
Sequence Neighborhoods Enable Reliable Prediction of Pathogenic Mutations in Cancer Genomes


Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Mutation Datasets for Building and Evaluating the Models
- Glioblastoma (GBM) and Ovarian Cancer (OVC) mutations reported in the COSMIC database only once;
- The reported mutations had no other mutations within 3bp of their position and were not part of either the training or test datasets for building the machine learning model (CanDrA).
2.2. Feature Extraction
2.2.1. Sequence-Based Features
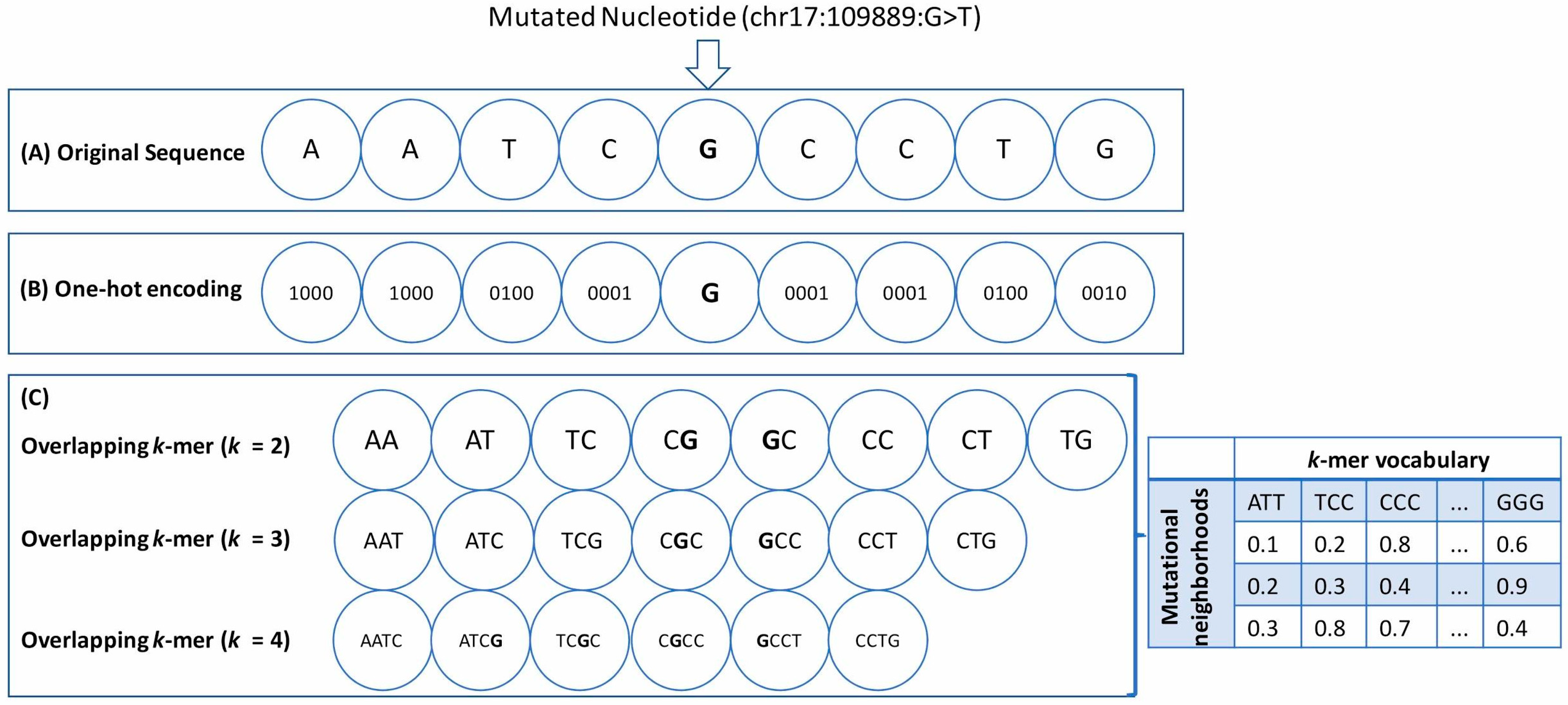
- One-hot encoding (OHE): Each neighboring nucleotide was represented as a binary vector of size 4 containing all zero values except the nucleotide index, marked as 1 (Figure 1A,B). Thus “A” was encoded as , “G” as , and so on.This particular feature representation resulted in a feature space of size , where represents the window sizes. We used the pandas get_dummies() to perform this task.
- Overlapping k-mers: In this type of feature representation, the neighboring nucleotide string sequences for a given window size were represented as overlapping k-mers of lengths 2, 3, and 4 (Figure 1C). For instance, an arbitrary sequence of window size 3 {ATTTGGA}, where ‘T’ is the wild-type base at the mutated position, can be decomposed into overlapping k-mers of size 2 {AT, TT, TT, TG, GG, GA}, 3 {ATT, TTT, TTG, TGG, GGA}, and 4 {ATTT, TTTG, TTGG, TGGA}, respectively.
2.2.2. Descriptive Genomic Features
2.3. Density Estimation
- One-hot encoding;
- Count Vectorizer (k-mer sizes of 2, 3 and 4);
- TF-IDF Vectorizer (k-mer sizes of 2, 3 and 4).
2.4. Classification Models
- The dataset was split using the cross-validation strategy;
- The training data was then split by label (driver/passenger);
- We fit a generative model for each class using the kernel density estimation method as described in the previous section. This provided us the likelihood that and , respectively, for a particular data point x;
- Next, the class prior, given by the number of examples of each class, or, and was calculated;
- Now, for a test data point x, the posterior probability was given by and . The label that maximized the posterior probabilities was the one assigned to x.
2.5. Model Selection and Tuning
Repeated Cross-Validation Experiments
2.6. Derivation of the Binary Classification Model to Distinguish between Driver and Passenger Mutations
2.7. Feature Selection
2.8. Hyperparameter Tuning and Classifier Threshold Selection
2.9. Performance Metrics
2.10. Comparison with Other Pan-Cancer Mutation Effect Predictors
3. Results
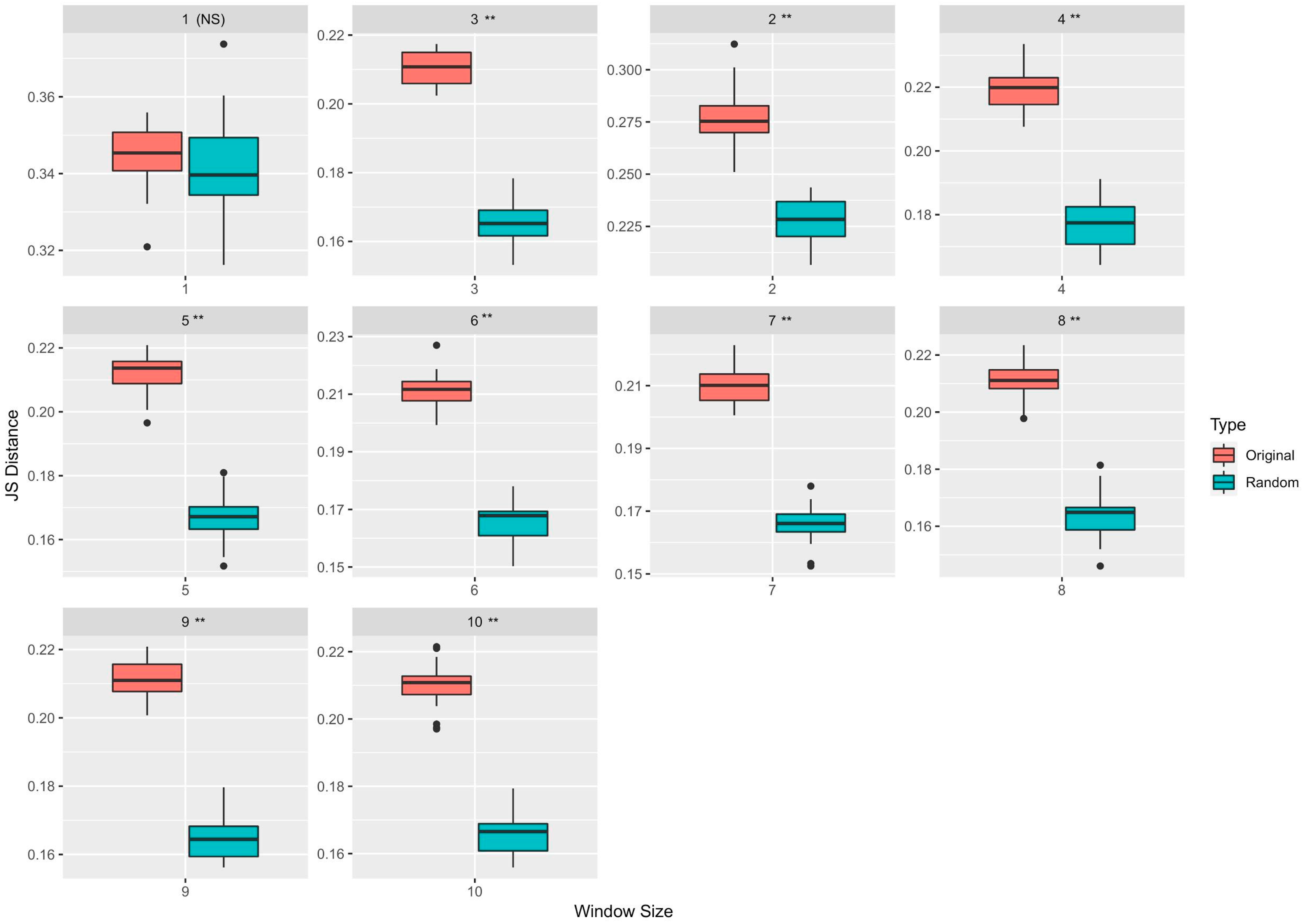
3.1. Neighborhood Sequences of Driver and Passenger Mutations Show Markedly Different Distributions
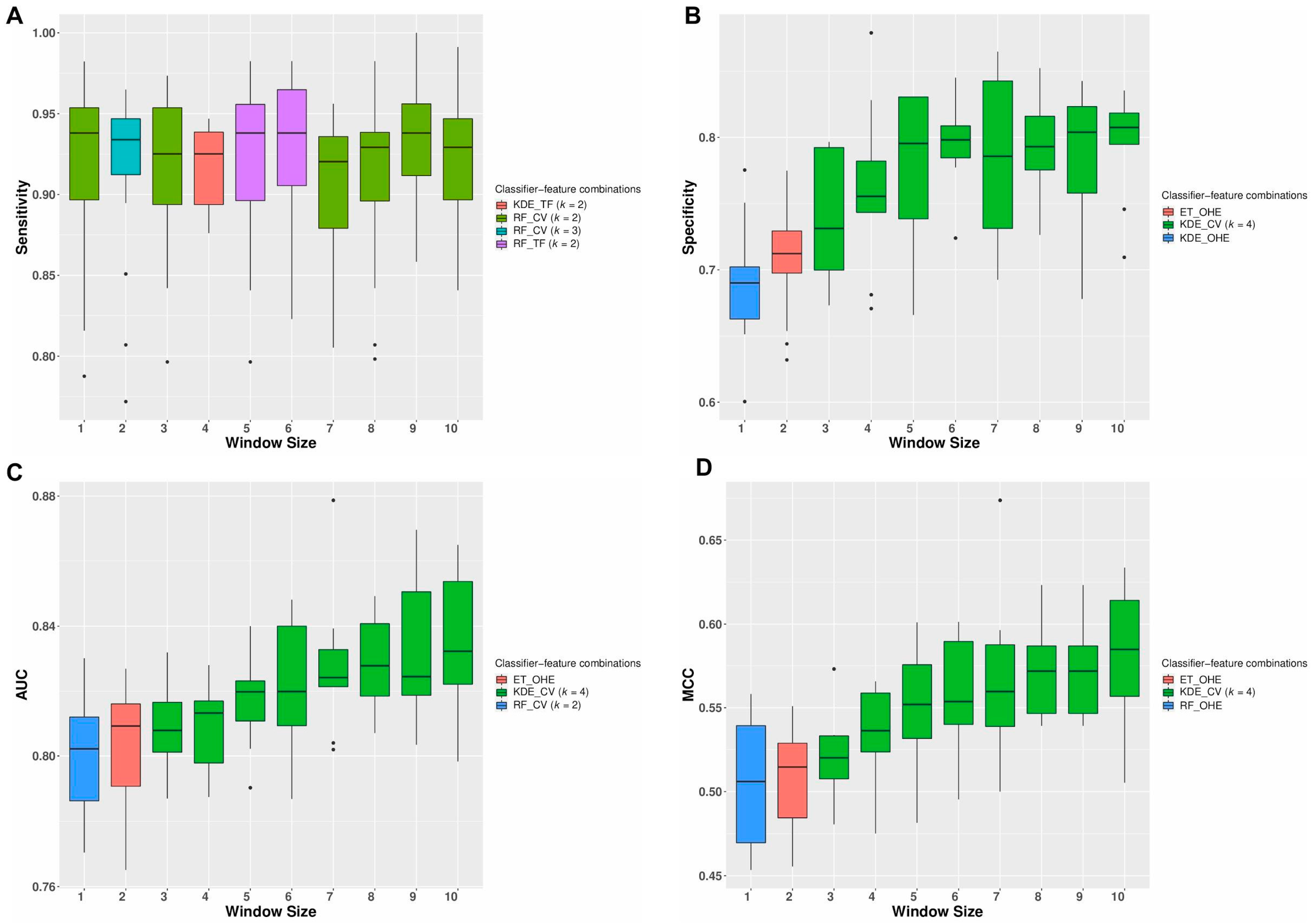
3.2. Repeated Cross-Validation Using Only Neighborhood Features Generates Robust Classification Models
3.3. Classification Models Provide Performances Comparable with Other State-of-the-Art Mutation Effect Predictors
3.4. Voting Ensemble of Prediction Algorithms Gives Better Classification Performances
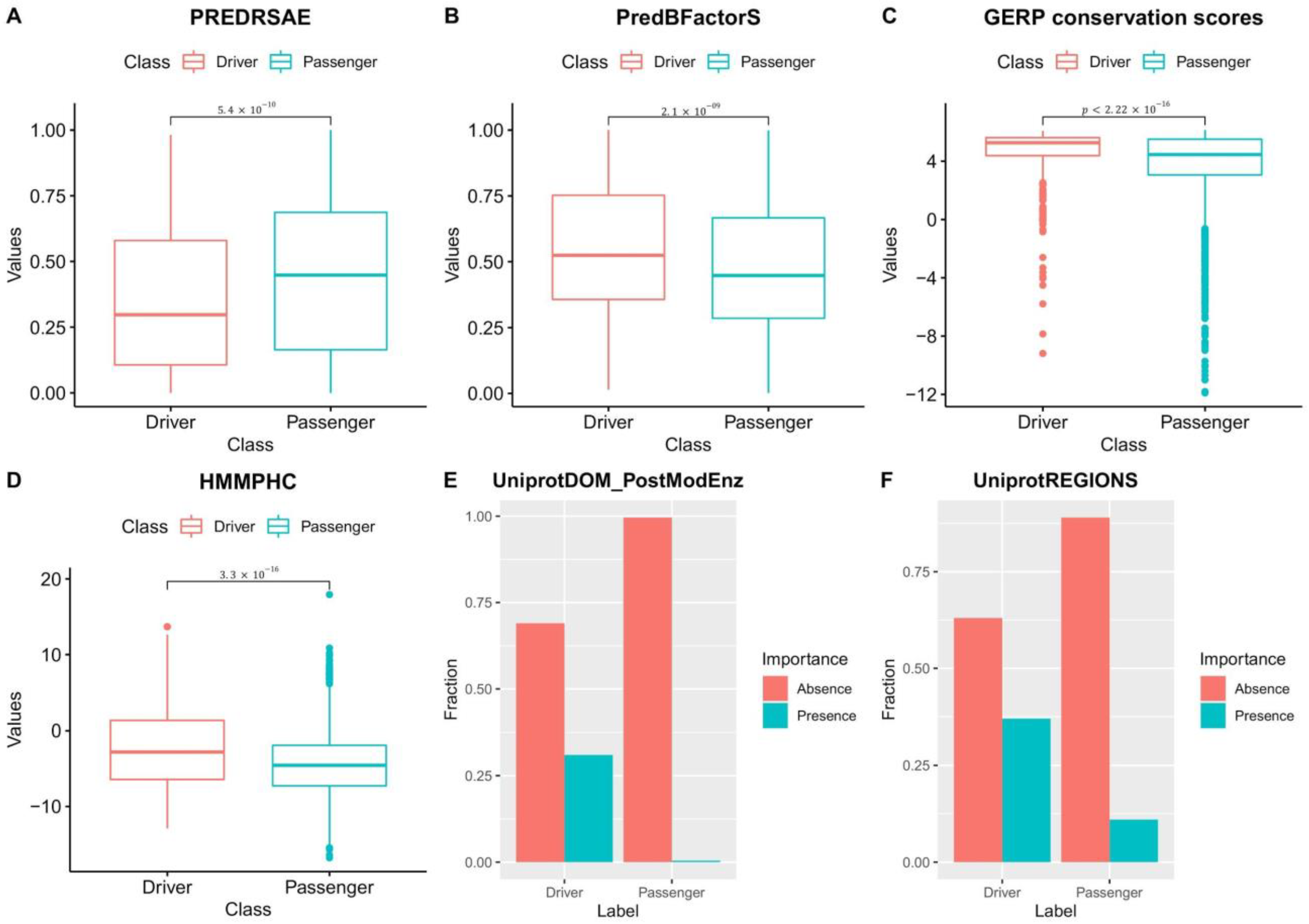
3.5. Driver and Passenger Mutations’ Features Used to Train NBDriver Are Significantly Different
3.6. Evaluation Using Previously Unseen Coding Mutation Data
3.7. Cancer Mutation Census
3.8. Cancer Genome Interpreter Database
3.9. Recurrent Driver Mutations
3.10. Rare Driver Mutations Found in Glioblastoma and Ovarian Cancer
3.11. Stratification of the Predicted Driver Genes Based on Literature Evidence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Samet, J.M. Radon and lung cancer. J. Natl. Cancer Inst. 1989, 81, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. Mutagenic mechanisms. Annu. Rev. Genet. 1969, 3, 247–268. [Google Scholar] [CrossRef]
- Zhu, W.; Wu, S.; Hannun, Y.A. Contributions of the Intrinsic Mutation Process to Cancer Mutation and Risk Burdens. EBioMedicine 2017, 24, 5–6. [Google Scholar] [CrossRef][Green Version]
- Raphael, B.J.; Dobson, J.R.; Oesper, L.; Vandin, F. Identifying driver mutations in sequenced cancer genomes: Computational approaches to enable precision medicine. Genome Med. 2014, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal—A one-stop shop for cancer genomics data. Database 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Ainscough, B.J.; Griffith, M.; Coffman, A.C.; Wagner, A.H.; Kunisaki, J.; Choudhary, M.N.; McMichael, J.F.; Fulton, R.S.; Wilson, R.K.; Griffith, O.L.; et al. DoCM: A database of curated mutations in cancer. Nat. Methods 2016, 13, 806–807. [Google Scholar] [CrossRef]
- Garraway, L.A. Genomics-driven oncology: Framework for an emerging paradigm. J. Clin. Oncol. 2013, 31, 1806–1814. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2. 0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, 1–14. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Cerami, E.; Demir, E.; Schultz, N.; Taylor, B.S.; Sander, C. Automated Network Analysis Identifies Core Pathways in Glioblastoma. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Vandin, F.; Upfal, E.; Raphael, B.J. Algorithms for detecting significantly mutated pathways in cancer. J. Comput. Biol. 2011, 18, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC Genom. 2013, 14, S3. [Google Scholar] [CrossRef] [PubMed]
- Tokheim, C.; Karchin, R. CHASMplus Reveals the Scope of Somatic Missense Mutations Driving Human Cancers. Cell Syst. 2019, 9, 9–23.e8. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Deu-Pons, J.; Lopez-Bigas, N. Improving the prediction of the functional impact of cancer mutations by baseline tolerance transformation. Genome Med. 2012, 4, 89. [Google Scholar] [CrossRef]
- Mao, Y.; Chen, H.; Liang, H.; Meric-Bernstam, F.; Mills, G.B.; Chen, K. CanDrA: Cancer-Specific Driver Missense Mutation Annotation with Optimized Features. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Hodgkinson, A.; Eyre-Walker, A. Variation in the mutation rate across mammalian genomes. Nat. Rev. Genet. 2011, 12, 756–766. [Google Scholar] [CrossRef]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rubin, A.F.; Green, P. Mutation patterns in cancer genomes. Proc. Nat. Acad. Sci. USA 2009, 106, 21766–21770. [Google Scholar] [CrossRef] [PubMed]
- Aggarwala, V.; Voight, B.F. An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nat. Genet. 2016, 48, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Boerwinkle, E. Neighboring-Nucleotide Effects on Single Nucleotide Polymorphisms: A Study of 2.6 Million Polymorphisms Across the Human Genome. Genome Res. 2002, 12, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef]
- Dietlein, F.; Weghorn, D.; Taylor-Weiner, A.; Richters, A.; Reardon, B.; Liu, D.; Lander, E.S.; Van Allen, E.M.; Sunyaev, S.R. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 2020, 52, 208–218. [Google Scholar] [CrossRef]
- Agajanian, S.; Oluyemi, O.; Verkhivker, G.M. Integration of Random Forest Classifiers and Deep Convolutional Neural Networks for Classification and Biomolecular Modeling of Cancer Driver Mutations. Front. Mol. Biosci. 2019. [Google Scholar] [CrossRef]
- Brown, A.-L.; Li, M.; Goncearenco, A.; Panchenko, A.R. Finding driver mutations in cancer: Elucidating the role of background mutational processes. PLoS Comput. Biol. 2019, 15, e1006981. [Google Scholar] [CrossRef]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.K.; Li, J.; Jeong, K.J.; Shao, S.; Chen, H.; Tsang, Y.H.; Sengupta, S.; Wang, Z.; Bhavana, V.H.; Tran, R.; et al. Systematic functional annotation of somatic mutations in cancer. Cancer Cell 2018, 33, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Young, D.L.; Islam, M.; Kitzman, J.O.; Gullingsrud, J.; Hause, R.J.; Fowler, D.M.; Parvin, J.D.; Shendure, J.; Fields, S. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics 2015, 200, 413–422. [Google Scholar] [CrossRef]
- Mahmood, K.; Jung, C.H.; Philip, G.; Georgeson, P.; Chung, J.; Pope, B.J.; Park, D.J. Variant effect prediction tools assessed using independent, functional assay-based datasets: Implications for discovery and diagnostics. Hum. Genom. 2017, 11, 10. [Google Scholar] [CrossRef]
- Martelotto, L.G.; Ng, C.K.; De Filippo, M.R.; Zhang, Y.; Piscuoglio, S.; Lim, R.S.; Shen, R.; Norton, L.; Reis-Filho, J.S.; Weigelt, B. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014, 15, 484. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, T.; Chong, Z.; Rohrdanz, M.A.; Melott, J.M.; Wakefield, C.; Zeng, J.; Weinstein, J.N.; Meric-Bernstam, F.; Mills, G.B.; et al. TransVar: A multilevel variant annotator for precision genomics. Nat. Methods 2015, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Tiao, G.; Hornshøj, H.; Hess, J.M.; Pedersen, R.I.; Feuerbach, L.; Sabarinathan, R.; Madsen, T.; et al. Discovery and characterization of coding and non-coding driver mutations in more than 2500 whole cancer genomes. BioRxiv 2017, 237313. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Lumley, T.; Johnson, R.C. gmodels: Various R programming tools for model fitting. R Package Version. 2015, 2. Available online: https://CRAN.R-project.org/package=gmodels (accessed on 20 November 2020).
- Wilson, D.L. Asymptotic properties of nearest neighbor rules using edited data. IEEE Trans. Syst. Man Cybern. 1972, 3, 408–421. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Pagel, K.A.; Kim, R.; Moad, K.; Busby, B.; Zheng, L.; Tokheim, C.; Ryan, M.; Karchin, R. Integrated informatics analysis of cancer-related variants. Jco Clin. Cancer Inform. 2020, 4, 310–317. [Google Scholar] [CrossRef]
- Ng, C.K.; Weigelt, B.; A’Hern, R.; Bidard, F.C.; Lemetre, C.; Swanton, C.; Shen, R.; Reis-Filho, J.S. Predictive Performance of Microarray Gene Signatures: Impact of Tumor Heterogeneity and Multiple Mechanisms of Drug Resistance. Cancer Res. 2014, 74, 2946–2961. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Schmidt, L.; Nakaigawa, N.; Webb, C.P.; Weirich, G.; Kishida, T.; Zbar, B.; Woude, G.F. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 11445–11450. [Google Scholar] [CrossRef] [PubMed]
- Akpınar, T.S.; Hançer, V.S.; Nalçacı, M.; Diz-Küçükkaya, R. MPL W515L/K Mutations in Chronic Myeloproliferative Neoplasms. Turk. J. Haematol. 2013, 30, 8–12. [Google Scholar] [CrossRef]
- Liang, D.C.; Shih, L.Y.; Hung, I.J.; Yang, C.P.; Chen, S.H.; Jaing, T.H.; Liu, H.C.; Wang, L.Y.; Chang, W.H. FLT3-TKD mutation in childhood acute myeloid leukemia. Leukemia 2003, 17, 883–886. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Fletcher, C.D.; Rubin, B.P.; Ashman, L.K.; Corless, C.L.; Heinrich, M.C. KIT gene mutations in gastrointestinal stromal tumors: More complex than previously recognized? Am. J. Pathol. 2002, 161, 737. [Google Scholar] [CrossRef]
- Yui, S.; Kurosawa, S.; Yamaguchi, H.; Kanamori, H.; Ueki, T.; Uoshima, N.; Mizuno, I.; Shono, K.; Usuki, K.; Chiba, S.; et al. D816 mutation of the KIT gene in core binding factor acute myeloid leukemia is associated with poorer prognosis than other KIT gene mutations. Ann. Hematol. 2017, 96, 1641–1652. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Lamba, S.; Leenstra, S.; Troost, D.; Hulsebos, T.; Vandertop, W.P.; Frattini, M.; Molinari, F.; Knowles, M.; Cerrato, A.; et al. IDH1 mutations at residue p. R132 (IDH1R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum. Mutat. 2009, 30, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Boeva, V.; Battistella, A.; Goundiam, O.; Smith, N.K.; Mueller, C.R.; Raynal, V.; Mariani, O.; Sastre-Garau, X.; et al. Ovarian cancers harboring inactivating mutations in CDK12 display a distinct genomic instability pattern characterized by large tandem duplications. Cancer Res. 2016, 76, 1882–1891. [Google Scholar] [CrossRef]
- Luo, H.; Xu, X.; Ye, M.; Sheng, B.; Zhu, X. The prognostic value of HER2 in ovarian cancer: A meta-analysis of observational studies. PLoS ONE 2018, 13, e0191972. [Google Scholar] [CrossRef]
- Zhao, C.; Li, S.; Zhao, M.; Zhu, H.; Zhu, X. Prognostic values of DNA mismatch repair genes in ovarian cancer patients treated with platinum-based chemotherapy. Arch. Gynecol. Obstet. 2018, 297, 153–159. [Google Scholar] [CrossRef]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3′-kinase p85α gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001, 61, 7426–7429. [Google Scholar]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal patterns of selection in cancer and somatic tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Moreno-Acosta, P.; Gamboa, O.; de Gomez, M.S.; Cendales, R.; Diaz, G.D.; Romero, A.; Serra, J.B.; Conrado, Z.; Levy, A.; Chargari, C.; et al. IGF1R Gene Expression as a Predictive Marker of Response to Ionizing Radiation for Patients with Locally Advanced HPV16-positive Cervical Cancer. Anticancer Res. 2012, 32, 4319–4325. [Google Scholar]
- Chen, J.S.; Huang, J.Q.; Luo, B.; Dong, S.H.; Wang, R.C.; Jiang, Z.K.; Xie, Y.K.; Yi, W.; Wen, G.M.; Zhong, J.F. PIK 3 CD induces cell growth and invasion by activating AKT/GSK-3β/β-catenin signaling in colorectal cancer. Cancer Sci. 2019, 110, 997–1011. [Google Scholar] [CrossRef]
- Pasche, B.; Pennison, M.J.; Jimenez, H.; Wang, M. TGFBR1 and cancer susceptibility. Trans. Am. Clin. Climatol. Assoc. 2014, 125, 300. [Google Scholar]
- Wang, Y.; Qi, X.; Wang, F.; Jiang, J.; Guo, Q. Association between TGFBR1 polymorphisms and cancer risk: A meta-analysis of 35 case-control studies. PLoS ONE 2012, 7, e42899. [Google Scholar] [CrossRef]
- Leone, P.E.; Mendiola, M.; Alonso, J.; Paz-y-Miño, C.; Pestaña, A. Implications of a RAD54L polymorphism (2290C/T) in human meningiomas as a risk factor and/or a genetic marker. Bmc Cancer 2003, 3, 6. [Google Scholar] [CrossRef]
- Geng, W.; Dong, K.; Pu, Q.; Lv, Y.; Gao, H. SHOC2 is associated with the survival of breast cancer cells and has prognostic value for patients with breast cancer. Mol. Med. Rep. 2020, 21, 867–875. [Google Scholar] [CrossRef]
- Tu, Q.; Hao, J.; Zhou, X.; Yan, L.; Dai, H.; Sun, B.; Yang, D.; An, S.; Lv, L.; Jiao, B.; et al. CDKN2B deletion is essential for pancreatic cancer development instead of unmeaningful co-deletion due to juxtaposition to CDKN2A. Oncogene 2018, 37, 128–138. [Google Scholar] [CrossRef]
- Park, D.J.; Lesueur, F.; Nguyen-Dumont, T.; Pertesi, M.; Odefrey, F.; Hammet, F.; Neuhausen, S.L.; John, E.M.; Andrulis, I.L.; Terry, M.B.; et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am. J. Hum. Genet. 2012, 90, 734–739. [Google Scholar] [CrossRef]
- Chen, L.; Liu, P.; Evans, T.C.; Ettwiller, L.M. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science 2017, 355, 752. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546. [Google Scholar] [CrossRef]
- Cheng, D.T.; Prasad, M.; Chekaluk, Y.; Benayed, R.; Sadowska, J.; Zehir, A.; Syed, A.; Wang, Y.E.; Somar, J.; Li, Y.; et al. Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med. Genom. 2017, 10, 33. [Google Scholar] [CrossRef]
- AACR Project Genie Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- VanderPlas, J. Python Data Science Handbook: Essential Tools for Working with Data; O’Reilly Media, Inc.: Newton, MA, USA, 2016. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Window Size | Feature Type | Median JS Distance (Original) | Median JS Distance (Randomized) | p-Value |
|---|---|---|---|---|
| 1 | TF (k = 2) | 0.345 | 0.339 | Not significant |
| 2 | OHE | 0.275 | 0.228 | <0.05 |
| 3 | CV (k = 2) | 0.219 | 0.177 | <0.05 |
| 4 | TF (k = 3) | 0.214 | 0.167 | <0.05 |
| 5 | CV (k = 3) | 0.211 | 0.166 | <0.05 |
| 6 | TF (k = 4) | 0.210 | 0.166 | <0.05 |
| 7 | CV (k = 2) | 0.211 | 0.165 | <0.05 |
| 8 | TF (k = 3) | 0.211 | 0.164 | <0.05 |
| 9 | TF (k = 3) | 0.211 | 0.166 | <0.05 |
| 10 | TF (k = 4) | 0.211 | 0.165 | <0.05 |
| Algorithm | Accuracy | Sensitivity | Specificity | PPV | NPV | CS | MCC |
|---|---|---|---|---|---|---|---|
| MutationTaster | 0.8857 | 0.9081 | 0.75 | 0.9566 | 0.5738 | 3.1885 | 0.590 |
| FATHMM (Cancer) | 0.91 | 0.9788 | 0.4929 | 0.9213 | 0.7931 | 3.1861 | 0.580 |
| CHASMplus (Pancancer) | 0.85 | 0.852 | 0.85 | 0.972 | 0.486 | 3.16 | 0.570 |
| NBDriver | 0.891 | 0.931 | 0.643 | 0.941 | 0.608 | 3.123 | 0.561 |
| Neighborhood-only model | 0.85 | 0.629 | 0.907 | 0.9744 | 0.285 | 2.7954 | 0.370 |
| Condel | 0.8584 | 0.9258 | 0.45 | 0.9108 | 0.5 | 2.7866 | 0.392 |
| FATHMM (missense) | 0.8251 | 0.8775 | 0.5071 | 0.9152 | 0.4057 | 2.7055 | 0.351 |
| PROVEAN | 0.7371 | 0.7444 | 0.6929 | 0.9363 | 0.3089 | 2.6825 | 0.327 |
| SIFT | 0.8099 | 0.861 | 0.5 | 0.9126 | 0.3723 | 2.6459 | 0.32 |
| Polyphen-2 | 0.7978 | 0.8422 | 0.5286 | 0.9155 | 0.3558 | 2.6421 | 0.317 |
| Mutation Assessor | 0.747 | 0.7665 | 0.6286 | 0.9259 | 0.3077 | 2.6287 | 0.3 |
| VEST | 0.7503 | 0.8269 | 0.2857 | 0.8753 | 0.2139 | 2.2018 | 0.1 |
| CanDrAplus (Cancer-in-general) | 0.592 | 0.857 | 0 | 0.99 | 0 | 1.847 | −0.03 |
| Ensemble ID | Algorithm | Accuracy | Sensitivity | Specificity | PPV | NPV | CS | MCC |
|---|---|---|---|---|---|---|---|---|
| Ensemble 1 (With NBDriver) | NBDriver + FATHMM (cancer) + MutationTaster + Condel | 0.9505 | 0.9953 | 0.6785 | 0.9494 | 0.9596 | 3.583 | 0.782 |
| Ensemble 1 (Without NBDriver) | FATHMM (cancer) + MutationTaster + Condel | 0.917 | 0.9941 | 0.5357 | 0.9285 | 0.9375 | 3.395 | 0.677 |
| Ensemble 2 (With NBDriver) | NBDriver + CHASMplus + FATHMM (cancer) + MutationTaster | 0.948 | 0.991 | 0.664 | 0.947 | 0.92 | 3.522 | 0.754 |
| Ensemble 2 (Without NBDriver) | CHASMplus + FATHMM (cancer) + MutationTaster | 0.917 | 0.939 | 0.756 | 0.963 | 0.679 | 3.367 | 0.682 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, S.; Raman, K.; Ravindran, B. Sequence Neighborhoods Enable Reliable Prediction of Pathogenic Mutations in Cancer Genomes. Cancers 2021, 13, 2366. https://doi.org/10.3390/cancers13102366
Banerjee S, Raman K, Ravindran B. Sequence Neighborhoods Enable Reliable Prediction of Pathogenic Mutations in Cancer Genomes. Cancers. 2021; 13(10):2366. https://doi.org/10.3390/cancers13102366
Chicago/Turabian StyleBanerjee, Shayantan, Karthik Raman, and Balaraman Ravindran. 2021. "Sequence Neighborhoods Enable Reliable Prediction of Pathogenic Mutations in Cancer Genomes" Cancers 13, no. 10: 2366. https://doi.org/10.3390/cancers13102366
APA StyleBanerjee, S., Raman, K., & Ravindran, B. (2021). Sequence Neighborhoods Enable Reliable Prediction of Pathogenic Mutations in Cancer Genomes. Cancers, 13(10), 2366. https://doi.org/10.3390/cancers13102366