MicroRNA-Mediated Metabolic Shaping of the Tumor Microenvironment




Abstract
Simple Summary
Abstract
1. Introduction
2. Cancer Metabolism
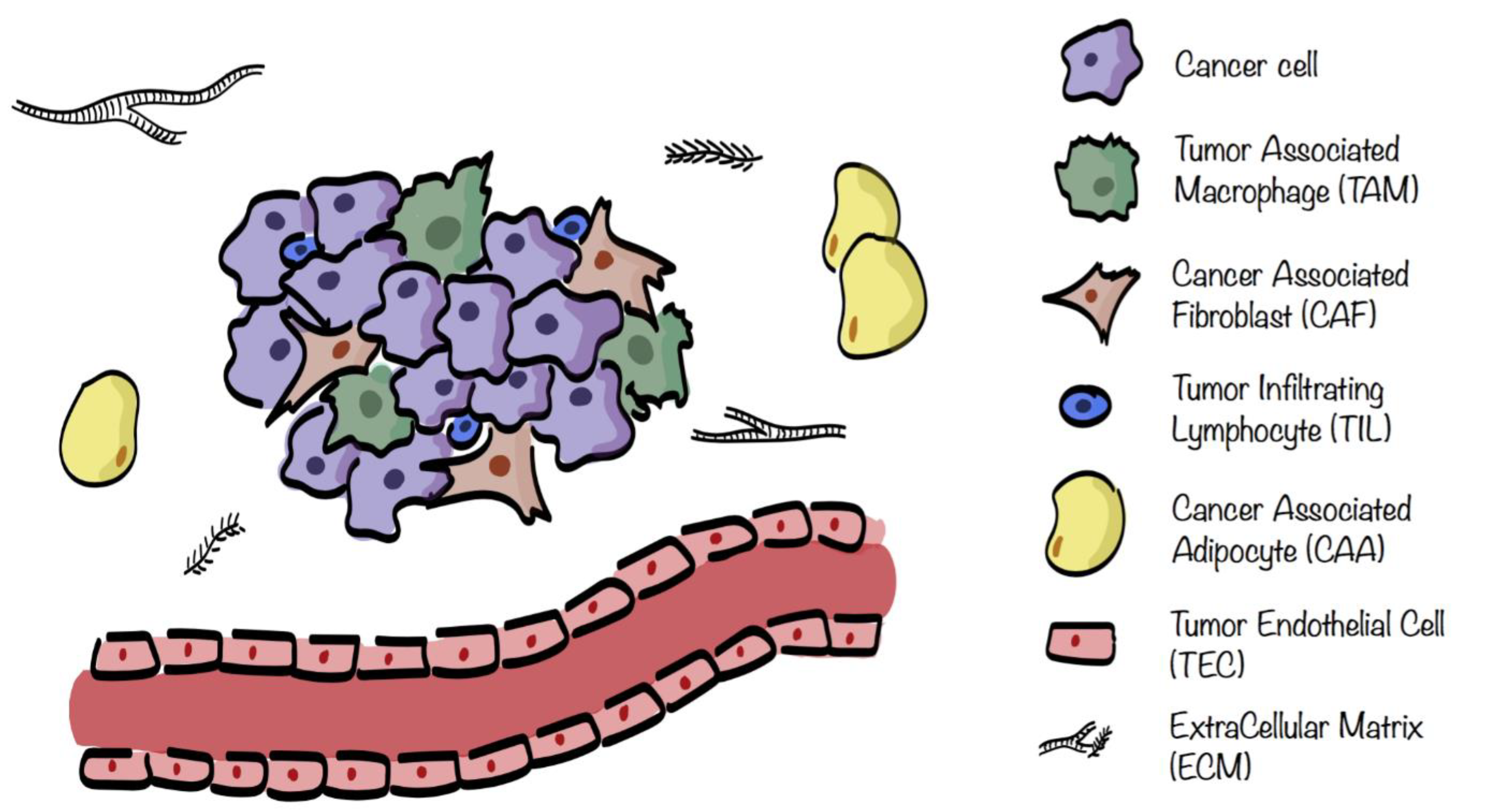
3. Tumor Microenvironment Metabolism
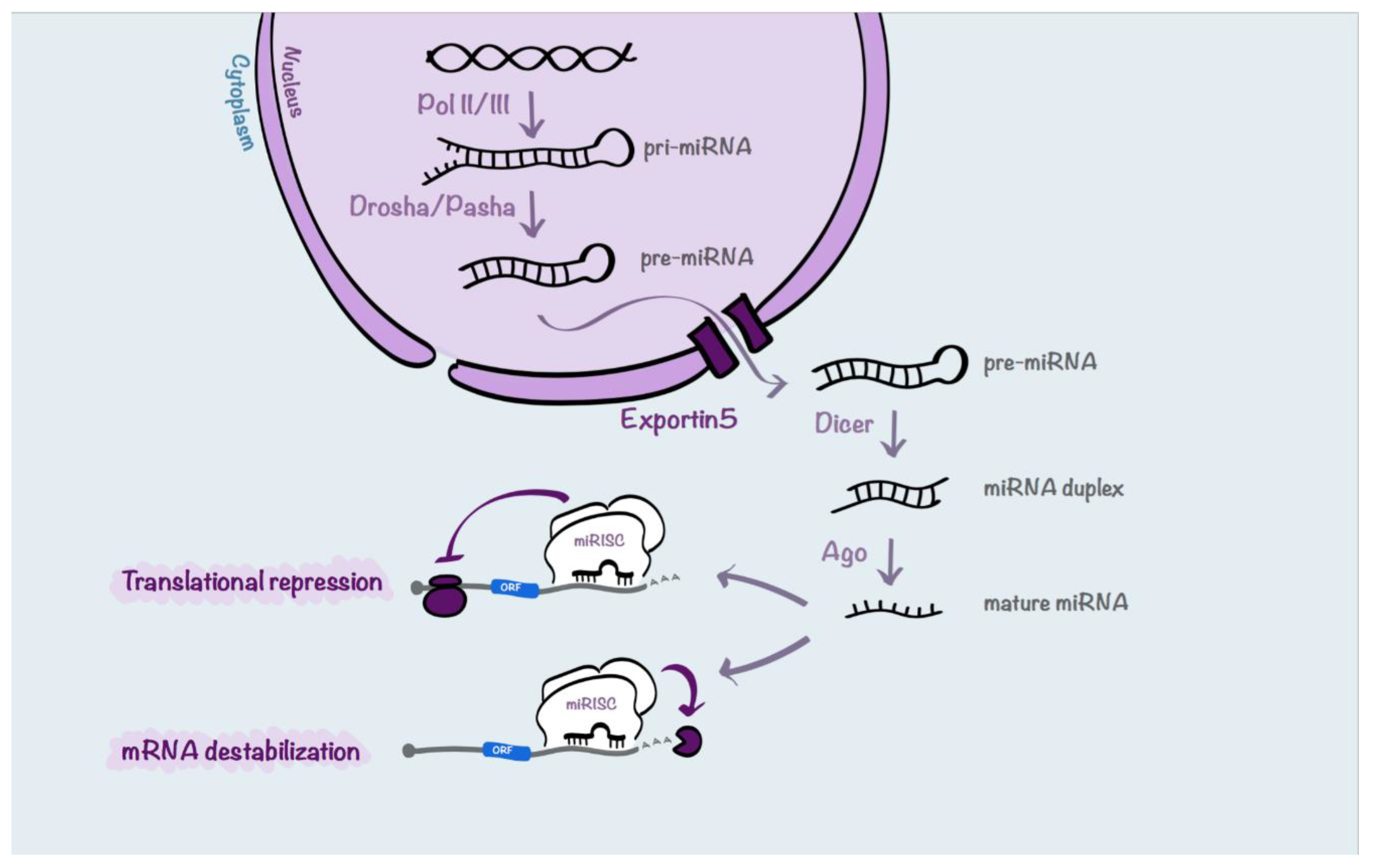
4. MicroRNAs (miRs)
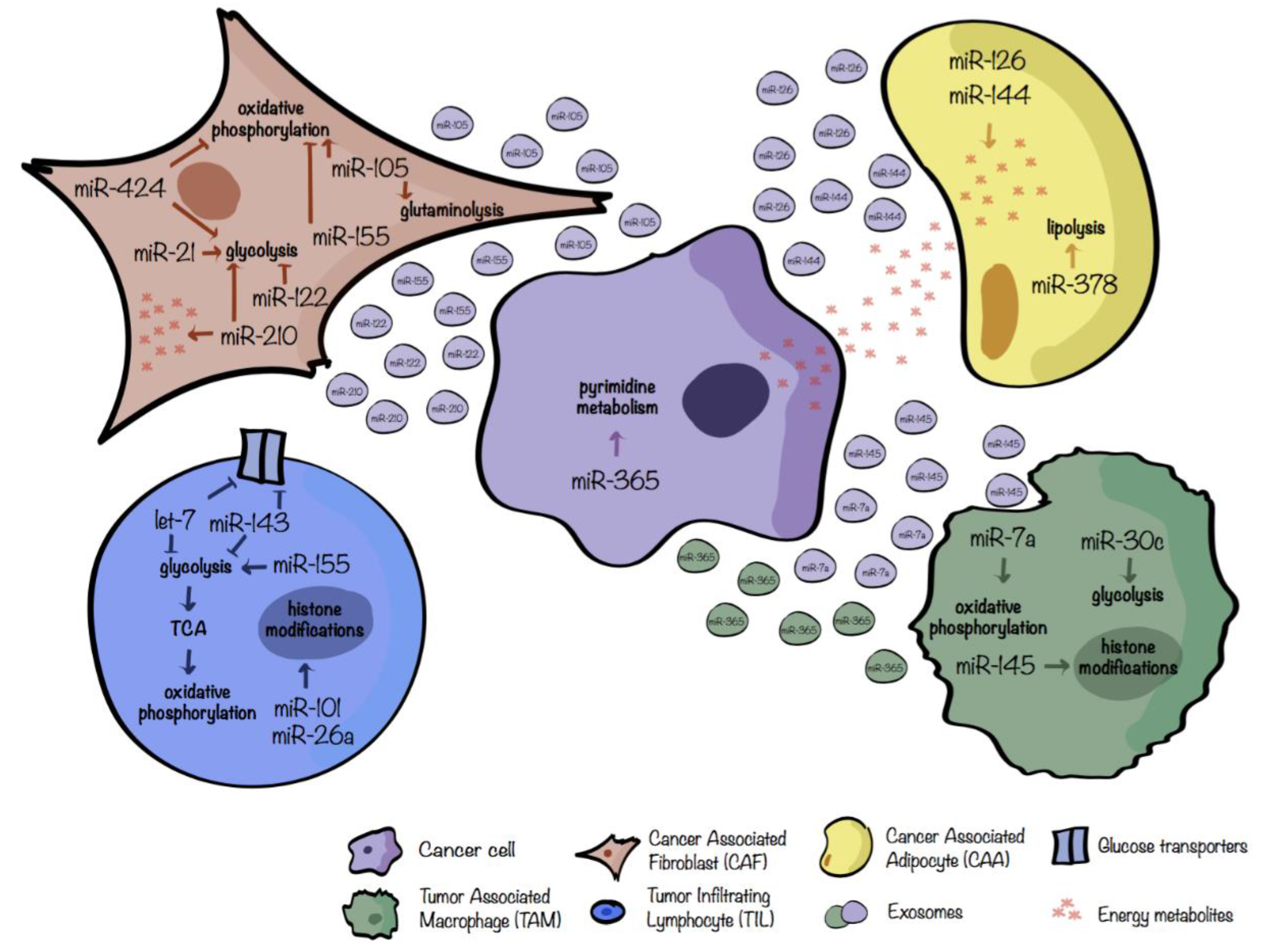
4.1. miRs in the Metabolic Crosstalk between Tumor Cells and CAFs
4.2. miRs in the Metabolic Crosstalk between Tumor Cells and CAAs
4.3. miRs in the Metabolic Crosstalk between Tumor Cells and TAMs
4.4. miRs in the Metabolic Crosstalk between Tumor Cells and TILs
5. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Riera-Domingo, C.; Audige, A.; Granja, S.; Cheng, W.C.; Ho, P.C.; Baltazar, F.; Stockmann, C.; Mazzone, M. Immunity, Hypoxia, and Metabolism-the Menage a Trois of Cancer: Implications for Immunotherapy. Physiol. Rev. 2020, 100, 1–102. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Yu, Y.R.; Ho, P.C. Sculpting tumor microenvironment with immune system: From immunometabolism to immunoediting. Clin. Exp. Immunol. 2019, 197, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.N.; Vidal, N.; Berthezene, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [PubMed]
- Virga, F.; Ehling, M.; Mazzone, M. Blood Vessel Proximity Shapes Cancer Cell Metabolism. Cell Metab. 2019, 30, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. 2019, 30, 201–211 e206. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Valerio, G.; Martinez-Lozano, M.; Bassani, N.; Vidal, A.; Ochoa-de-Olza, M.; Suarez, C.; Garcia-Del-Muro, X.; Carles, J.; Vinals, F.; Graupera, M.; et al. Resistance to Antiangiogenic Therapies by Metabolic Symbiosis in Renal Cell Carcinoma PDX Models and Patients. Cell Rep. 2016, 15, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Pisarsky, L.; Bill, R.; Fagiani, E.; Dimeloe, S.; Goosen, R.W.; Hagmann, J.; Hess, C.; Christofori, G. Targeting Metabolic Symbiosis to Overcome Resistance to Anti-angiogenic Therapy. Cell Rep. 2016, 15, 1161–1174. [Google Scholar] [CrossRef]
- Allen, E.; Mieville, P.; Warren, C.M.; Saghafinia, S.; Li, L.; Peng, M.W.; Hanahan, D. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef]
- Fei, X.; Qi, M.; Wu, B.; Song, Y.; Wang, Y.; Li, T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012, 586, 392–397. [Google Scholar] [CrossRef]
- Chan, B.; Manley, J.; Lee, J.; Singh, S.R. The emerging roles of microRNAs in cancer metabolism. Cancer Lett. 2015, 356, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Morandi, A.; Giannoni, E.; Chiarugi, P. Nutrient Exploitation within the Tumor-Stroma Metabolic Crosstalk. Trends Cancer 2016, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Santi, A.; Caselli, A.; Paoli, P.; Corti, D.; Camici, G.; Pieraccini, G.; Taddei, M.L.; Serni, S.; Chiarugi, P.; Cirri, P. The effects of CA IX catalysis products within tumor microenvironment. Cell Commun. Signal. 2013, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Giannoni, E.; Taddei, M.L.; Cirri, P.; Marini, A.; Pintus, G.; Nativi, C.; Richichi, B.; Scozzafava, A.; Carta, F.; et al. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle 2013, 12, 1791–1801. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Counillon, L.; Bouret, Y.; Marchiq, I.; Pouyssegur, J. Na(+)/H(+) antiporter (NHE1) and lactate/H(+) symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta 2016, 1863, 2465–2480. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Baltazar, F.; Pinheiro, C.; Morais-Santos, F.; Azevedo-Silva, J.; Queiros, O.; Preto, A.; Casal, M. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol. Histopathol. 2014, 29, 1511–1524. [Google Scholar] [CrossRef]
- Peppicelli, S.; Bianchini, F.; Calorini, L. Extracellular acidity, a “reappreciated” trait of tumor environment driving malignancy: Perspectives in diagnosis and therapy. Cancer Metastasis Rev. 2014, 33, 823–832. [Google Scholar] [CrossRef]
- Zhong, S.; Jeong, J.H.; Chen, Z.; Chen, Z.; Luo, J.L. Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl. Oncol. 2020, 13, 57–69. [Google Scholar] [CrossRef]
- Zhang, Y.; Dang, M.; Tian, Y.; Zhu, Y.; Liu, W.; Tian, W.; Su, Y.; Ni, Q.; Xu, C.; Lu, N.; et al. Tumor Acidic Microenvironment Targeted Drug Delivery Based on pHLIP-Modified Mesoporous Organosilica Nanoparticles. ACS Appl. Mater. Interfaces 2017, 9, 30543–30552. [Google Scholar] [CrossRef]
- Kanamala, M.; Wilson, W.R.; Yang, M.; Palmer, B.D.; Wu, Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials 2016, 85, 152–167. [Google Scholar] [CrossRef]
- Bosticardo, M.; Ariotti, S.; Losana, G.; Bernabei, P.; Forni, G.; Novelli, F. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur. J. Immunol. 2001, 31, 2829–2838. [Google Scholar] [CrossRef]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Araki, K.; Ahmed, R. Beyond adjuvants: Immunomodulation strategies to enhance T cell immunity. Vaccine 2015, 33 (Suppl. 2), B21–B28. [Google Scholar] [CrossRef]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM functions-it takes two to tango. FEBS J. 2018, 285, 700–716. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Calorini, L.; Chiarugi, P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid. Redox Signal. 2011, 14, 2361–2371. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.; et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell. Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Song, Z.; Wang, B.; Zhang, J.A. Emerging roles of microRNAs in the metabolic control of immune cells. Cancer Lett. 2018, 433, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, Y.; Yu, W.; Zheng, S.; Zhang, S.; Sun, L.; Ding, K. Small role with big impact: miRNAs as communicators in the cross-talk between cancer-associated fibroblasts and cancer cells. Int. J. Biol. Sci. 2017, 13, 339–348. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biol. Chem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Chiang, H.R.; Schoenfeld, L.W.; Ruby, J.G.; Auyeung, V.C.; Spies, N.; Baek, D.; Johnston, W.K.; Russ, C.; Luo, S.; Babiarz, J.E.; et al. Mammalian microRNAs: Experimental evaluation of novel and previously annotated genes. Genes Dev. 2010, 24, 992–1009. [Google Scholar] [CrossRef]
- Lim, L.P.; Glasner, M.E.; Yekta, S.; Burge, C.B.; Bartel, D.P. Vertebrate microRNA genes. Science 2003, 299, 1540. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grasser, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Orso, F.; Quirico, L.; Dettori, D.; Coppo, R.; Virga, F.; Ferreira, L.C.; Paoletti, C.; Baruffaldi, D.; Penna, E.; Taverna, D. Role of miRNAs in tumor and endothelial cell interactions during tumor progression. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Frediani, J.N.; Fabbri, M. Essential role of miRNAs in orchestrating the biology of the tumor microenvironment. Mol. Cancer 2016, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Kogure, A.; Kosaka, N.; Ochiya, T. Cross-talk between cancer cells and their neighbors via miRNA in extracellular vesicles: An emerging player in cancer metastasis. J. Biomed. Sci. 2019, 26, 7. [Google Scholar] [CrossRef]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Quirico, L.; Orso, F. The power of microRNAs as diagnostic and prognostic biomarkers in liquid biopsies. Cancer Drug Resist. China 2020, 3, 117–139. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Dettori, D.; Orso, F.; Penna, E.; Baruffaldi, D.; Brundu, S.; Maione, F.; Turco, E.; Giraudo, E.; Taverna, D. Therapeutic Silencing of miR-214 Inhibits Tumor Progression in Multiple Mouse Models. Mol. Ther. 2018, 26, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016, 380, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef]
- Shu, S.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef]
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R.; et al. Senescent stroma promotes prostate cancer progression: The role of miR-210. Mol. Oncol. 2014, 8, 1729–1746. [Google Scholar] [CrossRef]
- Chen, S.; Chen, X.; Shan, T.; Ma, J.; Lin, W.; Li, W.; Kang, Y. MiR-21-mediated Metabolic Alteration of Cancer-associated Fibroblasts and Its Effect on Pancreatic Cancer Cell Behavior. Int. J. Biol. Sci. 2018, 14, 100–110. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Attane, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Kulyte, A.; Lorente-Cebrian, S.; Gao, H.; Mejhert, N.; Agustsson, T.; Arner, P.; Ryden, M.; Dahlman, I. MicroRNA profiling links miR-378 to enhanced adipocyte lipolysis in human cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E267–E274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Zhihua, Y.; Yulin, T.; Yibo, W.; Wei, D.; Yin, C.; Jiahao, X.; Runqiu, J.; Xuezhong, X. Hypoxia decreases macrophage glycolysis and M1 percentage by targeting microRNA-30c and mTOR in human gastric cancer. Cancer Sci. 2019, 110, 2368–2377. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martin-Perez, R.; Serneels, J.; Prenen, H.; Ghesquiere, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; Ben David, G.; Shlomi, T.; Gil, Z. Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res. 2018, 78, 5287–5299. [Google Scholar] [CrossRef]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Kuranaga, Y.; Kumazaki, M.; Sugito, N.; Yoshikawa, Y.; Takai, T.; Taniguchi, K.; Ito, Y.; Akao, Y. Regulated Polarization of Tumor-Associated Macrophages by miR-145 via Colorectal Cancer-Derived Extracellular Vesicles. J. Immunol. 2017, 199, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.G.; Rabinovich, P.M. T Cell Reprogramming Against Cancer. Methods Mol. Biol. 2020, 2097, 3–44. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.C.; Daniels, K.A.; Angelou, C.C.; Fagerberg, E.; Burnside, A.S.; Markstein, M.; Alfandari, D.; Welsh, R.M.; Pobezinskaya, E.L.; Pobezinsky, L.A. Modulation of let-7 miRNAs controls the differentiation of effector CD8 T cells. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Gracias, D.T.; Stelekati, E.; Hope, J.L.; Boesteanu, A.C.; Doering, T.A.; Norton, J.; Mueller, Y.M.; Fraietta, J.A.; Wherry, E.J.; Turner, M.; et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat. Immunol. 2013, 14, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef]
- Monnot, G.C.; Martinez-Usatorre, A.; Lanitis, E.; Lopes, S.F.; Cheng, W.C.; Ho, P.C.; Irving, M.; Coukos, G.; Donda, A.; Romero, P. miR-155 Overexpression in OT-1 CD8(+) T Cells Improves Anti-Tumor Activity against Low-Affinity Tumor Antigen. Mol. Ther. Oncolytics 2020, 16, 111–123. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Li, F.; Ping, Y.; Qin, G.; Zhang, C.; Zhang, Y. miR-143 Regulates Memory T Cell Differentiation by Reprogramming T Cell Metabolism. J. Immunol. 2018, 201, 2165–2175. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P.; Vikas, P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience 2014, 8, 442. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Cufi, S.; Vazquez-Martin, A.; Torres-Garcia, V.Z.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: Induction of the tumor suppressor miRNA let-7a and suppression of the TGFbeta-induced oncomiR miRNA-181a. Cell Cycle 2011, 10, 1144–1151. [Google Scholar] [CrossRef]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev. Res. (Phila.) 2012, 5, 355–364. [Google Scholar] [CrossRef]
- Li, W.; Yuan, Y.; Huang, L.; Qiao, M.; Zhang, Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res. Clin. Pract. 2012, 96, 187–195. [Google Scholar] [CrossRef]
- Wang, F.; Xu, J.; Liu, H.; Liu, Z.; Xia, F. Metformin induces apoptosis by microRNA-26a-mediated downregulation of myeloid cell leukaemia-1 in human oral cancer cells. Mol. Med. Rep. 2016, 13, 4671–4676. [Google Scholar] [CrossRef]
- Yang, F.Q.; Wang, J.J.; Yan, J.S.; Huang, J.H.; Li, W.; Che, J.P.; Wang, G.C.; Liu, M.; Zheng, J.H. Metformin inhibits cell growth by upregulating microRNA-26a in renal cancer cells. Int. J. Clin. Exp. Med. 2014, 7, 3289–3296. [Google Scholar] [PubMed]
- Kang, M.; Lee, K.H.; Lee, H.S.; Jeong, C.W.; Ku, J.H.; Kim, H.H.; Kwak, C. Concurrent treatment with simvastatin and NF-kappaB inhibitor in human castration-resistant prostate cancer cells exerts synergistic anti-cancer effects via control of the NF-kappaB/LIN28/let-7 miRNA signaling pathway. PLoS ONE 2017, 12, e0184644. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miR | Position | TME Component | Cancer | Targets /Pathways | Metabolic Pathway | Ref |
|---|---|---|---|---|---|---|
| miR-424 | Xq26.3 | CAFs | melanoma | IDH3a | oxidative phosporylation | [73] |
| miR-122 | 18q21.31 | CAFs | breast | PKM2 | glycolysis | [74] |
| miR-105 | Xq28 | CAFs | breast | MYC | glutaminolysis/oxidative ohosphorylation | [75] |
| miR-155 | 21q21.3 | CAFs | melanoma | n/a | oxidative phosporylation | [76] |
| miR-210 | 11p15.5 | CAFs | melanoma | n/a | glycolysis | [76] |
| miR-210 | 11p15.5 | CAFs | prostate | n/a | energy rich compounds | [77] |
| miR-21 | 17q23.2 | CAFs | pancreatic | n/a | glycolysis | [78] |
| miR-144 | 17q11.2 | CAAs | breast | MAP3K/ERK1/2/PPARg | glycolysis | [79] |
| miR-126 | 9q34.3 | CAAs | breast | AMPK | glycolysis | [79] |
| miR-378 | 5q32 | CAAs | gastrointestinal cancer | n/a | lipoylisis | [80] |
| miR-30c | 6q13 | TAMs | gastric cancer | REDD1 | glycolysis | [81] |
| miR-365 | 16p13.12 | TAMs | pancreatic | CDA | pyrimidine metabolism (in cancer cells) | [82] |
| miR-7a | 9q21.32 | TAMs | melanoma | insulin-Akt-mTOR | oxidative phosporylation | [83] |
| miR-145 | 5q32 | TAMs | colorectal cancer | HDAC11 | histon acetylation | [84] |
| let-7 | 9q22.32 | TILs | mastocytoma | MYC EOMES | glycolysis | [85] |
| miR-155 | 21q21.3 | TILs | melanoma | INPP5D | glycolysis | [86] |
| miR-143 | 5q32 | TILs | esophagus | GLUT1 | glycolysis | [87] |
| miR-101 | 1p31.3 9p21.1 | TILs | ovarian | EZH2 | epigenetic modifications | [88] |
| miR-26a | 3p22.2 | TILs | ovarian | EZH2 | epigenetic modifications | [88] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virga, F.; Quirico, L.; Cucinelli, S.; Mazzone, M.; Taverna, D.; Orso, F. MicroRNA-Mediated Metabolic Shaping of the Tumor Microenvironment. Cancers 2021, 13, 127. https://doi.org/10.3390/cancers13010127
Virga F, Quirico L, Cucinelli S, Mazzone M, Taverna D, Orso F. MicroRNA-Mediated Metabolic Shaping of the Tumor Microenvironment. Cancers. 2021; 13(1):127. https://doi.org/10.3390/cancers13010127
Chicago/Turabian StyleVirga, Federico, Lorena Quirico, Stefania Cucinelli, Massimiliano Mazzone, Daniela Taverna, and Francesca Orso. 2021. "MicroRNA-Mediated Metabolic Shaping of the Tumor Microenvironment" Cancers 13, no. 1: 127. https://doi.org/10.3390/cancers13010127
APA StyleVirga, F., Quirico, L., Cucinelli, S., Mazzone, M., Taverna, D., & Orso, F. (2021). MicroRNA-Mediated Metabolic Shaping of the Tumor Microenvironment. Cancers, 13(1), 127. https://doi.org/10.3390/cancers13010127