Transcriptional Spatial Profiling of Cancer Tissues in the Era of Immunotherapy: The Potential and Promise

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. In Situ Hybridization
2.1. Fluorescence In Situ Hybridization
2.2. Single-Molecule FISH and RNAscope
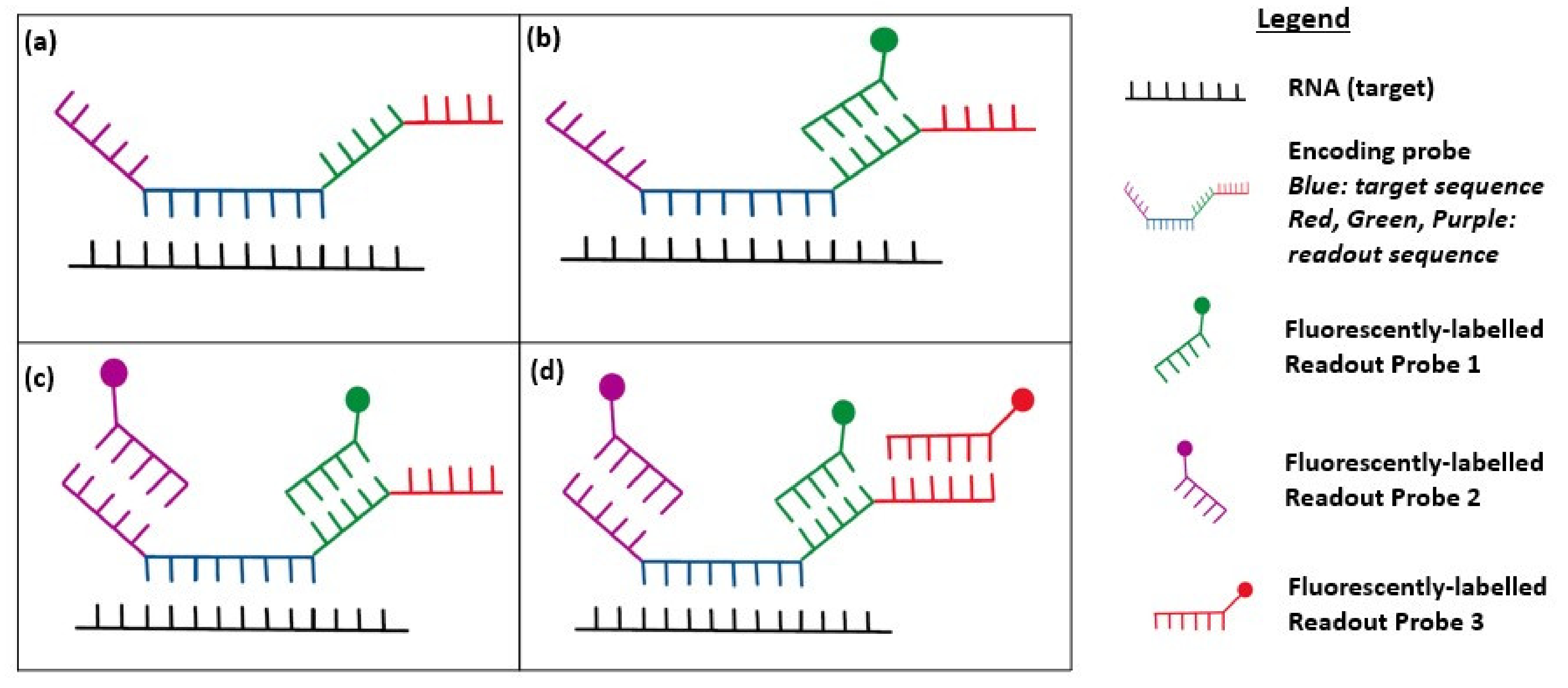
2.3. Multiplexed smFISH
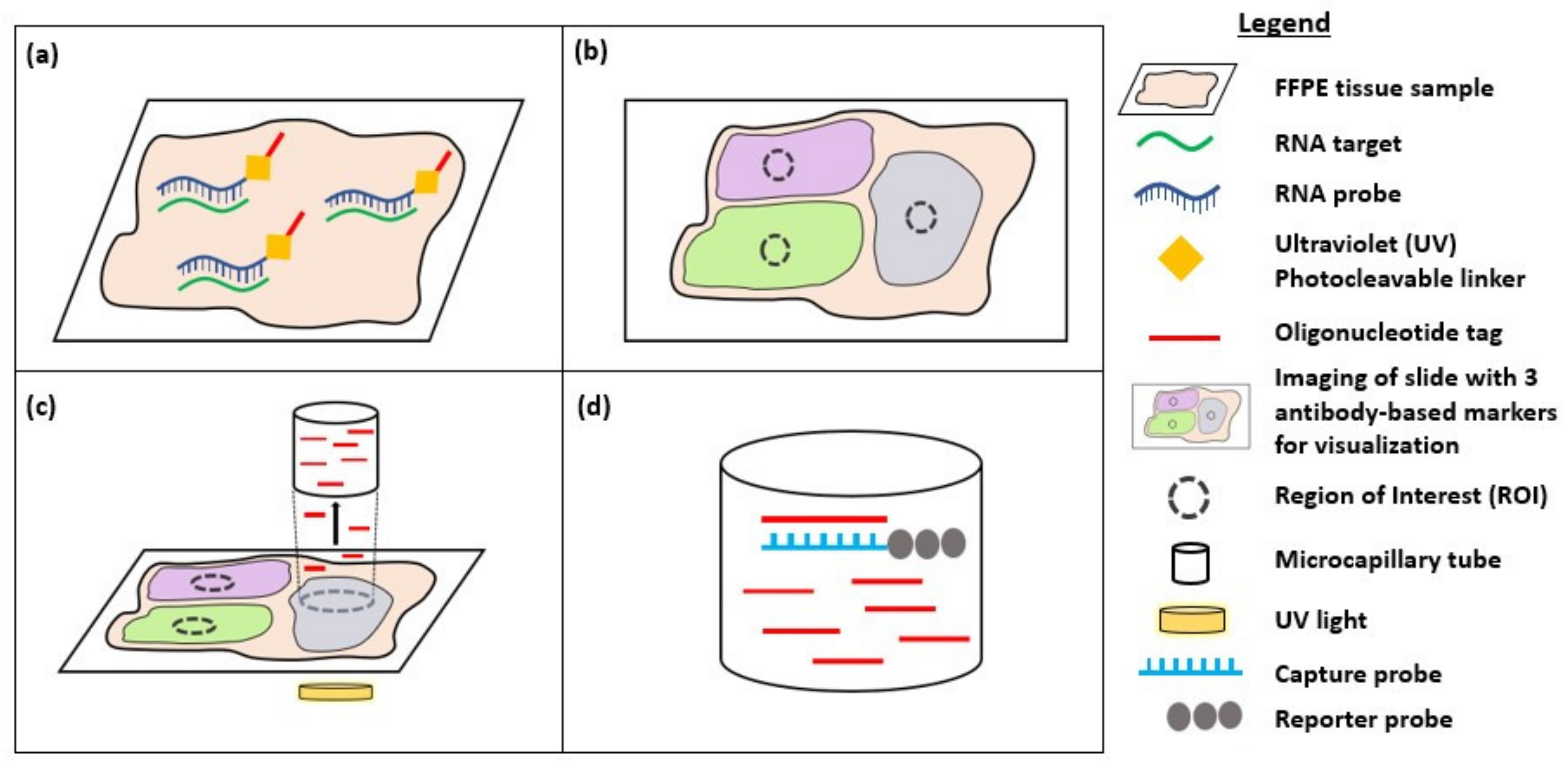
3. Digital Spatial Profiling
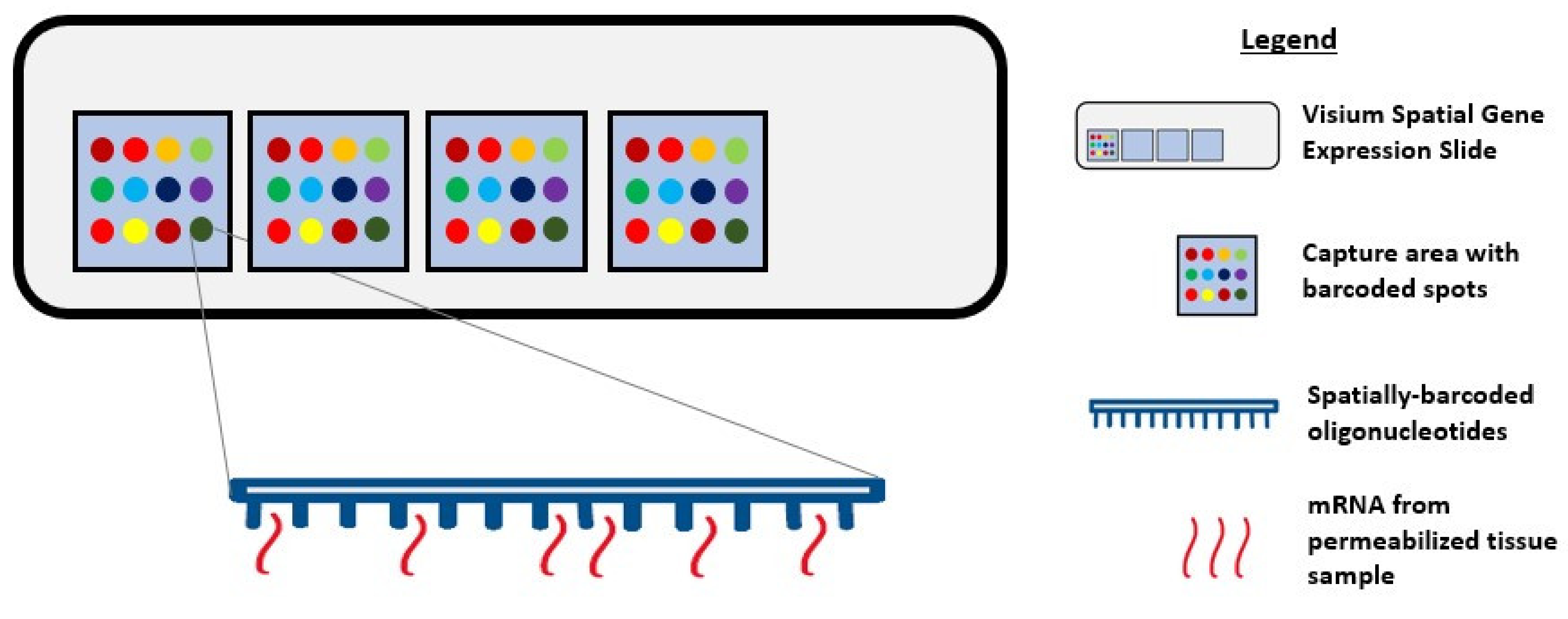
4. Spatial Transcriptomics
Visium Spatial Gene Expression Solution
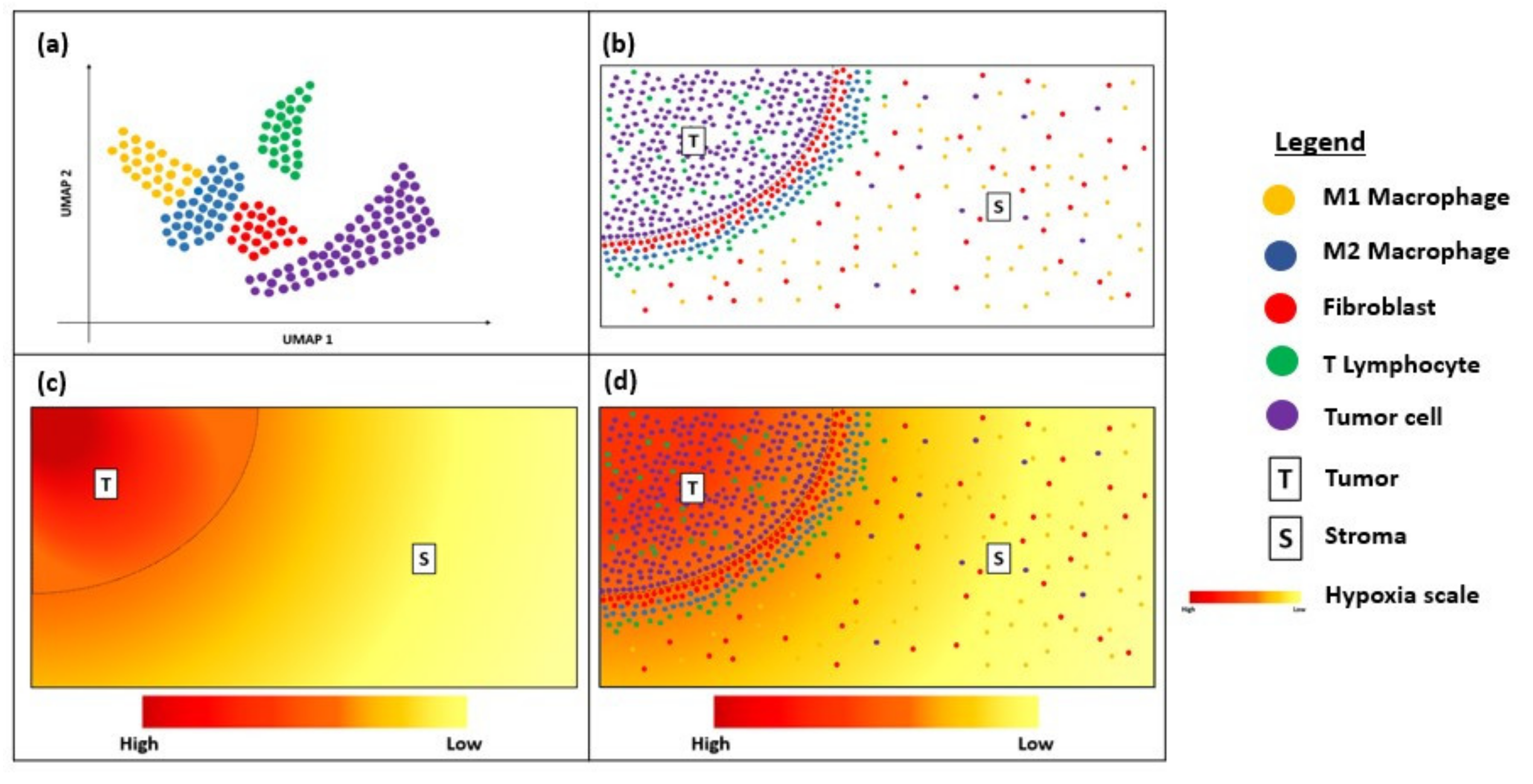
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Frankel, T.; Lanfranca, M.P.; Zou, W. The Role of Tumor Microenvironment in Cancer Immunotherapy. In Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy; Kalinski, P., Ed.; Springer International Publishing: Cham, Swizerland, 2017; pp. 51–64. ISBN 978-3-319-67577-0. [Google Scholar]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Lim, L.H.K.; Cheung, F.S.G. The role of the tumour microenvironment in immunotherapy. Endocr. Relat. Cancer 2017, 24, T283–T295. [Google Scholar] [CrossRef]
- Tang, H.; Qiao, J.; Fu, Y.-X. Immunotherapy and tumor microenvironment. Cancer Lett. 2016, 370, 85–90. [Google Scholar] [CrossRef]
- Chuah, S.; Chew, V. High-dimensional immune-profiling in cancer: Implications for immunotherapy. J. Immunother. Cancer 2020, 8, e000363. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Z.; Xu, X.; Yu, Z.; Mi, J. The influence of microenvironment on tumor immunotherapy. FEBS J. 2019, 286, 4160–4175. [Google Scholar] [CrossRef] [PubMed]
- Schwen, L.O.; Andersson, E.; Korski, K.; Weiss, N.; Haase, S.; Gaire, F.; Hahn, H.K.; Homeyer, A.; Grimm, O. Data-Driven Discovery of Immune Contexture Biomarkers. Front. Oncol. 2018, 8, 627. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L.; Murphy, A.G.; Weiss, M.J.; He, J.; Makary, M.A.; Burkhart, R.A.; Wolfgang, C.L.; Jaffee, E.M.; Zheng, L.; Thompson, E.D. Abstract 3102A: Analysis of spatial relationships between infiltrating immune cells within the tumor microenvironment following combinatorial immunotherapy. Cancer Res. 2019, 79, 3102A. [Google Scholar] [CrossRef]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Hofman, P.; Badoual, C.; Henderson, F.; Berland, L.; Hamila, M.; Long-Mira, E.; Lassalle, S.; Roussel, H.; Hofman, V.; Tartour, E.; et al. Multiplexed Immunohistochemistry for Molecular and Immune Profiling in Lung Cancer-Just About Ready for Prime-Time? Cancers 2019, 11, 283. [Google Scholar] [CrossRef]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1195–1204. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Eng. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Prat, A.; Navarro, A.; Paré, L.; Reguart, N.; Galván, P.; Pascual, T.; Martínez, A.; Nuciforo, P.; Comerma, L.; Alos, L.; et al. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res. 2017, 77, 3540–3550. [Google Scholar] [CrossRef]
- Merritt, C.; Ong, G.; Church, S.; Barker, K.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; Nguyen, K.; et al. High multiplex, digital spatial profiling of proteins and RNA in fixed tissue using genomic detection methods. bioRxiv 2019, 559021. [Google Scholar] [CrossRef]
- Wang, M.; Newsham, I.; Wu, Y.Q.; Dinh, H.; Kovar, C.; Santibanez, J.; Sabo, A.; Reid, J.; Bainbridge, M.; Boerwinkle, E.; et al. High-Throughput Next Generation Sequencing Methods and Applications. J Biomol. Tech. 2011, 22, S7. [Google Scholar]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Mignardi, M.; Hauling, T.; Nilsson, M. Fourth Generation of Next-Generation Sequencing Technologies: Promise and Consequences. Hum. Mutat. 2016, 37, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 2019, 20, 317. [Google Scholar] [CrossRef]
- Romanens, L.; Chaskar, P.; Tille, J.-C.; Ryser, S.; Liaudet, N.; Hu-Heimgartner, K.; Heimgartner, K.; Kaya, G.; Tsantoulis, P.; Labidi-Galy, I. Spatial transcriptomics of tumor microenvironment in formalin-fixed paraffin-embedded breast cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gall, J.G.; Pardue, M.L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. USA 1969, 63, 378–383. [Google Scholar] [CrossRef]
- John, H.A.; Birnstiel, M.L.; Jones, K.W. RNA-DNA hybrids at the cytological level. Nature 1969, 223, 582–587. [Google Scholar] [CrossRef]
- Buongiorno-Nardelli, M.; Amaldi, F. Autoradiographic detection of molecular hybrids between RNA and DNA in tissue sections. Nature 1970, 225, 946–948. [Google Scholar] [CrossRef]
- Dewald, G.W.; Schad, C.R.; Christensen, E.R.; Tiede, A.L.; Zinsmeister, A.R.; Spurbeck, J.L.; Thibodeau, S.N.; Jalal, S.M. The application of fluorescent in situ hybridization to detect Mbcr/abl fusion in variant Ph chromosomes in CML and ALL. Cancer Genet. Cytogenet. 1993, 71, 7–14. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, S.; Wang, L.; MacLennan, G.T.; Davidson, D.D. Fluorescence in situ hybridization in surgical pathology: Principles and applications. J. Pathol. Clin. Res. 2017, 3, 73–99. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.M.; Maher, V.E.; Bijwaard, K.E.; Becker, R.L.; Zhang, L.; Tang, S.W.; Song, P.; Liu, Q.; Marathe, A.; Gehrke, B.; et al. U.S. Food and Drug Administration approval: Crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin. Cancer. Res. 2014, 20, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Bozzetti, C.; Squadrilli, A.; Nizzoli, R.; Lagrasta, C.; Gasparro, D.; Majori, M.; Filippo, M.; Becchi, G.; Azzoni, C.; Campanini, N.; et al. Optimizing PD-L1 evaluation on cytological samples from advanced non-small-cell lung cancer. Immunotherapy 2020, 12, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Perottet, J.; Le Goff, E.; Legoupil, D.; Quere, G.; Schick, U.; Marcorelles, P.; Uguen, A. PD-L1 Copy Number Variation Does Not Correlate With PD-L1 Expression or Response to Anti-PD-1 Immunotherapy In Patients With Advanced Melanomas. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 161–165. [Google Scholar] [CrossRef]
- Gupta, S.; Cheville, J.C.; Jungbluth, A.A.; Zhang, Y.; Zhang, L.; Chen, Y.B.; Tickoo, S.K.; Fine, S.W.; Gopalan, A.; Al-Ahmadie, H.A.; et al. JAK2/PD-L1/PD-L2 (9p24.1) amplifications in renal cell carcinomas with sarcomatoid transformation: Implications for clinical management. Mod. Pathol. 2019, 32, 1344–1358. [Google Scholar] [CrossRef]
- Kamat, A.M.; Dickstein, R.J.; Messetti, F.; Anderson, R.; Pretzsch, S.M.; Gonzalez, G.N.; Katz, R.L.; Khanna, A.; Zaidi, T.; Wu, X.; et al. Use of fluorescence in situ hybridization to predict response to bacillus Calmette-Guerin therapy for bladder cancer: Results of a prospective trial. J. Urol. 2012, 187, 862–867. [Google Scholar] [CrossRef]
- Liem, E.; Baard, J.; Cauberg, E.C.C.; Bus, M.T.J.; de Bruin, D.M.; Laguna Pes, M.P.; de la Rosette, J.; de Reijke, T.M. Fluorescence in situ hybridization as prognostic predictor of tumor recurrence during treatment with Bacillus Calmette-Guerin therapy for intermediate- and high-risk non-muscle-invasive bladder cancer. Med. Oncol. 2017, 34, 172. [Google Scholar] [CrossRef]
- Lopez, M.E. Combined in situ Hybridization/Immunohistochemistry (ISH/Ich) on Free-floating Vibratome Tissue Sections. Bio. Protoc. 2014, 4. [Google Scholar] [CrossRef]
- Kwon, S. Single-molecule fluorescence in situ hybridization: Quantitative imaging of single RNA molecules. BMB Rep. 2013, 46, 65–72. [Google Scholar] [CrossRef]
- Wee, Y.T.F.; Alkaff, S.M.F.; Lim, J.C.T.; Loh, J.J.H.; Hilmy, M.H.; Ong, C.; Nei, W.L.; Jain, A.; Lim, A.; Takano, A.; et al. An integrated automated multispectral imaging technique that simultaneously detects and quantitates viral RNA and immune cell protein markers in fixed sections from Epstein-Barr virus-related tumours. Ann. Diagn. Pathol. 2018, 37, 12–19. [Google Scholar] [CrossRef]
- Schulz, D.; Zanotelli, V.R.T.; Fischer, J.R.; Schapiro, D.; Engler, S.; Lun, X.K.; Jackson, H.W.; Bodenmiller, B. Simultaneous Multiplexed Imaging of mRNA and Proteins with Subcellular Resolution in Breast Cancer Tissue Samples by Mass Cytometry. Cell Syst. 2018, 6, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Peskin, C.S.; Tranchina, D.; Vargas, D.Y.; Tyagi, S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006, 4, e309. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Lyubimova, A.; Itzkovitz, S.; Junker, J.P.; Fan, Z.P.; Wu, X.; van Oudenaarden, A. Single-molecule mRNA detection and counting in mammalian tissue. Nat. Protoc. 2013, 8, 1743–1758. [Google Scholar] [CrossRef]
- Annaratone, L.; Simonetti, M.; Wernersson, E.; Marchio, C.; Garnerone, S.; Scalzo, M.S.; Bienko, M.; Chiarle, R.; Sapino, A.; Crosetto, N. Quantification of HER2 and estrogen receptor heterogeneity in breast cancer by single-molecule RNA fluorescence in situ hybridization. Oncotarget 2017, 8, 18680–18698. [Google Scholar] [CrossRef]
- Wang, Z.; Portier, B.P.; Gruver, A.M.; Bui, S.; Wang, H.; Su, N.; Vo, H.T.; Ma, X.J.; Luo, Y.; Budd, G.T.; et al. Automated quantitative RNA in situ hybridization for resolution of equivocal and heterogeneous ERBB2 (HER2) status in invasive breast carcinoma. J. Mol. Diagn. 2013, 15, 210–219. [Google Scholar] [CrossRef]
- Wu, C.; Simonetti, M.; Rossell, C.; Mignardi, M.; Mirzazadeh, R.; Annaratone, L.; Marchio, C.; Sapino, A.; Bienko, M.; Crosetto, N.; et al. RollFISH achieves robust quantification of single-molecule RNA biomarkers in paraffin-embedded tumor tissue samples. Commun. Biol. 2018, 1, 209. [Google Scholar] [CrossRef]
- Erben, L.; Buonanno, A. Detection and Quantification of Multiple RNA Sequences Using Emerging Ultrasensitive Fluorescent In Situ Hybridization Techniques. Curr. Protoc. Neurosci. 2019, 87, e63. [Google Scholar] [CrossRef]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef]
- Chan, S.; Filezac de L’Etang, A.; Rangell, L.; Caplazi, P.; Lowe, J.B.; Romeo, V. A method for manual and automated multiplex RNAscope in situ hybridization and immunocytochemistry on cytospin samples. PLoS ONE 2018, 13, e0207619. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Zhang, B.; Miller, M.; Butko, E.; Wu, X.; Laver, T.; Kernag, C.; Kim, J.; Luo, Y.; Lamparski, H.; et al. Fully Automated RNAscope In Situ Hybridization Assays for Formalin-Fixed Paraffin-Embedded Cells and Tissues. J. Cell. Biochem. 2016, 117, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.; Bayraktar, O.A. Automation of Multiplexed RNAscope Single-Molecule Fluorescent In Situ Hybridization and Immunohistochemistry for Spatial Tissue Mapping. Methods Mol. Biol. 2020, 2148, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Bourne, T.D.; Wilson, J.D.; Saqqa, O.; Sharshir, M.A. Collapsing Glomerulopathy in a Patient With COVID-19. Kidney Int. Rep. 2020, 5, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Li, N.M.; Todorov, C.; Dikshit, A.; Ma, X.-J. Dissecting the cellular responses of immune cells and Lgr5+ stem cells in the inflamed mouse colon with the RNAscope in situ hybridization technology. J. Immunol. 2020, 204, 158.17. [Google Scholar]
- Pimentel, H.; Jarnagin, H.; Zong, H.; Todorov, C.; Anderson, C.M.; Zhang, B.; Bunker, C.; Ma, X.-J. Preclinical CAR-T cell target safety, biodistribution, and tumor infiltration analysis using in situ hybridization technology. J. Clin. Oncol. 2019, 37, 112. [Google Scholar] [CrossRef]
- Laeremans, A.; Li, N.; Rouault, M.; Luo, Y.; Ma, X.-J.; Park, E. Detection of RNA in the Central and Peripheral Nervous System Using the RNAscope® In Situ Hybridization Assay. Available online: https://cdn.technologynetworks.com/tn/Resources/pdf/detection-of-rna-in-the-central-and-peripheral-nervous-system-using-the-rnascope-in-situ.pdf (accessed on 4 July 2020).
- Shi, X.; Wu, S.; Sun, J.; Liu, Y.; Zeng, X.; Liang, Z. PD-L1 expression in lung adenosquamous carcinomas compared with the more common variants of non-small cell lung cancer. Sci. Rep. 2017, 7, 46209. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, J.; Zhu, Y.; Li, N.; Tian, T.; Li, Y.; Li, Y.; Li, Z.; Lai, Y.; Gao, J.; et al. Programmed death-ligand-1 expression in advanced gastric cancer detected with RNA in situ hybridization and its clinical significance. Oncotarget 2016, 7, 39671–39679. [Google Scholar] [CrossRef][Green Version]
- Guo, L.; Li, W.; Zhu, X.; Ling, Y.; Qiu, T.; Dong, L.; Fang, Y.; Yang, H.; Ying, J. PD-L1 expression and CD274 gene alteration in triple-negative breast cancer: Implication for prognostic biomarker. Springerplus 2016, 5, 805. [Google Scholar] [CrossRef]
- Yu, H.; Batenchuk, C.; Badzio, A.; Boyle, T.A.; Czapiewski, P.; Chan, D.C.; Lu, X.; Gao, D.; Ellison, K.; Kowalewski, A.A.; et al. PD-L1 Expression by Two Complementary Diagnostic Assays and mRNA In Situ Hybridization in Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 110–120. [Google Scholar] [CrossRef]
- Tretiakova, M.; Fulton, R.; Kocherginsky, M.; Long, T.; Ussakli, C.; Antic, T.; Gown, A. Concordance study of PD-L1 expression in primary and metastatic bladder carcinomas: Comparison of four commonly used antibodies and RNA expression. Mod. Pathol. 2018, 31, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Gafeer, M.M.; Hosny Mohammed, K.; Ormenisan-Gherasim, C.; Choudhary, F.; Siddiqui, M.T.; Cohen, C. Diagnostic Utility of PD-L1 Expression in Lung Adenocarcinoma: Immunohistochemistry and RNA In Situ Hybridization. Appl. Immunohistochem. Mol. Morphol. 2018, 26, e86–e90. [Google Scholar] [CrossRef] [PubMed]
- Franchini, D.M.; Lanvin, O.; Tosolini, M.; Patras de Campaigno, E.; Cammas, A.; Pericart, S.; Scarlata, C.M.; Lebras, M.; Rossi, C.; Ligat, L.; et al. Microtubule-Driven Stress Granule Dynamics Regulate Inhibitory Immune Checkpoint Expression in T Cells. Cell Rep. 2019, 26, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight 2018, 3, 1–24. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef]
- Boxberg, M.; Leising, L.; Steiger, K.; Jesinghaus, M.; Alkhamas, A.; Mielke, M.; Pfarr, N.; Gotz, C.; Wolff, K.D.; Weichert, W.; et al. Composition and Clinical Impact of the Immunologic Tumor Microenvironment in Oral Squamous Cell Carcinoma. J. Immunol. 2019, 202, 278–291. [Google Scholar] [CrossRef]
- Du, Z.; Abedalthagafi, M.; Aizer, A.A.; McHenry, A.R.; Sun, H.H.; Bray, M.A.; Viramontes, O.; Machaidze, R.; Brastianos, P.K.; Reardon, D.A.; et al. Increased expression of the immune modulatory molecule PD-L1 (CD274) in anaplastic meningioma. Oncotarget 2015, 6, 4704–4716. [Google Scholar] [CrossRef]
- Xiao, Q.; Wu, J.; Wang, W.J.; Chen, S.; Zheng, Y.; Yu, X.; Meeth, K.; Sahraei, M.; Bothwell, A.L.M.; Chen, L.; et al. DKK2 imparts tumor immunity evasion through beta-catenin-independent suppression of cytotoxic immune-cell activation. Nat. Med. 2018, 24, 262–270. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Perez-Gracia, J.L.; Schalper, K.A.; Fusco, J.P.; Gonzalez, A.; Rodriguez-Ruiz, M.E.; Onate, C.; Perez, G.; Alfaro, C.; Martin-Algarra, S.; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer. Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.X.; Singh, R.; Choi, E.E.; Ruella, M.; Nunez-Cruz, S.; Mansfield, K.G.; Bennett, P.; Barton, N.; Wu, Q.; Zhang, J.; et al. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget 2018, 9, 25764–25780. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Hao, J.; Wang, G.; Chen, K.H.; Babcock, H.P.; Zhuang, X. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. USA 2016, 113, 11046–11051. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Babcock, H.P.; Moffitt, J.R.; Zhuang, X. Multiplexed detection of RNA using MERFISH and branched DNA amplification. Sci. Rep. 2019, 9, 7721. [Google Scholar] [CrossRef]
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Shah, S.; Takei, Y.; Zhou, W.; Lubeck, E.; Yun, J.; Eng, C.L.; Koulena, N.; Cronin, C.; Karp, C.; Liaw, E.J.; et al. Dynamics and Spatial Genomics of the Nascent Transcriptome by Intron seqFISH. Cell 2018, 174, 363–376. [Google Scholar] [CrossRef]
- Zhou, W.; Yui, M.A.; Williams, B.A.; Yun, J.; Wold, B.J.; Cai, L.; Rothenberg, E.V. Single-Cell Analysis Reveals Regulatory Gene Expression Dynamics Leading to Lineage Commitment in Early T Cell Development. Cell Syst. 2019, 9, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Voith von Voithenberg, L.; Fomitcheva Khartchenko, A.; Huber, D.; Schraml, P.; Kaigala, G.V. Spatially multiplexed RNA in situ hybridization to reveal tumor heterogeneity. Nucleic Acids Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Zhuang, X. RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). Methods Enzymol. 2016, 572, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Hilscher, M.M.; Laxman, N.; Svedlund, J.; Wu, C.; Yokota, C.; Nilsson, M. Placing RNA in context and space – methods for spatially resolved transcriptomics. FEBS J. 2019, 286, 1468–1481. [Google Scholar] [CrossRef] [PubMed]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays 2020, E1900221. [Google Scholar] [CrossRef]
- Gupta, S.; Zugazagoitia, J.; Martinez-Morilla, S.; Fuhrman, K.; Rimm, D.L. Digital quantitative assessment of PD-L1 using digital spatial profiling. Lab. Investig. 2020. [Google Scholar] [CrossRef]
- Jeyasekharan, A.D.; Hoppe, M.M.; Fan, S.; VanSchoiack, A.; Xin, L.; De Mel, S.; Poon, M.; Chan, E.; Lee, J.; Chee, Y.L.; et al. Digital Spatial Profiling of Immune Markers in R-chop Treated Diffuse Large B-cell Lymphoma Reveals a Dominant Prognostic Significance of M2 Macrophage Infiltration. Hematol. Oncol. 2019, 37, 356–357. [Google Scholar] [CrossRef]
- McCart Reed, A.E.; Bennett, J.; Kutasovic, J.R.; Kalaw, E.; Ferguson, K.; Yeong, J.; Simpson, P.T.; Lakhani, S.R. Digital spatial profiling application in breast cancer: A user’s perspective. Virchows Arch. 2020. [Google Scholar] [CrossRef]
- Ilie, M.; Hofman, P. Reproducibility of PD-L1 assessment in non-small cell lung cancer-know your limits but never stop trying to exceed them. Transl. Lung Cancer Res. 2017, 6, S51–S54. [Google Scholar] [CrossRef]
- Koppel, C.; Schwellenbach, H.; Zielinski, D.; Eckstein, S.; Martin-Ortega, M.; D’Arrigo, C.; Schildhaus, H.-U.; Rüschoff, J.; Jasani, B. Optimization and validation of PD-L1 immunohistochemistry staining protocols using the antibody clone 28-8 on different staining platforms. Mod. Pathol. 2018, 31, 1630–1644. [Google Scholar] [CrossRef] [PubMed]
- Toki, M.I.; Merritt, C.R.; Wong, P.F.; Smithy, J.W.; Kluger, H.M.; Syrigos, K.N.; Ong, G.T.; Warren, S.E.; Beechem, J.M.; Rimm, D.L. High-Plex Predictive Marker Discovery for Melanoma Immunotherapy–Treated Patients Using Digital Spatial Profiling. Clin. Cancer Res. 2019, 25, 5503–5512. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Gupta, S.; Liu, Y.; Fuhrman, K.; Gettinger, S.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Biomarkers associated with beneficial PD-1 checkpoint blockade in non-small-cell lung cancer (NSCLC) identified using high-plex digital spatial profiling. Clin. Cancer Res. 2020, 26, 4360–4368. [Google Scholar] [CrossRef] [PubMed]
- Koldej, R.M.; Ritchie, D.S. High multiplex analysis of the immune microenvironment in bone marrow trephine samples using GeoMX™ digital spatial profiling. IOTECH 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Decalf, J.; Albert, M.L.; Ziai, J. New tools for pathology: A user’s review of a highly multiplexed method for in situ analysis of protein and RNA expression in tissue. J. Pathol. 2019, 247, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Wilbrey-Clark, A.; Roberts, K.; Teichmann, S.A. Cell Atlas technologies and insights into tissue architecture. Biochem. J. 2020, 477, 1427–1442. [Google Scholar] [CrossRef]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-omics: Novel approaches to probe cell heterogeneity and extracellular matrix biology. Matrix Biol. 2020, 91–92, 152–166. [Google Scholar] [CrossRef]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Ståhl, P.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- 10x Genomics Acquires Spatial Transcriptomics. Available online: https://www.10xgenomics.com/news/10x-genomics-acquires-spatial-transcriptomics/ (accessed on 16 April 2020).
- Rao, N.; Clark, S.; Habern, O. Bridging Genomics and Tissue Pathology. Genet. Eng. Biotechnol. News 2020, 40, 50–51. [Google Scholar] [CrossRef]
- Jemt, A.; Salmén, F.; Lundmark, A.; Mollbrink, A.; Fernández Navarro, J.; Ståhl, P.L.; Yucel-Lindberg, T.; Lundeberg, J. An automated approach to prepare tissue-derived spatially barcoded RNA-sequencing libraries. Sci. Rep. 2016, 6, 37137. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Sjöstrand, J.; Salmén, F.; Lundeberg, J.; Ståhl, P.L. ST Pipeline: An automated pipeline for spatial mapping of unique transcripts. Bioinformatics 2017, 33, 2591–2593. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res. 2018, 78, 5970. [Google Scholar] [CrossRef]
- Willis, E.F.; MacDonald, K.P.A.; Nguyen, Q.H.; Garrido, A.L.; Gillespie, E.R.; Harley, S.B.R.; Bartlett, P.F.; Schroder, W.A.; Yates, A.G.; Anthony, D.C.; et al. Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell 2020, 180, 833–846.e16. [Google Scholar] [CrossRef]
- 10x Genomics Begins Shipments of Visium Spatial Gene Expression Solution. Available online: https://investors.10xgenomics.com/news-releases/news-release-details/10x-genomics-begins-shipments-visium-spatial-gene-expression (accessed on 17 April 2020).
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Catallini, J.L.; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. bioRxiv 2020, 2020. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Han, A.P. Integrated Single-Cell RNA-Seq, Array Spatial Transcriptomics Analysis Puts Cells into Context. Available online: https://www.genomeweb.com/sequencing/integrated-single-cell-rna-seq-array-spatial-transcriptomics-analysis-puts-cells-context#.XphXjC17HUo (accessed on 22 April 2020).
- Visium Spatial Gene Expression Optimized Tissues. Available online: https://support.10xgenomics.com/spatial-gene-expression/tissue-optimization/doc/specifications-visium-spatial-gene-expression-optimized-tissues (accessed on 30 April 2020).
- Villacampa, E.G.; Larsson, L.; Kvastad, L.; Andersson, A.; Carlson, J.; Lundeberg, J. Genome-wide Spatial Expression Profiling in FFPE Tissues. bioRxiv 2020. [Google Scholar] [CrossRef]
- Visium Spatial Gene Expression. Available online: https://www.10xgenomics.com/products/spatial-gene-expression/ (accessed on 30 April 2020).
- Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.; Liu, S.D.; Zhao, S.; et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.A.; Ryall, K.A.; Wilm, C.; Caldara, J.; Grote, H.J.; Patterson-Kane, J.C. PD-L1 immunostaining scoring for non-small cell lung cancer based on immunosurveillance parameters. PLoS ONE 2018, 13, e0196464. [Google Scholar] [CrossRef]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef]
- Ascierto, M.L.; McMiller, T.L.; Berger, A.E.; Danilova, L.; Anders, R.A.; Netto, G.J.; Xu, H.; Pritchard, T.S.; Fan, J.; Cheadle, C.; et al. The Intratumoral Balance between Metabolic and Immunologic Gene Expression Is Associated with Anti-PD-1 Response in Patients with Renal Cell Carcinoma. Cancer Immunol. Res. 2016, 4, 726–733. [Google Scholar] [CrossRef]
- Alfaro, J.A.; Sinha, A.; Kislinger, T.; Boutros, P.C. Onco-proteogenomics: Cancer proteomics joins forces with genomics. Nat. Methods 2014, 11, 1107–1113. [Google Scholar] [CrossRef]
- Cieślik, M.; Chinnaiyan, A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2017, 19, 93–109. [Google Scholar] [CrossRef]
- De Vries, N.L.; Mahfouz, A.; Koning, F.; de Miranda, N.F.C.C. Unraveling the Complexity of the Cancer Microenvironment With Multidimensional Genomic and Cytometric Technologies. Front. Oncol. 2020, 10, 1254. [Google Scholar] [CrossRef] [PubMed]
- Feder, M.; Walser, J.-C. The biological limitations of transcriptomics in elucidating stress and stress responses. J. Evol. Boil. 2005, 18, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Jehan, Z.; Uddin, S.; Al-Kuraya, K.S. In-situ hybridization as a molecular tool in cancer diagnosis and treatment. Curr. Med. Chem. 2012, 19, 3730–3738. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Neumeister, V.; McGuire, J.; Song, Y.S.; Acs, B.; Ho, K.; Weidler, J.; Wong, W.; Rhees, B.; Bates, M.; et al. Quantitative assessments and clinical outcomes in HER2 equivocal 2018 ASCO/CAP ISH group 4 breast cancer. NPJ Breast Cancer 2019, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.; Tefferi, A. Fluorescent in situ hybridization in the diagnosis, prognosis, and treatment monitoring of chronic myeloid leukemia. Leuk. Lymphoma 2006, 47, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Morilla, S.; McGuire, J.; Gaule, P.; Moore, L.; Acs, B.; Cougot, D.; Gown, A.M.; Yaziji, H.; Wang, W.L.; Cartun, R.W.; et al. Quantitative assessment of PD-L1 as an analyte in immunohistochemistry diagnostic assays using a standardized cell line tissue microarray. Lab. Investig. 2020, 100, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mashock, M.; Tong, Z.; Mu, X.; Chen, H.; Zhou, X.; Zhang, H.; Zhao, G.; Liu, B.; Li, X. Changing Technologies of RNA Sequencing and Their Applications in Clinical Oncology. Front. Oncol. 2020, 10, 447. [Google Scholar] [CrossRef]
- Visium Spatial Gene Expression with Immunofluorescence. Available online: https://www.10xgenomics.com/products/spatial-proteomics/ (accessed on 25 June 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Imaging Modalities | |||
|---|---|---|---|---|
| RNAscope | MERFISH | DSP | Visium | |
| Vendor | Advanced Cell Diagnostics | Vizgen | NanoString | 10× Genomics |
| Year of Launch | 2012 | 2015–2016 | 2018–2019 | 2019 |
| Target molecules | RNA | DNA or RNA | RNA or protein | mRNA |
| Methodology | Hybridization of branched DNA probes followed by signal amplification | Hybridization of fluorochrome-labelled barcoded DNA probes followed by signal amplification | Oligonucleotide-tagged RNA or antibody probes followed by photocleavage and sequencing or hybridization | On-slide cDNA barcoding followed by sequencing |
| FFPE validation | √ | × | √ | × |
| Maximum multiplexed capacity | 12 RNA species | 10,000+ mRNA species | 96 proteins and 1400+ RNA | 100,000+ unique molecule identifiers |
| Turnaround time | 30 slides/11–14 h | 1 slide/2–3 days | 10–20 slides/48 h | 4 capture areas/slide/day |
| Whole slide imaging | √ | × | Possible but very costly and time consuming | √ |
| Resolution | <1 μm | <1 μm | 10 μm | 55 μm |
| Key equipment required | Standard bright-field or fluorescence microscope | Microscope integrated with an automatic fluid handling system | Pressure cooker (for manual slide prep) or automated stainer (for Leica slide prep), GeoMx Digital Spatial Profiler | Cryostat, microscope, sequencer |
| Analytic software | HALO® | 3D-daoSTORM or MERlin (for decoding) | Bundled software | Bundled software |
| Commercialized | √ | In progress | √ | √ |
| Cost | $$ | $ | $$$ | $$ |
| Publications | 2000+ | 10+ | 15+ | 4 |
| Advantages |
|
|
|
|
| Disadvantages |
|
|
|
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nerurkar, S.N.; Goh, D.; Cheung, C.C.L.; Nga, P.Q.Y.; Lim, J.C.T.; Yeong, J.P.S. Transcriptional Spatial Profiling of Cancer Tissues in the Era of Immunotherapy: The Potential and Promise. Cancers 2020, 12, 2572. https://doi.org/10.3390/cancers12092572
Nerurkar SN, Goh D, Cheung CCL, Nga PQY, Lim JCT, Yeong JPS. Transcriptional Spatial Profiling of Cancer Tissues in the Era of Immunotherapy: The Potential and Promise. Cancers. 2020; 12(9):2572. https://doi.org/10.3390/cancers12092572
Chicago/Turabian StyleNerurkar, Sanjna Nilesh, Denise Goh, Chun Chau Lawrence Cheung, Pei Qi Yvonne Nga, Jeffrey Chun Tatt Lim, and Joe Poh Sheng Yeong. 2020. "Transcriptional Spatial Profiling of Cancer Tissues in the Era of Immunotherapy: The Potential and Promise" Cancers 12, no. 9: 2572. https://doi.org/10.3390/cancers12092572
APA StyleNerurkar, S. N., Goh, D., Cheung, C. C. L., Nga, P. Q. Y., Lim, J. C. T., & Yeong, J. P. S. (2020). Transcriptional Spatial Profiling of Cancer Tissues in the Era of Immunotherapy: The Potential and Promise. Cancers, 12(9), 2572. https://doi.org/10.3390/cancers12092572