The Inducible Role of Ambient Particulate Matter in Cancer Progression via Oxidative Stress-Mediated Reactive Oxygen Species Pathways: A Recent Perception

Simple Summary
Abstract
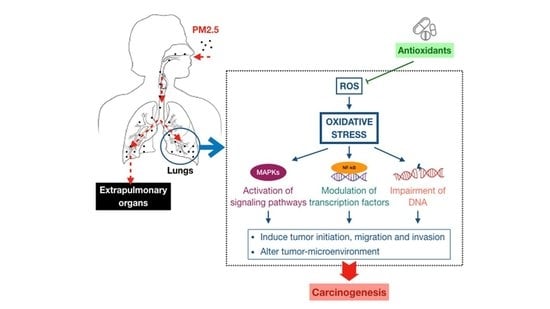
1. Introduction
2. Associations between Fine Particulate Matter and Cancer Progression
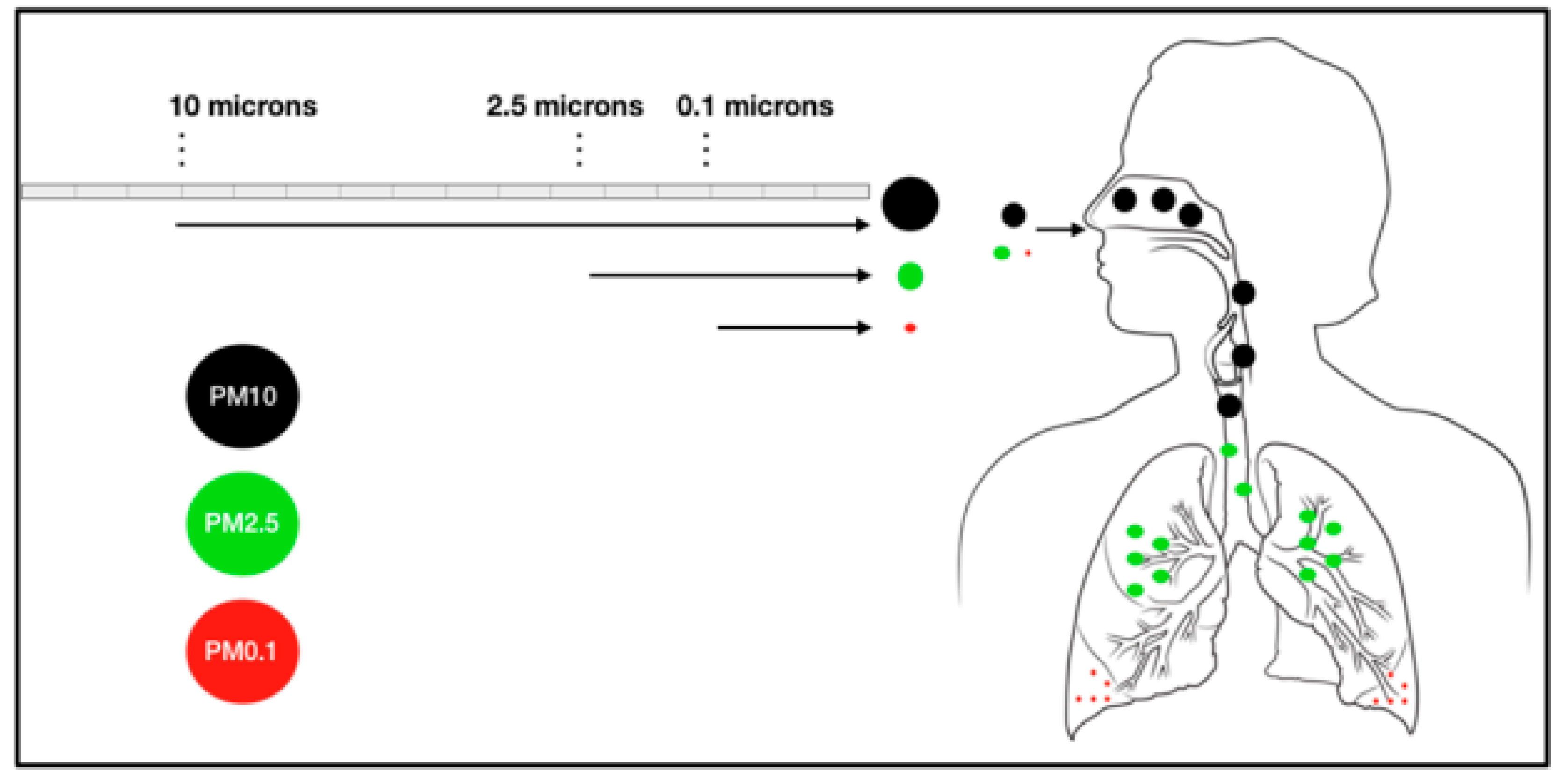
2.1. Epidemiological Evidences of PM2.5-Associated Lung Cancer
2.2. Experimental Evidences of PM2.5-Associated Lung Cancer
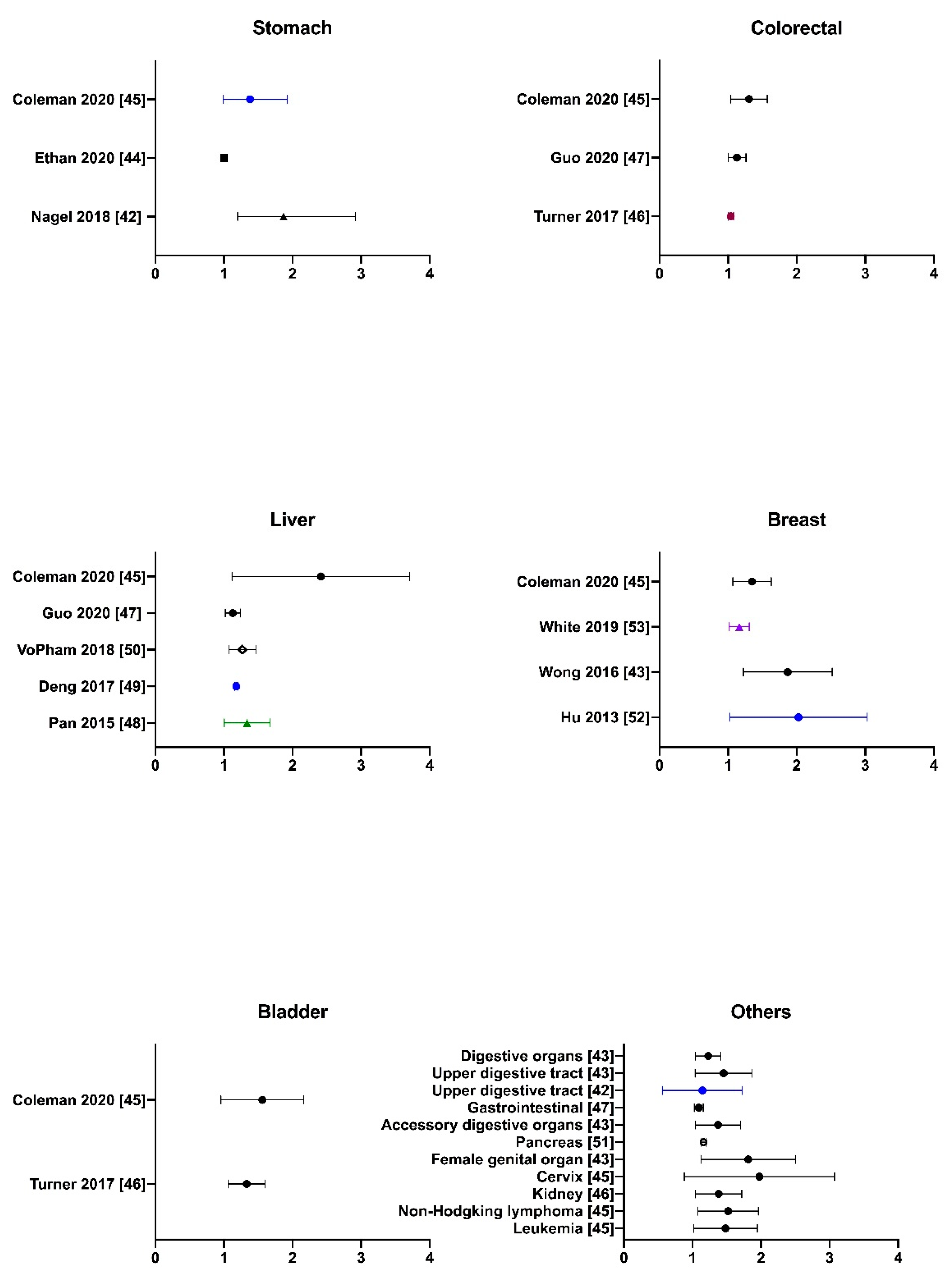
2.3. Literature Evidence of PM2.5-Associated Non-Lung Cancer
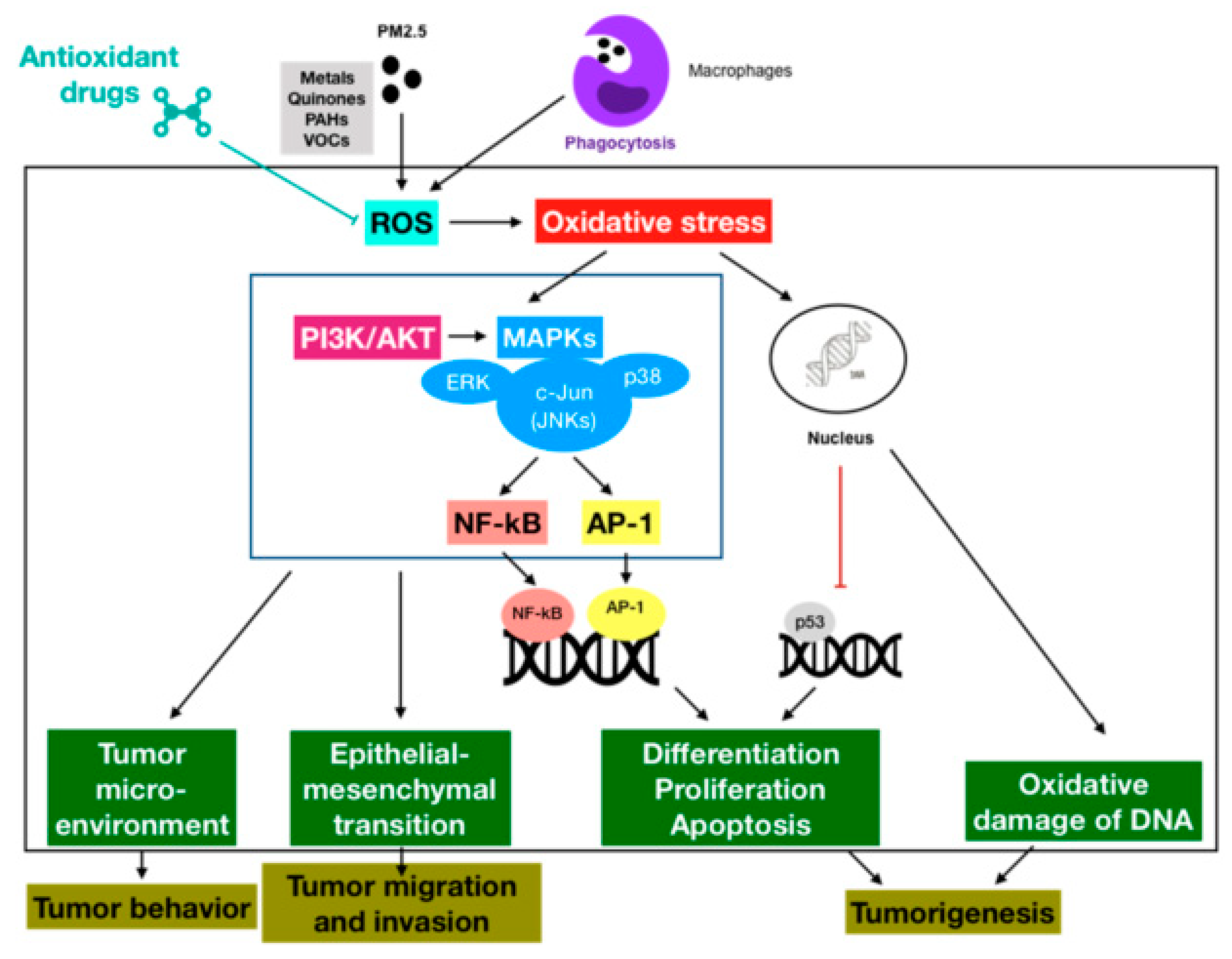
3. Association between Fine Particulate Matter Induced-Oxidative Stress Mechanisms and Lung Cancer Process
3.1. Oxidative Stress-Activated Signaling Pathways
3.2. Oxidative Stress-Modulated Transcription Factors
3.3. Oxidative Stress-Impaired DNA
4. Antioxidant Therapeutics against PM2.5-Induced Lung Cancer
5. Conclusions
Funding
Conflicts of Interest
References
- International Agency for Research on Cancer. Outdoor Air Pollution a Leading Environmental Cause of Cancer Deaths. Available online: https://www.iarc.fr/wp-content/uploads/2018/07/pr221_E.pdf (accessed on 3 June 2020).
- World Health Organization. Ambient (Outdoor) Air Pollution. Available online: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health (accessed on 4 June 2020).
- Nääv, Å.; Erlandsson, L.; Isaxon, C.; Åsander Frostner, E.; Ehinger, J.; Sporre, M.K.; Krais, A.M.; Strandberg, B.; Lundh, T.; Elmér, E.; et al. Urban PM2.5 induces cellular toxicity, hormone dysregulation, oxidative damage, inflammation, and mitochondrial interference in the HRT8 trophoblast cell line. Front. Endocrinol. (Lausanne) 2020, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Wang, X.; Zhang, N.; Zheng, K.; Du, K.; Zheng, M.; Li, Y.; Chang, Y.; Zhang, L.; Bachert, C. The effect of fine particulate matter on the inflammatory responses in human upper airway mucosa. Am. J. Respir. Crit. Care Med. 2019, 200, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- DeMarini, D.M.; Warren, S.H.; Lavrich, K.; Flen, A.; Aurell, J.; Mitchell, W.; Greenwell, D.; Preston, W.; Schmid, J.E.; Linak, W.P.; et al. Mutagenicity and oxidative damage induced by an organic extract of the particulate emissions from a simulation of the deepwater horizon surface oil burns. Environ. Mol. Mutagenesis 2017, 58, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Vattanasit, U.; Navasumrit, P.; Khadka, M.B.; Kanitwithayanun, J.; Promvijit, J.; Autrup, H.; Ruchirawat, M. Oxidative DNA damage and inflammatory responses in cultured human cells and in humans exposed to traffic-related particles. Int. J. Hyg. Environ. Health 2014, 217, 23–33. [Google Scholar] [CrossRef]
- Dergham, M.; Lepers, C.; Verdin, A.; Cazier, F.; Billet, S.; Courcot, D.; Shirali, P.; Garçon, G. Temporal-spatial variations of the physicochemical characteristics of air pollution particulate matter (PM2.5-0.3) and toxicological effects in human bronchial epithelial cells (BEAS-2B). Environ. Res. 2015, 137, 256–267. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Harari, S.; Martinelli, I.; Franchini, M. Effects on health of air pollution: A narrative review. Intern. Emerg. Med. 2015, 10, 657–662. [Google Scholar] [CrossRef]
- Mo, J.H. Association of particulate matter with ENT diseases. Clin. Exp. Otorhinolaryngol. 2019, 12, 237–238. [Google Scholar] [CrossRef]
- Schraufnagel, D.E. The health effects of ultrafine particles. Exp. Mol. Med. 2020, 52, 311–317. [Google Scholar] [CrossRef]
- Fantke, P.; Jolliet, O.; Evans, J.S.; Apte, J.S.; Cohen, A.J.; Hänninen, O.O.; Hurley, F.; Jantunen, M.J.; Jerrett, M.; Levy, J.I.; et al. Health effects of fine particulate matter in life cycle impact assessment: Findings from the basel guidance workshop. Int. J. Life Cycle Assess. 2015, 20, 276–288. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, Y.; Hou, T.; Liao, J.; Zhang, C.; Sun, C.; Wang, G. PM2.5 induces EMT and promotes CSC properties by activating Notch pathway in vivo and vitro. Ecotoxicol. Environ. Saf. 2019, 178, 159–167. [Google Scholar] [CrossRef]
- Wang, N.; Mengersen, K.; Tong, S.; Kimlin, M.; Zhou, M.; Wang, L.; Yin, P.; Xu, Z.; Cheng, J.; Zhang, Y.; et al. Short-term association between ambient air pollution and lung cancer mortality. Environ. Res. 2019, 179, 108748. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Shim, J.Y.; Park, B.; Lee, Y.J. Long-term exposure to air pollution and the risk of non-lung cancer: A meta-analysis of observational studies. Perspect. Public Health 2019, 140, 222–231. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018. Available online: https://www.who.int/cancer/PRGlobocanFinal.pdf?ua=1 (accessed on 4 June 2020).
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Dubey, A.; Saini, D.; Singh, M.; Prasad, C.P.; Roy, S.; Bharati, S.J.; Rinki, M.; Singh, N.; Seth, T.; et al. Environmental and occupational determinants of lung cancer. Transl. Lung Cancer Res. 2019, 8, S31–S49. [Google Scholar] [CrossRef]
- National Research Council. Analysis of Cancer Risks in Populations Near Nuclear Facilities: Phase 1; The National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- National Cancer Institute. SEER Cancer Stat Facts: Lung and Bronchus Cancer. Available online: http://seer.cancer.gov/statfacts/html/lungb.html (accessed on 5 June 2020).
- Hamra, G.B.; Guha, N.; Cohen, A.; Laden, F.; Raaschou-Nielsen, O.; Samet, J.M.; Vineis, P.; Forastiere, F.; Saldiva, P.; Yorifuji, T.; et al. Outdoor particulate matter exposure and lung cancer: A systematic review and meta-analysis. Environ. Health Perspect. 2014, 122, 906–911. [Google Scholar] [CrossRef]
- Cui, P.; Huang, Y.; Han, J.; Song, F.; Chen, K. Ambient particulate matter and lung cancer incidence and mortality: A meta-analysis of prospective studies. Eur. J. Public Health 2015, 25, 324–329. [Google Scholar] [CrossRef]
- Kim, H.B.; Shim, J.Y.; Park, B.; Lee, Y.J. Long-term exposure to air pollutants and cancer mortality: A meta-analysis of cohort studies. Int. J. Environ. Res. Public Health 2018, 15, 2608. [Google Scholar] [CrossRef]
- Hystad, P.; Demers, P.A.; Johnson, K.C.; Carpiano, R.M.; Brauer, M. Long-term residential exposure to air pollution and lung cancer risk. Epidemiology 2013, 24, 762–772. [Google Scholar] [CrossRef]
- Puett, R.C.; Hart, J.E.; Yanosky, J.D.; Spiegelman, D.; Wang, M.; Fisher, J.A.; Hong, B.; Laden, F. Particulate matter air pollution exposure, distance to road, and incident lung cancer in the nurses’ health study cohort. Environ. Health Perspect. 2014, 122, 926–932. [Google Scholar] [CrossRef]
- Raaschou-Nielsen, O.; Andersen, Z.J.; Beelen, R.; Samoli, E.; Stafoggia, M.; Weinmayr, G.; Hoffmann, B.; Fischer, P.; Nieuwenhuijsen, M.J.; Brunekreef, B.; et al. Air pollution and lung cancer incidence in 17 European cohorts: Prospective analyses from the European Study of Cohorts for Air Pollution Effects (ESCAPE). Lancet Oncol. 2013, 14, 813–822. [Google Scholar] [CrossRef]
- Huang, F.; Pan, B.; Wu, J.; Chen, E.; Chen, L. Relationship between exposure to PM2.5 and lung cancer incidence and mortality: A meta-analysis. Oncotarget 2017, 8, 43322–43331. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, W.; Wu, J. Ambient PM2.5 and annual lung cancer incidence: A nationwide study in 295 Chinese counties. Int. J. Environ. Res. Public Health 2020, 17, 1481. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, A.; Miller, A.B.; Weichenthal, S.A.; To, T.; Wall, C.; van Donkelaar, A.; Martin, R.V.; Crouse, D.L.; Villeneuve, P.J. Long-term exposure to fine particulate matter air pollution and the risk of lung cancer among participants of the canadian national breast screening study. Int. J. Cancer 2016, 139, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- International Commission on Radiological Protection. Human Respiratory Tract Model for Radiological Protection, ICRP Publication 66; Elsevier Science: Tarrytown, NY, USA, 1994. [Google Scholar]
- Wang, G.; Huang, L.; Gao, S.; Gao, S.; Wang, L. Measurements of PM10 and PM2.5 in urban area of Nanjing, China and the assessment of pulmonary deposition of particle mass. Chemosphere 2002, 48, 689–695. [Google Scholar] [CrossRef]
- Nag, S.; Gupta, A.K.; Mukhopadhyay, U.K. Size distribution of atmospheric aerosols in Kolkata, India and the assessment of pulmonary deposition of particle mass. Indoor Built Environ. 2005, 14, 381–389. [Google Scholar] [CrossRef]
- Stuart, B.O. Deposition and clearance of inhaled particles. Environ. Health Perspect. 1984, 55, 369–390. [Google Scholar] [CrossRef]
- Hofmann, W. Modelling particle deposition in human lungs: Modelling concepts and comparison with experimental data. Biomarkers 2009, 14, 59–62. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Li, G.; Zhang, Y.; Li, J.; Chen, H. Fluorescent reconstitution on deposition of PM2.5 in lung and extrapulmonary organs. Proc. Natl. Acad. Sci. USA 2019, 116, 2488–2493. [Google Scholar] [CrossRef]
- Yang, B.; Chen, D.; Zhao, H.; Xiao, C. The effects for PM2.5 exposure on non-small-cell lung cancer induced motility and proliferation. Springerplus 2016, 5, 2059. [Google Scholar] [CrossRef]
- Wei, H.; Liang, F.; Cheng, W.; Zhou, R.; Wu, X.; Feng, Y.; Wang, Y. The mechanisms for lung cancer risk of PM2.5: Induction of epithelial-mesenchymal transition and cancer stem cell properties in human non-small cell lung cancer cells. Environ. Toxicol. 2017, 32, 2341–2351. [Google Scholar] [CrossRef]
- Xu, H.; Jiao, X.; Wu, Y.; Li, S.; Cao, L.; Dong, L. Exosomes derived from PM2.5-treated lung cancer cells promote the growth of lung cancer via the Wnt3a/β-catenin pathway. Oncol. Rep. 2019, 41, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ma, M.; Zhou, W.; Yang, B.; Xiao, C. Inhibition of miR-32 activity promoted EMT induced by PM2.5 exposure through the modulation of the Smad1-mediated signaling pathways in lung cancer cells. Chemosphere 2017, 184, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, Y.; Yu, Z.; Ding, H.; Ma, Z. The influence of PM2.5 on lung injury and cytokines in mice. Exp. Ther. Med. 2019, 18, 2503–2511. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Xiao, C. PM2.5 exposure significantly improves the exacerbation of A549 tumor-bearing CB17-SCID mice. Environ. Toxicol. Pharmacol. 2018, 60, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.H.; Kao, S.W.; Tantoh, D.M.; Ko, P.C.; Lan, S.J.; Liaw, Y.P. Association between fine particulate matter and oral cancer among Taiwanese men. J. Investig. Med. 2019, 67, 34–38. [Google Scholar] [CrossRef]
- Nagel, G.; Stafoggia, M.; Pedersen, M.; Andersen, Z.J.; Galassi, C.; Munkenast, J.; Jaensch, A.; Sommar, J.; Forsberg, B.; Olsson, D.; et al. Air pollution and incidence of cancers of the stomach and the upper aerodigestive tract in the European Study of Cohorts for Air Pollution Effects (ESCAPE). Int. J. Cancer 2018, 143, 1632–1643. [Google Scholar] [CrossRef]
- Wong, C.M.; Tsang, H.; Lai, H.K.; Thomas, G.N.; Lam, K.B.; Chan, K.P.; Zheng, Q.; Ayres, J.G.; Lee, S.Y.; Lam, T.H.; et al. Cancer mortality risks from long-term exposure to ambient fine particle. Cancer Epidemiol. Biomark. 2016, 25, 839–845. [Google Scholar] [CrossRef]
- Ethan, C.J.; Mokoena, K.K.; Yu, Y.; Shale, K.; Fan, Y.; Rong, J.; Liu, F. Association between PM2.5 and mortality of stomach and colorectal cancer in Xi’an: A time-series study. Environ. Sci. 2020, 1, 1–11. [Google Scholar] [CrossRef]
- Coleman, N.C.; Burnett, R.T.; Higbee, J.D.; Lefler, J.S.; Merrill, R.M.; Ezzati, M.; Marshall, J.D.; Kim, S.Y.; Bechle, M.; Robinson, A.L.; et al. Cancer mortality risk, fine particulate air pollution, and smoking in a large, representative cohort of US adults. Cancer Causes Control 2020, 31, 767–776. [Google Scholar] [CrossRef]
- Turner, M.C.; Krewski, D.; Diver, W.R.; Pope, C.A., III; Burnett, R.T.; Jerrett, M.; Marshall, J.D.; Gapstur, S.M. Ambient air pollution and cancer mortality in the cancer prevention study II. Environ. Health Perspect. 2017, 125, 087013. [Google Scholar] [CrossRef]
- Guo, C.; Chan, T.C.; Teng, Y.C.; Lin, C.; Bo, Y.; Chang, L.Y.; Lau, A.; Tam, T.; Wong, M.; Qian Lao, X. Long-term exposure to ambient fine particles and gastrointestinal cancer mortality in Taiwan: A cohort study. Environ. Int. 2020, 138, 105640. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.C.; Wu, C.D.; Chen, M.J.; Huang, Y.T.; Chen, C.J.; Su, H.J.; Yang, H.I. Fine particle pollution, alanine transaminase, and liver cancer: A Taiwanese prospective cohort study (REVEAL-HBV). J. Natl. Cancer. Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Wan, X.; Sun, Y.; Cheng, K.; Zhao, X.; Zheng, Y.; Yang, G.; Wang, L. Spatiotemporal analysis of PM2.5 and pancreatic cancer mortality in China. Environ. Res. 2018, 164, 132–139. [Google Scholar] [CrossRef] [PubMed]
- VoPham, T.; Bertrand, K.A.; Tamimi, R.M.; Laden, F.; Hart, J.E. Ambient PM2.5 air pollution exposure and hepatocellular carcinoma incidence in the United States. Cancer Causes Control 2018, 29, 563–572. [Google Scholar] [CrossRef]
- Hu, H.; Dailey, A.B.; Kan, H.; Xu, X. The effect of atmospheric particulate matter on survival of breast cancer among US females. Breast Cancer Res. Treat. 2013, 139, 217–226. [Google Scholar] [CrossRef]
- White, A.J.; Keller, J.P.; Zhao, S.; Carroll, R.; Kaufman, J.D.; Sandler, D.P. Air pollution, clustering of particulate matter components, and breast cancer in the sister study: A US-wide cohort. Environ. Health Perspect. 2019, 127, 107002. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Z.; Ren, J.; Liu, Y.; Chang, L.T.; Cheung, K.; Feng, Y.; Li, Y. An urban-rural and sex differences in cancer incidence and mortality and the relationship with PM2.5 exposure: An ecological study in the southeastern side of Hu line. Chemosphere 2019, 216, 766–773. [Google Scholar] [CrossRef]
- Cheng, I.; Tseng, C.; Wu, J.; Yang, J.; Conroy, S.M.; Shariff-Marco, S.; Li, L.; Hertz, A.; Gomez, S.L.; Le Marchand, L.; et al. Association between ambient air pollution and breast cancer risk: The multiethnic cohort study. Int. J. Cancer 2020, 146, 699–711. [Google Scholar] [CrossRef]
- Andersen, Z.J.; Pedersen, M.; Weinmayr, G.; Stafoggia, M.; Galassi, C.; Jørgensen, J.T.; Sommar, J.N.; Forsberg, B.; Olsson, D.; Oftedal, B.; et al. Long-term exposure to ambient air pollution and incidence of brain tumor: The European Study of Cohorts for Air Pollution Effects (ESCAPE). Neuro. Oncol. 2018, 20, 420–432. [Google Scholar] [CrossRef]
- Weinmayr, G.; Pedersen, M.; Stafoggia, M.; Andersen, Z.J.; Galassi, C.; Munkenast, J.; Jaensch, A.; Oftedal, B.; Krog, N.H.; Aamodt, G.; et al. Particulate matter air pollution components and incidence of cancers of the stomach and the upper aerodigestive tract in the European Study of Cohorts of Air Pollution Effects (ESCAPE). Environ. Int. 2018, 120, 163–171. [Google Scholar] [CrossRef]
- Pedersen, M.; Stafoggia, M.; Weinmayr, G.; Andersen, Z.J.; Galassi, C.; Sommar, J.; Forsberg, B.; Olsson, D.; Oftedal, B.; Krog, N.H.; et al. Is there an association between ambient air pollution and bladder cancer incidence? Analysis of 15 European cohorts. Eur. Urol. Focus 2018, 4, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Andersen, Z.J.; Stafoggia, M.; Weinmayr, G.; Galassi, C.; Sørensen, M.; Eriksen, K.T.; Tjønneland, A.; Loft, S.; Jaensch, A.; et al. Ambient air pollution and primary liver cancer incidence in four European cohorts within the ESCAPE project. Environ. Res. 2017, 154, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative Stress: Introductory Remarks; Oxidative Stress, Academic Press: London, UK, 1985; pp. 1–8. [Google Scholar]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Lee, T.L.; Chen, Y.C.; Liang, C.J.; Wang, S.H.; Lue, J.H.; Tsai, J.S.; Lee, S.W.; Chen, S.H.; Yang, Y.F.; et al. PM2.5-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6/AKT/STAT3/NF-κB-dependent pathway. Part. Fibre Toxicol. 2018, 15, 4. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Wang, L.; Chen, C.; Yang, D.; Jin, M.; Bai, C.; Song, Y. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-κB signaling pathway. J. Thorac. Dis. 2017, 9, 4398–4412. [Google Scholar] [CrossRef]
- Pourazar, J.; Mudway, I.S.; Samet, J.M.; Helleday, R.; Blomberg, A.; Wilson, S.J.; Frew, A.J.; Kelly, F.J.; Sandström, T. Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L724–L730. [Google Scholar] [CrossRef]
- Zhou, W.; Tian, D.; He, J.; Wang, Y.; Zhang, L.; Cui, L.; Jia, L.; Zhang, L.; Li, L.; Shu, Y.; et al. Repeated PM2.5 exposure inhibits BEAS-2B cell p53 expression through ROS-Akt-DNMT3B pathway-mediated promoter hypermethylation. Oncotarget 2016, 7, 20691–20703. [Google Scholar] [CrossRef]
- Niu, B.Y.; Li, W.K.; Li, J.S.; Hong, Q.H.; Khodahemmati, S.; Gao, J.F.; Zhou, Z.X. Effects of DNA damage and oxidative stress in human bronchial epithelial cells exposed to PM2.5 from Beijing, China, in Winter. Int. J. Environ. Res. Public Health 2020, 17, 4874. [Google Scholar] [CrossRef]
- Veerappan, I.; Sankareswaran, S.K.; Palanisamy, R. Morin protects human respiratory cells from PM2.5 induced genotoxicity by mitigating ROS and reverting altered miRNA expression. Int. J. Environ. Res. Public Health 2019, 16, 2389. [Google Scholar] [CrossRef]
- Ren, X.; Tang, Y.; Sun, J.; Feng, J.; Chen, L.; Chen, H.; Zeng, S.; Chen, C.; Li, X.; Zhu, H.; et al. Flavone protects HBE cells from DNA double-strand breaks caused by PM2.5. Hum. Cell. 2018, 31, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhao, L.; Zhang, L.; Chen, M.; Shi, J.; Dong, C.; Cai, Z. Effects of ambient PM2.5 and 9-nitroanthracene on DNA damage and repair, oxidative stress and metabolic enzymes in the lungs of rats. Toxicol. Res. (Camb.) 2017, 6, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Huang, H.B.; Chang, Y.C.; Su, T.Y.; Wang, Y.C.; Wang, G.C.; Chen, J.E.; Tang, C.S.; Wu, T.N.; Liou, S.H. Exposure to fine particulate matter causes oxidative and methylated DNA damage in young adults: A longitudinal study. Sci. Total Environ. 2017, 598, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Lu, S.; Wang, Y.; Zhu, Y.; Shi, T.; Lin, M.; Deng, Z.; Wang, Z.; Song, N.; Li, S.; et al. Long-term exposure to high air pollution induces cumulative DNA damages in traffic policemen. Sci. Total Environ. 2017, 593–594, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Chen, Y.; Li, X.; Zhang, Z.; Chu, H. Ambient fine particulate matter (PM2.5) induces oxidative stress and pro-inflammatory response via up-regulating the expression of CYP1A1/1B1 in human bronchial epithelial cells in vitro. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 839, 40–48. [Google Scholar] [CrossRef]
- He, M.; Ichinose, T.; Yoshida, S.; Ito, T.; He, C.; Yoshida, Y.; Arashidani, K.; Takano, H.; Sun, G.; Shibamoto, T. PM2.5-induced lung inflammation in mice: Differences of inflammatory response in macrophages and type II alveolar cells. J. Appl. Toxicol. 2017, 37, 1203–1218. [Google Scholar] [CrossRef]
- Riva, D.R.; Magalhães, C.B.; Lopes, A.A.; Lanças, T.; Mauad, T.; Malm, O.; Valença, S.S.; Saldiva, P.H.; Faffe, D.S.; Zin, W.A. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhal. Toxicol. 2011, 23, 257–267. [Google Scholar] [CrossRef]
- Shi, Y.; Zhao, T.; Yang, X.; Sun, B.; Li, Y.; Duan, J.; Sun, Z. PM2.5-induced alteration of DNA methylation and RNA-transcription are associated with inflammatory response and lung injury. Sci. Total Environ. 2019, 650, 908–921. [Google Scholar] [CrossRef]
- Lee, C.W.; Chi, M.C.; Hsu, L.F.; Yang, C.M.; Hsu, T.H.; Chuang, C.C.; Lin, W.N.; Chu, P.M.; Lee, I.T. Carbon monoxide releasing molecule-2 protects against particulate matter-induced lung inflammation by inhibiting TLR2 and 4/ROS/NLRP3 inflammasome activation. Mol. Immunol. 2019, 112, 163–174. [Google Scholar] [CrossRef]
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019, 459, 30–40. [Google Scholar] [CrossRef]
- Li, R.; Zhou, R.; Zhang, J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol. Lett. 2018, 15, 7506–7514. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ding, W.; Deng, X. PM2.5, fine particulate matter: A novel player in the epithelial-mesenchymal transition? Front. Physiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Refsnes, M.; Låg, M.; Holme, J.A.; Schwarze, P.E. Activation of proinflammatory responses in cells of the airway mucosa by particulate matter: Oxidant- and non-oxidant-mediated triggering mechanisms. Biomolecules 2015, 5, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Iles, K.E.; Dickinson, D.A.; Watanabe, N.; Iwamoto, T.; Forman, H.J. AP-1 activation through endogenous H2O2 generation by alveolar macrophages. Free Radic. Biol. Med. 2002, 32, 1304–1313. [Google Scholar] [CrossRef]
- Kastan, M.B. DNA damage responses: Mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture. Mol. Cancer Res. 2008, 6, 517–524. [Google Scholar] [CrossRef]
- Wang, K.; Li, L.; Zhang, Y.; Gao, D. Crosstalk between signaling pathways and DNA damage response. Genome Instab. Dis. 2020, 1, 81–91. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Flagg, E.W.; Coates, R.J.; Greenberg, R.S. Epidemiologic studies of antioxidants and cancer in humans. J. Am. Coll. Nutr. 1995, 14, 419–427. [Google Scholar] [CrossRef]
- Zhang, Y.; Darland, D.; He, Y.; Yang, L.; Dong, X.; Chang, Y. Reduction of PM2.5 toxicity on human alveolar epithelial cells A549 by tea polyphenols. J. Food Biochem. 2018, 42, e12496. [Google Scholar] [CrossRef]
- Mughal, M.H. Turmeric polyphenols: A comprehensive review. Integr. Food Nutr. Metab. 2019, 6. [Google Scholar] [CrossRef]
- Lelli, D.; Sahebkar, A.; Johnston, T.P.; Pedone, C. Curcumin use in pulmonary diseases: State of the art and future perspectives. Pharmacol. Res. 2017, 115, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Oztay, F.; Kayalar, O.; Yildirim, M. Pulmonary oxidative stress and antioxidant defence system in the lung ageing and fibrotic and diabetic lungs. Oxidative Stress Lung Dis. 2019, 325–353. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year | Study Design | Experimental Model | Toxicological Effects of PM2.5 | Conclusions |
|---|---|---|---|---|
| Yang, B. et al. 2016 [35] | In vitro | Human lung carcinoma cell lines A549 and H1299 | Enhanced proliferation and motility in both cell lines. Up-regulated IL-1β and MMP-1 genes which were involved in the cell proliferation as well as the progression of invasion and metastasis. Induced mRNA expression of regulated transcripts in response to MAPK signaling pathway. | PM2.5 might induce crosstalk among pathways that promote survival, proliferation, invasion, and migration of cancer cells. MAPK signaling pathway played a crucial role. |
| Wei, H. et al. 2017 [36] | In vitro | A549 cells | Enhanced cell migration and invasion. Decreased mRNA expression of epithelial markers and increased mRNA expression of mesenchymal markers. Caused changes in the levels of stemness-associated microRNAs. Induced EMT markers and morphology as well as CSC properties. | Chronic PM2.5 exposure possibly induced malignant behaviors as well as EMT and CSC properties in vitro. Stemness-associated microRNAs were likely to be involved. |
| Xu, H. et al. 2019 [37] | In vitro and In vivo | A549 cells A549 tumor model in female nude mice | Induced secretion of exosomes containing high levels of Wnt3a. Promoted A549 cell proliferation via Wnt3a/β-catenin pathway. Enhanced tumor growth in vivo in Wnt3a-dependent manner. | Exosomes derived from PM2.5 exposure appeared to promote the growth of lung tumor in vitro and in vivo via Wnt3a/β-catenin pathway. |
| Yang, D. et al. 2017 [38] | In vitro | Human lung carcinoma cell lines A549 and H292 | Induced EMT in both cell lines. Up-regulated the activity of Smad1-mediated pathway and down-regulated the expression of Smad6 and Smad7 which were associated with EMT in A549 cells. | PM2.5 might induce EMT through Smad1-mediated signaling pathway. |
| Yang, J. et al. 2019 [39] | In vivo | Female Kunming mice | Induced lung damage. Increased the number of inflammatory cells, particularly M1 and M2 macrophages. Increased the levels cytokines such as IL-4, TNF-α, TGF-β1 in sera and tissues. | PM2.5 capably activated the inflammatory responses and compromised immune function, leading to ultrastructural damage in mice lungs. |
| Yang, B. et al. 2018 [40] | In vivo | A549 tumor model in male CB17-SCID mice | Increased the number of tumor nodules. Increased the levels of total protein in BALF. Enhanced the expression of MMP-1, IL-1β and VEGF which are hallmarkers in cancer progression. Increased the levels of angiogenesis factors in blood serum. | PM2.5 possibly promoted lung tumor progression in tumor-bearing mice. |
| Liu, C.W. et al. 2018 [62] | In vitro In vivo Cross-sectional study | A549 cells WT and IL-6 KO mice COPD patients and Healthy subjects | Induced cytotoxicity, ROS generation and monocyte adherence in A549 cells. Increased the expression of ICAM-1 via IL-6/AKT/STAT3/NF-κB-dependent pathway in A549 cells. Induced the expression of ICAM-1 and IL-6 in lung tissues and plasma of WT mice but not IL-6 KO mice. Higher levels of white blood cell count, CRP, sICAM-1 and IL-6 in COPD patients. | Oxidative stress mediated by PM2.5-induced ROS production might increase the expression of ICAM-1 both in vitro and in vivo. The ICAM-1 expression was regulated through IL-6/AKT/STAT3/NF-κB-dependent pathway in lung epithelial cells. |
| Wang, J. et al. 2017 [63] | In vitro and In vivo | HBECs Male C57 mice model of PM-induced acute lung inflammation | Enhanced ROS generation in vitro and in vivo. Induced inflammatory responses in vitro through increased the expression of IL-1β, IL-6, IL-8, MMP-9 and COX-2. Activated MAPK (ERK, JNK, p38 MAPK) and NF-κB pathways involving in inflammation in vitro. Increased oxidant stress in lung tissues, infiltration of inflammatory cells, the number of total cells and inflammatory cells as well as the concentrations of IL-1β, IL-6, IL-8, MMP-9 in BALF. | PM2.5 capably induced inflammatory responses both in vitro and in vivo via ROS/MAPK/NF-κB signaling pathway. |
| Pourazar, J. et al. 2005 [64] | Prospective study | Nonatopic nonsmokers exposed to diesel exhaust | Increased total immunoreactivity of phosphorylated p38 MAPK. Increased nuclear translocation of p65 subunit of NF-κB, c-Jun subunit of AP-1, phosphorylated JNK, and phosphorylated p38 MAPK. | Exposure of healthy subjects to diesel exhaust, a major component of PM2.5, might activate the redox-sensitive transcription factors (NF-κB and AP-1) and related signaling pathways (JNK and p38 MAPK) in bronchial epithelium. |
| Zhou, W. et al. 2016 [65] | In vitro | HBECs (BEAS-2B) | Induced p53 promoter hypermethylation and down-regulated p53 expression via DNMT3B up-regulation. Activated ROS-mediated PI3K/AKT pathway which involved in DNMT3B up-regulation. | PM2.5 could induce epigenetic silencing of p53 through ROS/AKT/DNMT3B pathway-mediated promoter hypermethylation in BEAS-2B cells. |
| Niu, B.Y. et al. 2020 [66] | In vitro | HBECs (16HBE) | Induced cytotoxicity. Generated oxidative stress through increased levels of oxidants such as ROS and decreased level of antioxidant GSH. Caused DNA damage including DNA strand breaks, oxidative DNA damage and chromatin damage. Altered the expression of DNA repair genes. | PM2.5 capably exerted cytotoxicity and genotoxicity in 16HBE cells through the combined effects of generation of oxidative stress-induced DNA damage and alteration of DDR. |
| Veerappan, I. et al. 2019 [67] | In vitro | A549 cells | Induced lipid peroxidation and oxidative stress via ROS generation. Induced cytotoxicity and DNA damage. Altered the expression of miRNAs related to oxidative stress, inflammation and DNA damage. | PM2.5 possibly induced genotoxicity in A549 cells by increased production of ROS generation and the alteration of miRNA expression associated to oxidative stress, inflammation and DNA damage. |
| Ren, X. et al. 2018 [68] | In vitro | HBECs | Decreased cell viability. Induced oxidative DNA double-strand breaks. | PM2.5 might cause cytotoxicity and genotoxicity in HBECs by oxidative manner. |
| Li, R. et al. 2017 [69] | In vivo | Male Wistar rats | Caused pathological damage in rat lungs. Increased the levels of DNA damage markers. Regulated the expression of DNA repair genes (↑OGG1, ↓MTH1, ↓XRCC1). Altered the levels of oxidative stress markers (↑GADD153, ↑HO-1, ↑MDA, ↓SOD). Increased the levels of metabolic enzymes (CYP450, CYP1A1, CYP1A2, GST). | PM2.5 and higher dosage 9-NA (a typical compound of NPAHs in PM2.5) possibly induced genotoxicity in rat lungs through three major ways including induction of DNA damage combined with inhibition of DNA repair process, production of oxidative stress, and disturbance of biotransformation via metabolic enzymes activation. |
| Lai, C.H. et al. 2017 [70] | Longitudinal study | Healthy students | Significant relationship between biomarker of urinary exposure (1-OHP) and biomarkers of urinary oxidative and methylated DNA damage (8-oxoG and N7-MeG). N7-MeG increased by 8.1% per 10 μg/m3 increment in PM2.5. | PM2.5 might cause oxidative and methylated DNA damage in healthy and young adults. |
| Tan, C. et al. 2017 [71] | Cross-sectional study | Traffic policemen and Office policemen | Significant correlation between cumulative intersection duty time with biomarkers of oxidative stress (↓GSH) and DNA damage (↑8-OHdG, ↑tail DNA, ↑MN frequency) in traffic policemen. The levels of biomarkers of oxidative stress in traffic policemen were lower than those in office policemen, but of DNA damage were higher. | Long-term exposure to high concentrations of PM2.5 could induce cumulative DNA damage via oxidative stress pathway. |
| Yuan, Q. et al. 2019 [72] | In vitro | HBECs (HBE and BEAS-2B) | Reduced cell viability. Increased the level of cellular ROS. Induced cellular pro-inflammation through increased levels of IL-6, IL-8. Up-regulated the expression of CYP1A1 and CYP1B1 genes which involved in generation of oxidative stress and pro-inflammation. Inhibition of STAT3/P-STAT3 hampered ROS production and IL6/IL-8 secretion by PM2.5. | PM2.5 capably induced oxidative stress and pro-inflammatory response via up-regulating the expression of CYP1A1/1B1 in two main cell lines of HBECs. JAK/STAT3 pathway might be associated. |
| He, M. et al. 2017 [73] | In vitro and In vivo | RAW264.7 cells BMDMs of WT and MyD88 deficient BALB/c mice Mouse alveolar cell line MLE-12 BALB/c mice | Induced NF-κB, p38 MAPK and ERK phosphorylation, increased proinflammatory gene and protein expressions, and induced oxidative stress marker HO-1 gene in RAW264.7 cells. Caused increase of proinflammatory mediators in BMDMs in which WT cells were higher than MyD88 cells. Increased proinflammatory gene expressions and HO-1 gene as well as induced intracellular ROS generation in MLE-12 cells. Caused sever alveolitis and bronchitis, and increased the number of macrophages and neutrophils as well as the levels of proinflammatory mediators in mice BALF. | Macrophages might preferentially release proinflammatory mediators due to LPS presenting in PM2.5 via LPS/MyD88 pathway, whereas type II alveolar cell could be prone to PM2.5-induced oxidative stress to cause inflammatory response. PM2.5 possibly induced oxidative stress-dependent inflammation, leading to lung injury. |
| Riva, D.R. et al. 2019 [74] | In vivo | BALB/c mice | Impaired lung function characterized by increased elastic and viscoelastic components of lung mechanics. Induced lung inflammation via increase in MPO activity, neutrophil infiltration and proinflammatory cytokine expressions (IL-6, TNF-α). Caused oxidative damage expressed by increased reactive substances to thiobarbituric acid and 8-isoprostane. Induced oxidative stress through increase in CAT and reduction in GSH/GSSG. | Acute exposure to low dose of PM2.5 might induce oxidative stress, inflammation and functional impairment in healthy mice lungs. |
| Shi, Y. et al. 2019 [75] | In vitro and In vivo | HBECs (BEAS-2B) Male Sprague-Dawley rats | Induced genome wide DNA methylation and RNA transcription alterations in BEAS-2B cells. Caused pathological changes such as inflammatory cell infiltration and alveolar wall thickening in rat lung tissue. Enhanced the secretion of cytotoxicity markers and inflammatory cytokines in BALF. Up-regulated the expression levels of inflammatory cytokine genes and NF-κB in BEAS-2B cells and rat lung tissue. | PM2.5 capably induced genome wide DNA methylation and RNA transcription changes as well as inflammatory responses, contributing to pulmonary toxicity and pathogenesis. |
| Lee, C.W. et al. 2019 [76] | In vitro and In vivo | HPAEpiCs Male BALB/c mice | Induced CRP expression, NLRP3 inflammasome activation, IL-1β secretion, and caspse-1 activation in HPAEpiCs. Induced inflammatory responses via TLR2 and TLR4 in HPAEpiCs. Induced intracellular and mitochondrial ROS generation in HPAEpiCs. Increased the expression of CRP, NLRP3 and ASC protein lung tissue, the levels of IL-1β in serum, and leukocyte count in BALF in mice. | PM2.5 possibly induced lung inflammation via TLR2 and 4/ ROS/NLRPH3 signaling pathway. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-W.; Vo, T.T.T.; Wu, C.-Z.; Chi, M.-C.; Lin, C.-M.; Fang, M.-L.; Lee, I.-T. The Inducible Role of Ambient Particulate Matter in Cancer Progression via Oxidative Stress-Mediated Reactive Oxygen Species Pathways: A Recent Perception. Cancers 2020, 12, 2505. https://doi.org/10.3390/cancers12092505
Lee C-W, Vo TTT, Wu C-Z, Chi M-C, Lin C-M, Fang M-L, Lee I-T. The Inducible Role of Ambient Particulate Matter in Cancer Progression via Oxidative Stress-Mediated Reactive Oxygen Species Pathways: A Recent Perception. Cancers. 2020; 12(9):2505. https://doi.org/10.3390/cancers12092505
Chicago/Turabian StyleLee, Chiang-Wen, Thi Thuy Tien Vo, Ching-Zong Wu, Miao-Ching Chi, Chieh-Mo Lin, Mei-Ling Fang, and I-Ta Lee. 2020. "The Inducible Role of Ambient Particulate Matter in Cancer Progression via Oxidative Stress-Mediated Reactive Oxygen Species Pathways: A Recent Perception" Cancers 12, no. 9: 2505. https://doi.org/10.3390/cancers12092505
APA StyleLee, C.-W., Vo, T. T. T., Wu, C.-Z., Chi, M.-C., Lin, C.-M., Fang, M.-L., & Lee, I.-T. (2020). The Inducible Role of Ambient Particulate Matter in Cancer Progression via Oxidative Stress-Mediated Reactive Oxygen Species Pathways: A Recent Perception. Cancers, 12(9), 2505. https://doi.org/10.3390/cancers12092505