
Substance P Antagonism as a Novel Therapeutic Option to Enhance Efficacy of Cisplatin in Triple Negative Breast Cancer and Protect PC12 Cells against Cisplatin-Induced Oxidative Stress and Apoptosis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Cisplatin Increases NK1R Levels in Both PC12 Neuronal Cells and TNBC Cells
2.2. NK1R Antagonism Protects Rat Neuronal PC12 Cells from Loss of Viability Induced by Cisplatin
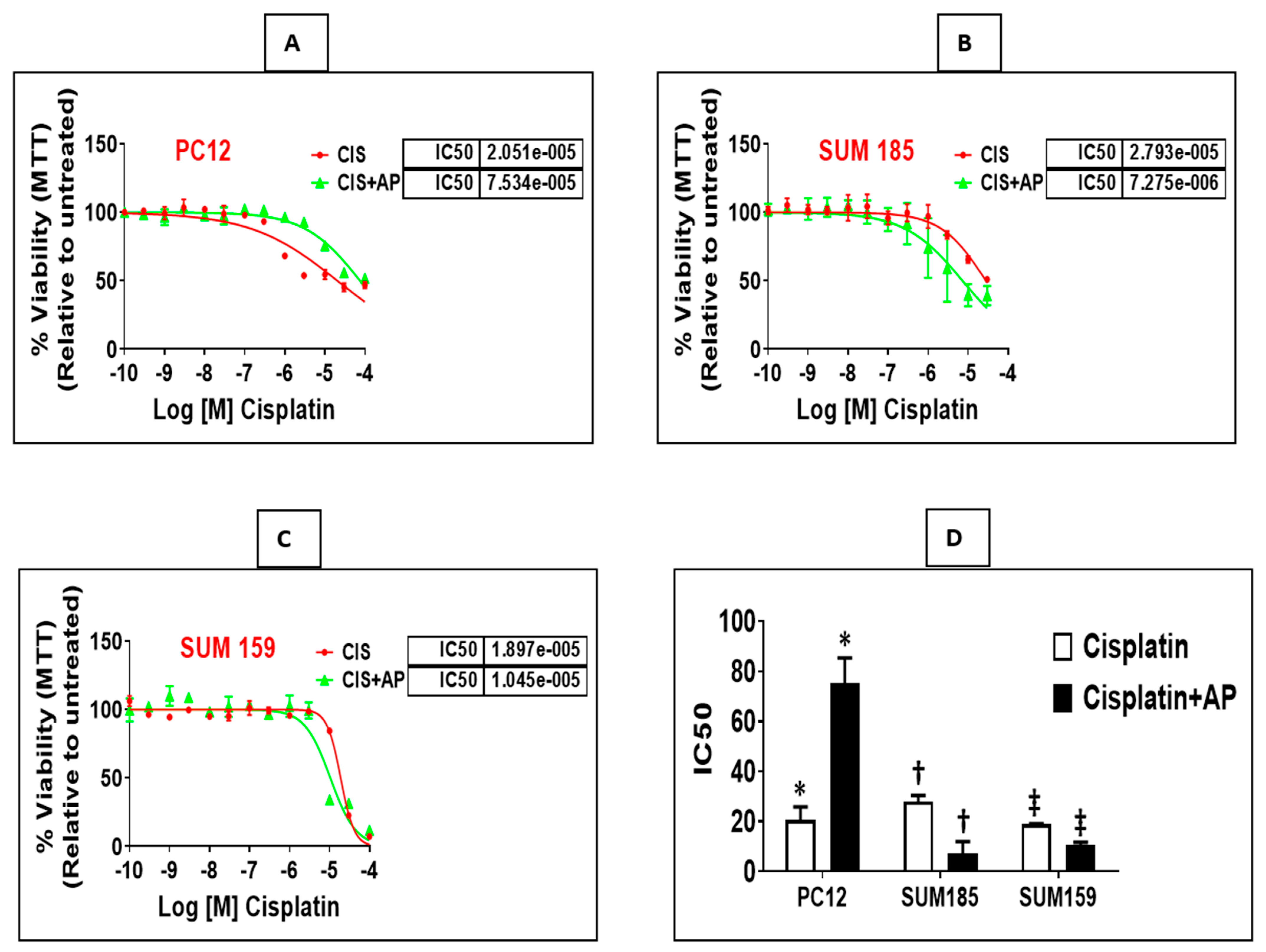
2.3. NK1R Antagonism Enhances Efficacy of Cisplatin in Two Triple Negative Breast Cancer (TNBC) Cells
2.4. NK1R Antagonism Attenuates ROS Production Induced by Cisplatin in PC12 Cells
2.5. Effect of NK1R Antagonism on ROS Production Induced by Cisplatin in TNBC Cells
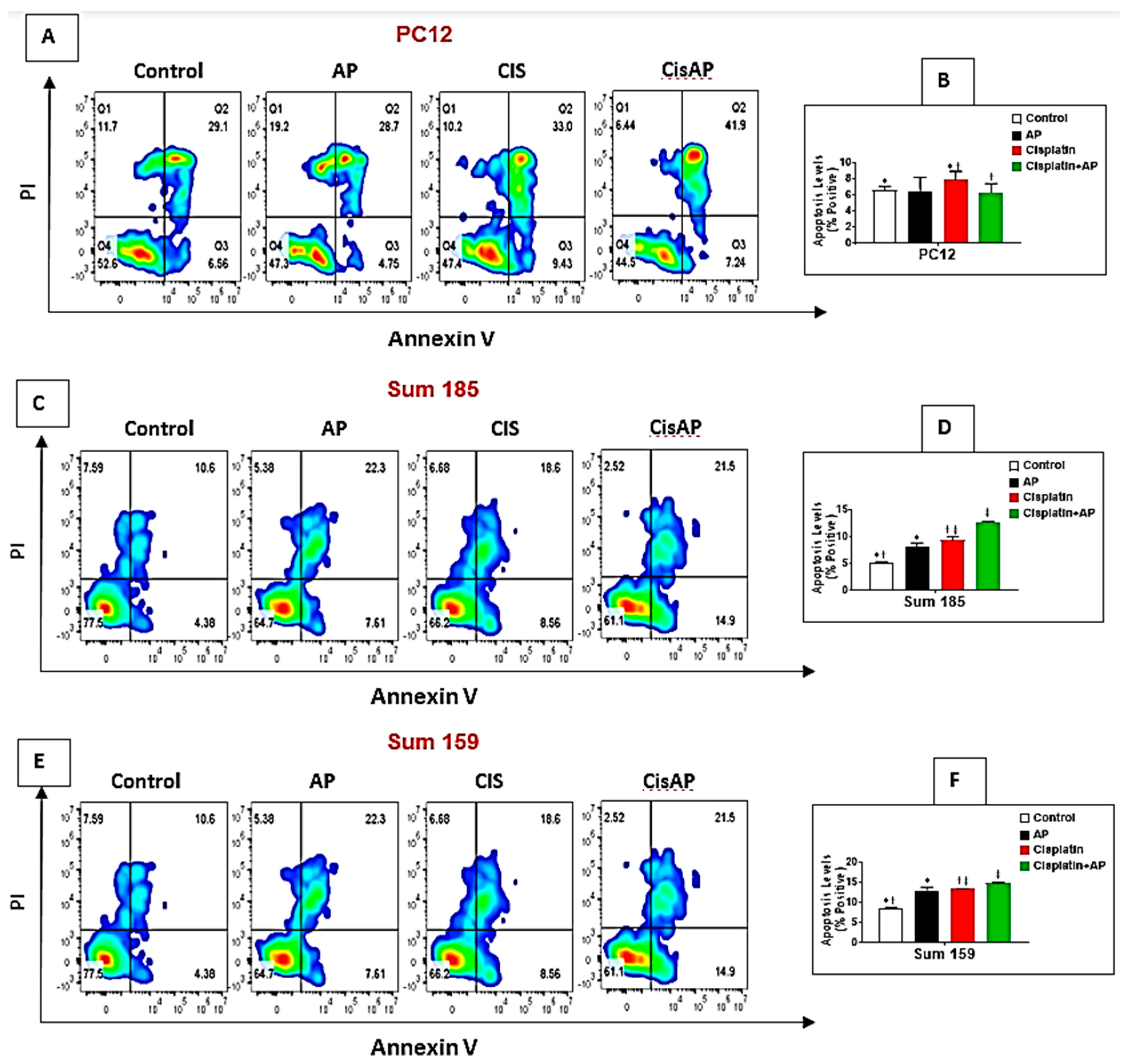
2.6. NK1R Antagonism Led to Decreased Levels of Apoptosis of PC12 Cells
2.7. NK1R Antagonism Led to Increased Levels of Apoptosis of TNBC Cells
2.8. Effects of NK1R Antagonism on the Transcriptome in TNBC Cells
3. Material and Methods
3.1. Cell Culture
3.2. Viability Assay
3.3. ROS Measurement
3.4. Flow Cytometry
3.5. RNA Isolation and Library Preparation and RNA-Seq Data Analysis
3.6. Statistical Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer Targets Ther. 2016, 8, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Parise, C.A.; Caggiano, V. Risk factors associated with the triple-negative breast cancer subtype within four race/ethnicities. Breast Cancer Res. Treat. 2017, 163, 151–158. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Hu, X.; Wang, B.; Wang, L.; Yang, W.; Liu, Y.; Liu, G.; Di, G.; Hu, Z.; et al. Cisplatin and gemcitabine as the first line therapy in metastatic triple negative breast cancer. Int. J. Cancer 2015, 136, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci Ther 2019, 11, 97. [Google Scholar]
- Tsimberidou, A.M.; Braiteh, F.; Stewart, D.J.; Kurzrock, R. Ultimate fate of oncology drugs approved by the us food and drug administration without a randomized Trial. J. Clin. Oncol. 2009, 27, 6243–6250. [Google Scholar] [CrossRef]
- Dhar, S.; Kolishetti, N.; Lippard, S.J.; Farokhzad, O.C. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 1850–1855. [Google Scholar] [CrossRef] [Green Version]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol. Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Munoz, M.; Rosso, M.; Covenas, R. A new frontier in the treatment of cancer: NK-1 receptor antagonists. Curr. Med. Chem. 2010, 17, 504–516. [Google Scholar] [CrossRef]
- Munoz, M.; Rosso, M.; Covenas, R. The NK-1 receptor: A new target in cancer therapy. Curr. Drug Targets 2011, 12, 909–921. [Google Scholar] [CrossRef]
- Hennig, I.M.; Laissue, J.A.; Horisberger, U.; Reubi, J.C. Substance-P receptors in human primary neoplasms: Tumoral and vascular localization. Int. J. Cancer 1995, 61, 786–792. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013, 48, 1–9. [Google Scholar] [CrossRef]
- Munoz, M.; Gonzalez-Ortega, A.; Salinas-Martin, M.V.; Carranza, A.; Garcia-Recio, S.; Almendro, V.; Covenas, R. The neurokinin-1 receptor antagonist aprepitant is a promising candidate for the treatment of breast cancer. Int. J. Oncol. 2014, 45, 1658–1672. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Moles, M.A.; Ramos-Garcia, P.; Esteban, F. Significance of the Overexpression of Substance P and Its Receptor NK-1R in Head and Neck Carcinogenesis: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 1349. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R. The Neurokinin-1 Receptor Antagonist Aprepitant: An Intelligent Bullet against Cancer? Cancers 2020, 12, 2682. [Google Scholar] [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Dery, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.; Yang, J.; Tong, L.; Yuan, S.; Tian, Y.; Hong, L.; Wang, W.; Zhang, H. Substance P immunoreactive nerve fibres are related to gastric cancer differentiation status and could promote proliferation and migration of gastric cancer cells. Cell Biol. Int. 2011, 35, 623–629. [Google Scholar] [CrossRef]
- Lewis, K.M.; Harford-Wright, E.; Vink, R.; Nimmo, A.J.; Ghabriel, M.N. Walker 256 tumour cells increase substance P immunoreactivity locally and modify the properties of the blood-brain barrier during extravasation and brain invasion. Clin. Exp. Metastasis 2013, 30, 1–12. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R.; Esteban, F.; Redondo, M. The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J. Biosci. 2015, 40, 441–463. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R. Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 2014, 46, 1727–1750. [Google Scholar] [CrossRef] [PubMed]
- Tejero-Taldo, M.I.; Kramer, J.H.; Mak Iu, T.; Komarov, A.M.; Weglicki, W.B. The nerve-heart connection in the pro-oxidant response to Mg-deficiency. Heart Fail. Rev. 2006, 11, 35–44. [Google Scholar] [CrossRef]
- Sterner-Kock, A.; Braun, R.K.; van der Vliet, A.; Schrenzel, M.D.; McDonald, R.J.; Kabbur, M.B.; Vulliet, P.R.; Hyde, D.M. Substance P primes the formation of hydrogen peroxide and nitric oxide in human neutrophils. J. Leukoc. Biol. 1999, 65, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Kasembeli, M.; Bharadwaj, U.; Engineer, N.; Eckols, K.T.; Tweardy, D.J. Substance P Receptor Signaling Mediates Doxorubicin-Induced Cardiomyocyte Apoptosis and Triple-Negative Breast Cancer Chemoresistance. Biomed. Res. Int. 2016, 2016, 1959270. [Google Scholar] [CrossRef] [Green Version]
- Mitro, N.; Cermenati, G.; Brioschi, E.; Abbiati, F.; Audano, M.; Giatti, S.; Crestani, M.; De Fabiani, E.; Azcoitia, I.; Garcia-Segura, L.M.; et al. Neuroactive steroid treatment modulates myelin lipid profile in diabetic peripheral neuropathy. J. Steroid Biochem. Mol. Biol. 2014, 143, 115–121. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, F.; Mao, J.; Lu, Y.; Li, J.; Ma, W.; Fan, S.; Zhang, C.; Li, Q.; Wang, B.; et al. Protein tyrosine phosphatase receptor-type delta acts as a negative regulator suppressing breast cancer. Oncotarget 2017, 8, 98798–98811. [Google Scholar] [CrossRef]
- Teider, N.; Scott, D.K.; Neiss, A.; Weeraratne, S.D.; Amani, V.M.; Wang, Y.; Marquez, V.E.; Cho, Y.J.; Pomeroy, S.L. Neuralized1 causes apoptosis and downregulates Notch target genes in medulloblastoma. Neuro Oncol. 2010, 12, 1244–1256. [Google Scholar] [CrossRef] [Green Version]
- Sopariwala, D.H.; Likhite, N.; Pei, G.; Haroon, F.; Lin, L.; Yadav, V.; Zhao, Z.; Narkar, V.A. Estrogen-related receptor alpha is involved in angiogenesis and skeletal muscle revascularization in hindlimb ischemia. FASEB J. 2021, 35, e21480. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Starobova, H.; Vetter, I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 174. [Google Scholar] [CrossRef]
- Tardaguila, M.; Mira, E.; Garcia-Cabezas, M.A.; Feijoo, A.M.; Quintela-Fandino, M.; Azcoitia, I.; Lira, S.A.; Manes, S. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013, 73, 4461–4473. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Ouzounova, M.; Piranlioglu, R.; Ma, M.T.; Guzel, M.; Marasco, D.; Chadli, A.; Gestwicki, J.E.; Cowell, J.K.; Wicha, M.S.; et al. The pleiotropic effects of TNFalpha in breast cancer subtypes is regulated by TNFAIP3/A20. Oncogene 2019, 38, 469–482. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 2012, 24, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Fingleton, B. Matrix metalloproteinases: Roles in cancer and metastasis. Front. Biosci. 2006, 11, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44-46, 200–206. [Google Scholar] [CrossRef]
- Johnson, B.; Mahadevan, D. Emerging Role and Targeting of Carcinoembryonic Antigen-related Cell Adhesion Molecule 6 (CEACAM6) in Human Malignancies. Clin. Cancer Drugs 2015, 2, 100–111. [Google Scholar] [CrossRef]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Oshi, M.; Butash, A.L.; Katsuta, E.; Tachibana, K.; Saito, K.; Okayama, H.; Peng, X.; Yan, L.; Kono, K.; et al. Triple-Negative Breast Cancer with High Levels of Annexin A1 Expression Is Associated with Mast Cell Infiltration, Inflammation, and Angiogenesis. Int. J. Mol. Sci 2019, 20, 4197. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Shi, Z.; Bai, Y.; Liu, L.; Cheng, K. Prognostic significance of systemic immune-inflammation index in triple-negative breast cancer. Cancer Manag Res. 2019, 11, 4471–4480. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Gaber, A.; Allaoui, R.; Mehmeti, M.; Jirstrom, K.; Leanderson, T.; Leandersson, K. S100A9 expressed in ER(-)PgR(-) breast cancers induces inflammatory cytokines and is associated with an impaired overall survival. Br. J. Cancer 2015, 113, 1234–1243. [Google Scholar] [CrossRef] [Green Version]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Korner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol 2019, 10, 757. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, Z.; Lu, W.; Chen, Z.; Chen, L.; Han, S.; Wu, X.; Cai, T.; Cai, Y. Chemokines and chemokine receptors: A new strategy for breast cancer therapy. Cancer Med. 2020, 9, 3786–3799. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, R.M.C.; Gibbs, C.R.; Kim, S.; Lee, E.S.; Adunyah, S.E.; Son, D.S. Serum amyloid A predisposes inflammatory tumor microenvironment in triple negative breast cancer. Oncotarget 2019, 10, 511–526. [Google Scholar] [CrossRef] [Green Version]
- Marzec, K.A.; Baxter, R.C.; Martin, J.L. Targeting Insulin-Like Growth Factor Binding Protein-3 Signaling in Triple-Negative Breast Cancer. Biomed. Res. Int. 2015, 2015, 638526. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Yuan, P.; Du, F.; Hong, R.; Ding, X.; Shi, X.; Fan, Y.; Wang, J.; Luo, Y.; Ma, F.; et al. SPARC overexpression in primary tumors correlates with disease recurrence and overall survival in patients with triple negative breast cancer. Oncotarget 2016, 7, 76628–76634. [Google Scholar] [CrossRef]
- Wiedemann, S.M.; Mildner, S.N.; Bonisch, C.; Israel, L.; Maiser, A.; Matheisl, S.; Straub, T.; Merkl, R.; Leonhardt, H.; Kremmer, E.; et al. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell Biol. 2010, 190, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Hojo, T.; Akiyama, Y.; Nagasaki, K.; Maruyama, K.; Kikuchi, K.; Ikeda, T.; Kitajima, M.; Yamaguchi, K. Association of maspin expression with the malignancy grade and tumor vascularization in breast cancer tissues. Cancer Lett. 2001, 171, 103–110. [Google Scholar] [CrossRef]
- Xia, W.; Lau, Y.K.; Hu, M.C.; Li, L.; Johnston, D.A.; Sheng, S.; El-Naggar, A.; Hung, M.C. High tumoral maspin expression is associated with improved survival of patients with oral squamous cell carcinoma. Oncogene 2000, 19, 2398–2403. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.G.; Kim, W.J.; Noh, M.G.; Chun, K.H.; Kim, S.J. SPON2 Is Upregulated through Notch Signaling Pathway and Promotes Tumor Progression in Gastric Cancer. Cancers 2020, 12, 1439. [Google Scholar] [CrossRef]
- Robertson, D.M.; Pruysers, E.; Jobling, T. Inhibin as a diagnostic marker for ovarian cancer. Cancer Lett. 2007, 249, 14–17. [Google Scholar] [CrossRef]
- Lasham, A.; Mehta, S.Y.; Fitzgerald, S.J.; Woolley, A.G.; Hearn, J.I.; Hurley, D.G.; Ruza, I.; Algie, M.; Shelling, A.N.; Braithwaite, A.W.; et al. A novel EGR-1 dependent mechanism for YB-1 modulation of paclitaxel response in a triple negative breast cancer cell line. Int. J. Cancer 2016, 139, 1157–1170. [Google Scholar] [CrossRef] [Green Version]
- Ozols, R.F.; Young, R.C. High-dose cisplatin therapy in ovarian cancer. Semin. Oncol. 1985, 12, 21–30. [Google Scholar]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Cersosimo, R.J. Cisplatin neurotoxicity. Cancer Treat. Rev. 1989, 16, 195–211. [Google Scholar] [CrossRef]
- Li, D.W.; Sun, J.Y.; Wang, K.; Zhang, S.; Hou, Y.J.; Yang, M.F.; Fu, X.Y.; Zhang, Z.Y.; Mao, L.L.; Yuan, H.; et al. Attenuation of Cisplatin-Induced Neurotoxicity by Cyanidin, a Natural Inhibitor of ROS-Mediated Apoptosis in PC12 Cells. Cell Mol. Neurobiol. 2015, 35, 995–1001. [Google Scholar] [CrossRef]
- Un, H.; Ugan, R.A.; Kose, D.; Bayir, Y.; Cadirci, E.; Selli, J.; Halici, Z. A novel effect of Aprepitant: Protection for cisplatin-induced nephrotoxicity and hepatotoxicity. Eur. J. Pharmacol. 2020, 880, 173168. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, E.; Pei, G.; Zhao, Z.; Kim, S.T.; German, A.; Robinson, P. Substance P Antagonism as a Novel Therapeutic Option to Enhance Efficacy of Cisplatin in Triple Negative Breast Cancer and Protect PC12 Cells against Cisplatin-Induced Oxidative Stress and Apoptosis. Cancers 2021, 13, 3871. https://doi.org/10.3390/cancers13153871
Rodriguez E, Pei G, Zhao Z, Kim ST, German A, Robinson P. Substance P Antagonism as a Novel Therapeutic Option to Enhance Efficacy of Cisplatin in Triple Negative Breast Cancer and Protect PC12 Cells against Cisplatin-Induced Oxidative Stress and Apoptosis. Cancers. 2021; 13(15):3871. https://doi.org/10.3390/cancers13153871
Chicago/Turabian StyleRodriguez, Emma, Guangsheng Pei, Zhongming Zhao, Sang T. Kim, Alexis German, and Prema Robinson. 2021. "Substance P Antagonism as a Novel Therapeutic Option to Enhance Efficacy of Cisplatin in Triple Negative Breast Cancer and Protect PC12 Cells against Cisplatin-Induced Oxidative Stress and Apoptosis" Cancers 13, no. 15: 3871. https://doi.org/10.3390/cancers13153871
APA StyleRodriguez, E., Pei, G., Zhao, Z., Kim, S. T., German, A., & Robinson, P. (2021). Substance P Antagonism as a Novel Therapeutic Option to Enhance Efficacy of Cisplatin in Triple Negative Breast Cancer and Protect PC12 Cells against Cisplatin-Induced Oxidative Stress and Apoptosis. Cancers, 13(15), 3871. https://doi.org/10.3390/cancers13153871