. Investig. Dermatol. 1992, 98, 2S–7S. [Google Scholar] [CrossRef]
- Garnier, A.; Ilmer, M.; Becker, K.; Häberle, B.; von Schweinitz, D.; Kappler, R.; Berger, M. Truncated neurokinin-1 receptor is an ubiquitous antitumor target in hepatoblastoma, and its expression is independent of tumor biology and stage. Oncol. Lett. 2016, 11, 870–878. [Google Scholar] [CrossRef][Green Version]
- Lai, J.-P.; Lai, S.; Tuluc, F.; Tansky, M.F.; Kilpatrick, L.E.; Leeman, S.E.; Douglas, S.D. Differences in the length of the carboxyl terminus mediate functional properties of neurokinin-1 receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 12605–12610. [Google Scholar] [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Déry, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a β-arrestin dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef]
- Tuluc, F.; Lai, J.P.; Kilpatrick, L.E.; Evans, D.; Douglas, S.D. Neurokinin 1 receptor isoforms and the control of innate immunity. Trends Immunol. 2009, 30, 271–276. [Google Scholar] [CrossRef]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30. [Google Scholar]
- Muñoz, M.; Crespo, J.C.; Crespo, J.P.; Coveñas, R. Neurokinin-1 receptor antagonist aprepitant and radiotherapy, a successful combination therapy in a patient with lung cancer: A case report. Mol. Clin. Oncol. 2019, 11, 50–54. [Google Scholar]
- Berger, M.; Neth, O.; Ilmer, M.; Garnier, A.; Salinas-Martin, M.V.; de Agustín Ascencio, J.C.; von Schweinitz, D.; Kappler, R.; Muñoz, M. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J. Hepatol. 2014, 60, 985–994. [Google Scholar] [CrossRef]
- Garnier, A.; Vykoukal, J.; Hubertus, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M.; Ilmer, M. Targeting the neurokinin-1 receptor inhibits growth of human colon cancer cells. Int. J. Oncol. 2015, 47, 151–160. [Google Scholar] [CrossRef]
- Ilmer, M.; Garnier, A.; Vykoukal, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M. Targeting the Neurokinin-1 Receptor Compromises Canonical Wnt Signaling in Hepatoblastoma. Mol. Cancer Ther. 2015, 14, 2712–2721. [Google Scholar] [CrossRef]
- Ilmer, M.; Boiles, A.R.; Regel, I.; Yokoi, K.; Michalski, C.W.; Wistuba, I.I.; Rodriguez, J.; Alt, E.; Vykoukal, J. RSpo2 enhances canonical Wnt signaling to confer stemness associated traits to susceptible pancreatic cancer cells. Cancer Res. 2015, 75, 1883–1896. [Google Scholar] [CrossRef]
- Kolorz, J.; Demir, S.; Gottschlich, A.; Beirith, I.; Ilmer, M.; Lüthy, D.; Walz, C.; Dorostkar, M.; von Schweinitz, D.; Kobold, S.; et al. The Neurokinin-1 receptor is a target in pediatric rhabdoid tumors. Department of Pediatric Surgery, Research Laboratories, Dr. von Hauner Children’s Hospital, Ludwig-Maximilians-University Munich, 80337 Munich, Germany. Unpublished work. 2021. [Google Scholar]
- Monkman, J.H.; Thompson, E.W.; Nagaraj, S.H. Targeting Epithelial Mesenchymal Plasticity in Pancreatic Cancer: A Compendium of Preclinical Discovery in a Heterogeneous Disease. Cancers 2019, 11, 1745. [Google Scholar] [CrossRef]
- Dijk, F.; Veenstra, V.L.; Soer, E.C.; Dings, M.P.G.; Zhao, L.; Halfwerk, J.B.; Hooijer, G.K.; Damhofer, H.; Marzano, M.; Steins, A.; et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020, 10, 337. [Google Scholar] [CrossRef]
- Sinha, A.; Cherba, D.; Bartlam, H.; Lenkiewicz, E.; Evers, L.; Barrett, M.T.; Haab, B.B. Mesenchymal-like pancreatic cancer cells harbor specific genomic alterations more frequently than their epithelial-like counterparts. Mol. Oncol. 2014, 8, 1253–1265. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M. The NK-1 receptor antagonist aprepitant as a broad spectrum antitumor drug. Investig. New Drugs 2010, 28, 187–193. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumor metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, V.; Vilas Jain, M. In Vitro Tumorigenic Assay: Colony Forming Assay for Cancer Stem Cells. Methods Mol. Biol. 2018, 1692, 89–95. [Google Scholar] [PubMed]
- Zhou, Y.; Zhao, L.; Xiong, T.; Chen, X.; Zhang, Y.; Yu, M.; Yang, J.; Yao, Z. Roles of full-length and truncated neurokinin-1 receptors on tumor progression and distant metastasis in human breast cancer. Breast Cancer Res. Treat. 2013, 140, 49–61. [Google Scholar] [CrossRef]
- Spitsin, S.; Pappa, V.; Douglas, S.D. Truncation of neurokinin-1 receptor—Negative regulation of substance P signaling. J. Leukoc. Biol. 2018, 103, 1043–1051. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M.; Conveñas, R. The NK-1 Receptor is Involved in the Antitumoral Action of L-733,060 and in the Mitogenic Action of Substance P on Human Pancreatic Cancer Cell Lines. Lett. Drug Des. Discov. 2006, 3, 323–329. [Google Scholar] [CrossRef]
- Friess, H.; Zhu, Z.; Liard, V.; Shi, X.; Shrikhande, S.V.; Wang, L.; Lieb, K.; Korc, M.; Palma, C.; Zimmermann, A.; et al. Neurokinin-1 receptor expression and its potential effects on tumor growth in human pancreatic cancer. Lab Investig. 2003, 83, 731–742. [Google Scholar] [CrossRef]
- Wong, M.L.; Medrano, J.F. Real-time PCR form RNA quantification. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef]
- Edfors, F.; Danielsson, F.; Hallström, B.M.; Käll, L.; Lundberg, E.; Pontén, F.; Forsström, B.; Uhlén, M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst Biol. 2016, 12, 883. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, P.; Lei, S.; Deng, F.; Xiao, G.G.; Liu, Y.; Chen, X.; Li, L.; Wu, S.; Chen, Y.; et al. How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytes. Acta Biochim. Biophys. Sin. 2008, 40, 426–436. [Google Scholar] [CrossRef]
- Freire, V.S.; Burkhard, F.C.; Kessler, T.M.; Kuhn, A.; Draegger, A.; Monastyrskaya, K. MicroRNAs May Mediate the Down-Regulation of Neurokinin-1 Receptor in Chronic Bladder Pain Syndrome. Am. J. Pathol. 2010, 176, 288–303. [Google Scholar] [CrossRef]
- Timaner, M.; Tsai, K.K.; Shaked, Y. The multifaceted role of mesenchymal stem cells in cancer. Semin. Cancer Biol. 2020, 60, 225–237. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef]
- Jin, Y.; Hong, H.S.; So, Y. Substance P enhances mesenchymal stem cells-mediated immune modulation. Cytokine 2015, 71, 145–153. [Google Scholar] [CrossRef]
- Ferdek, P.E.; Jakubowska, M.A. Biology of pancreatic stellate cells-more than just pancreatic cancer. Pflug. Arch. 2017, 469, 1039–1050. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer. 2019, 5, 128–142. [Google Scholar] [CrossRef]
- Pojawa-Golab, M.; Jaworecka, K.; Reich, A. NK-1 Receptor Antagonists and Pruritus: Review of Current Literature. Dermatol. Ther. 2019, 9, 391–405. [Google Scholar] [CrossRef]
- Hayashi, H.; Kohno, T.; Hiraoka, N.; Sakamoto, Y.; Kondo, S.; Morizane, C.; Saito, M.; Shimada, K.; Ichikawa, H.; Komatsu, Y.; et al. Gene Mutation Profile Of Pancreatic Cancer Obtained Using Targeted Deep Sequencing And Its Association With Prognosis. Ann. Oncol. 2014, 25, iv210–iv253. [Google Scholar] [CrossRef][Green Version]
- Yan, W.; Liu, X.; Wang, Y.; Shuqing, H.; Wang, F.; Liu, X.; Xiao, F.; Guang, H. Identifying Drug Targets in Pancreatic Ductal Adenocarcinoma Through Machine Learning, Analyzing Biomolecular Networks, and Structural Modeling. Front. Pharmacol. 2020, 11, 534. [Google Scholar] [CrossRef]
- Melisi, D.; Calvetti, L.; Frizziero, M.; Tortora, G. Pancreatic cancer: Systemic combination therapies for a heterogeneous disease. Curr. Pharm. Des. 2014, 20, 6660–6669. [Google Scholar] [CrossRef]
- David, M.; Lepage, C.; Jouve, J.-L.; Jooste, V.; Chauvenet, M.; Faivre, J.; Bouvier, A.M. Management and prognosis of pancreatic cancer over a 30-year period. Br. J. Cancer 2009, 101, 215–218. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDAC Cell Line | EMT State | TAC1 | TACR1 |
|---|---|---|---|
| BxPC-3 | epithelial | + | + |
| Capan-1 | epithelial | + | + |
| DanG | epithelial | − − | − − |
| HuP-T3 | epithelial/mesenchymal | − − | + + |
| Panc-1 | epithelial/mesenchymal | + + | + + |
| MIA PaCa-2 | mesenchymal | + | − |
| PSN-1 | mesenchymal | + | − |
| AsPC-1 | mesenchymal | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beirith, I.; Renz, B.W.; Mudusetti, S.; Ring, N.S.; Kolorz, J.; Koch, D.; Bazhin, A.V.; Berger, M.; Wang, J.; Angele, M.K.; et al. Identification of the Neurokinin-1 Receptor as Targetable Stratification Factor for Drug Repurposing in Pancreatic Cancer. Cancers 2021, 13, 2703. https://doi.org/10.3390/cancers13112703
Beirith I, Renz BW, Mudusetti S, Ring NS, Kolorz J, Koch D, Bazhin AV, Berger M, Wang J, Angele MK, et al. Identification of the Neurokinin-1 Receptor as Targetable Stratification Factor for Drug Repurposing in Pancreatic Cancer. Cancers. 2021; 13(11):2703. https://doi.org/10.3390/cancers13112703
Chicago/Turabian StyleBeirith, Iris, Bernhard W. Renz, Shristee Mudusetti, Natalja Sergejewna Ring, Julian Kolorz, Dominik Koch, Alexandr V. Bazhin, Michael Berger, Jing Wang, Martin K. Angele, and et al. 2021. "Identification of the Neurokinin-1 Receptor as Targetable Stratification Factor for Drug Repurposing in Pancreatic Cancer" Cancers 13, no. 11: 2703. https://doi.org/10.3390/cancers13112703
APA StyleBeirith, I., Renz, B. W., Mudusetti, S., Ring, N. S., Kolorz, J., Koch, D., Bazhin, A. V., Berger, M., Wang, J., Angele, M. K., D’Haese, J. G., Guba, M. O., Niess, H., Andrassy, J., Werner, J., & Ilmer, M. (2021). Identification of the Neurokinin-1 Receptor as Targetable Stratification Factor for Drug Repurposing in Pancreatic Cancer. Cancers, 13(11), 2703. https://doi.org/10.3390/cancers13112703