Pre- and Post-Zygotic TP53 De Novo Mutations in SHH-Medulloblastoma

 , , ,
, , ,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Results
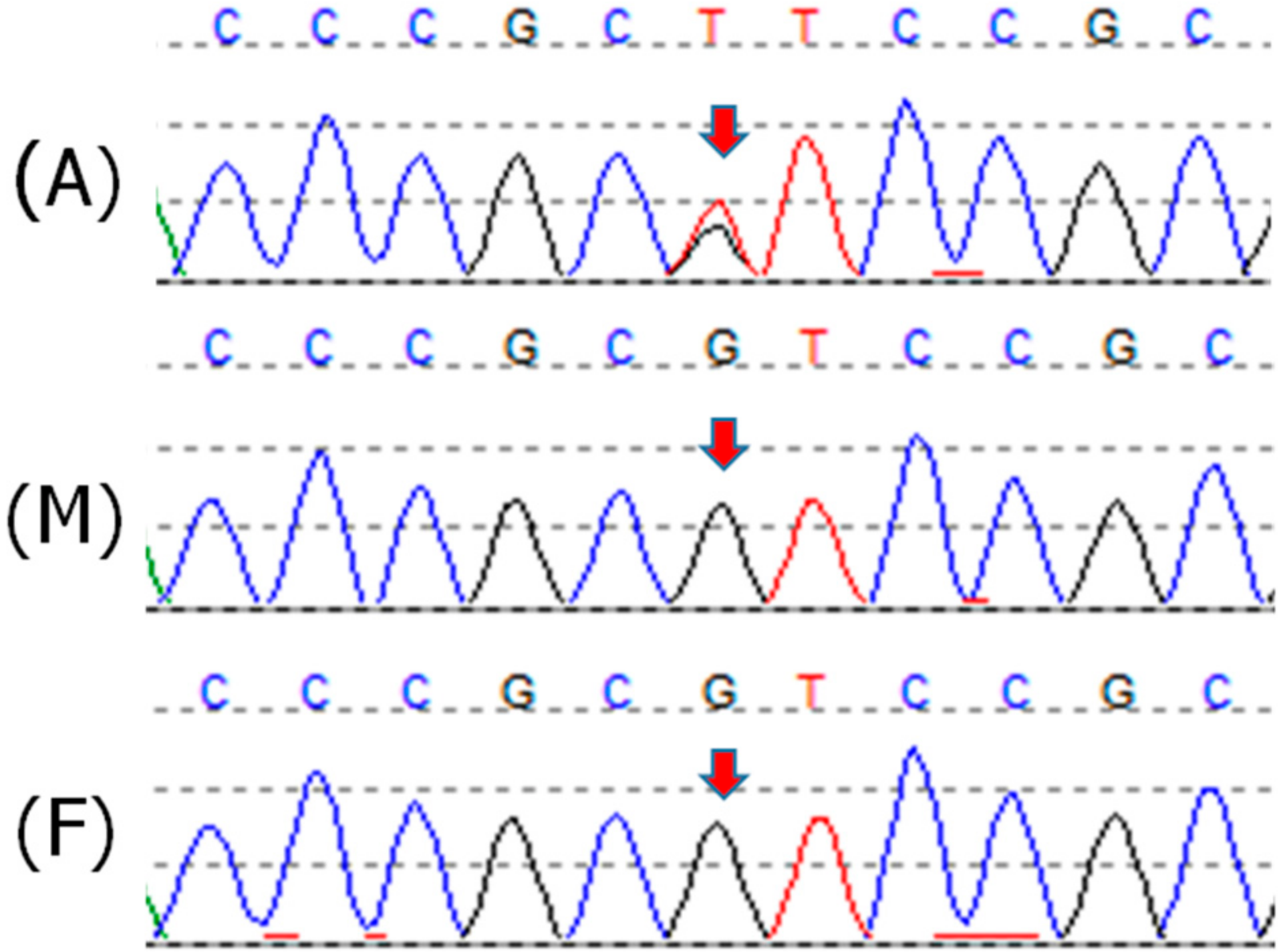
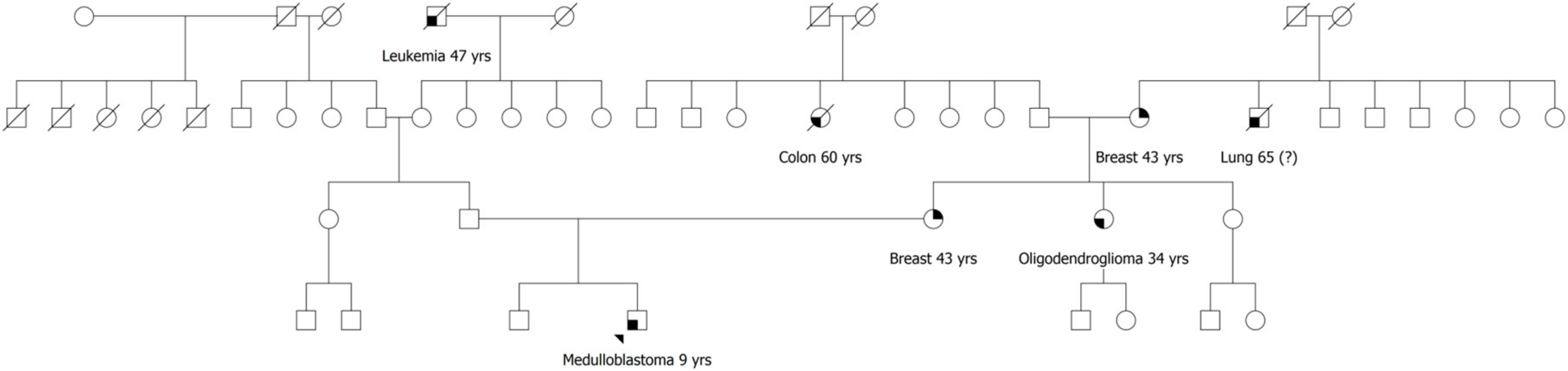
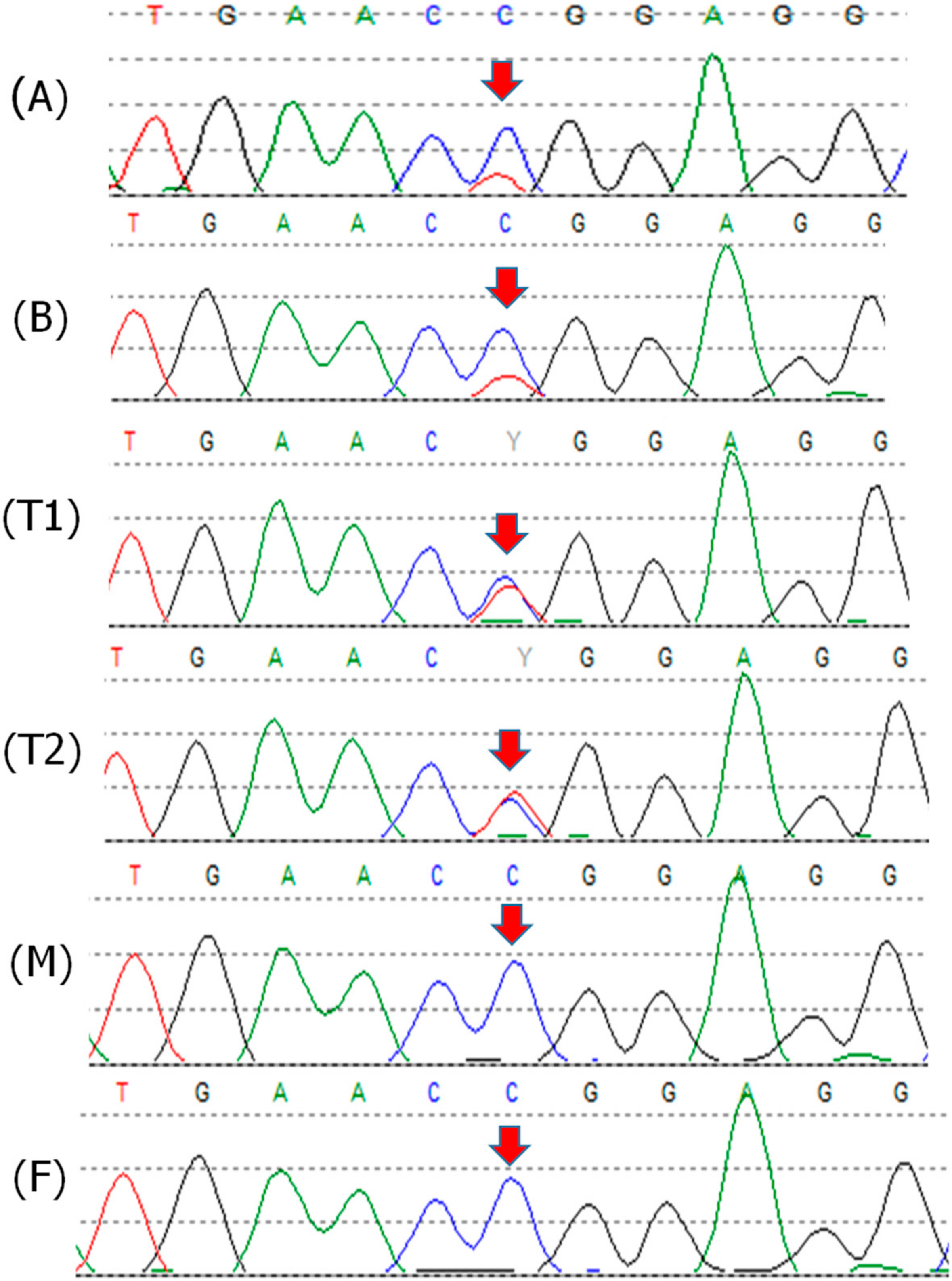
2.1. Patient 1
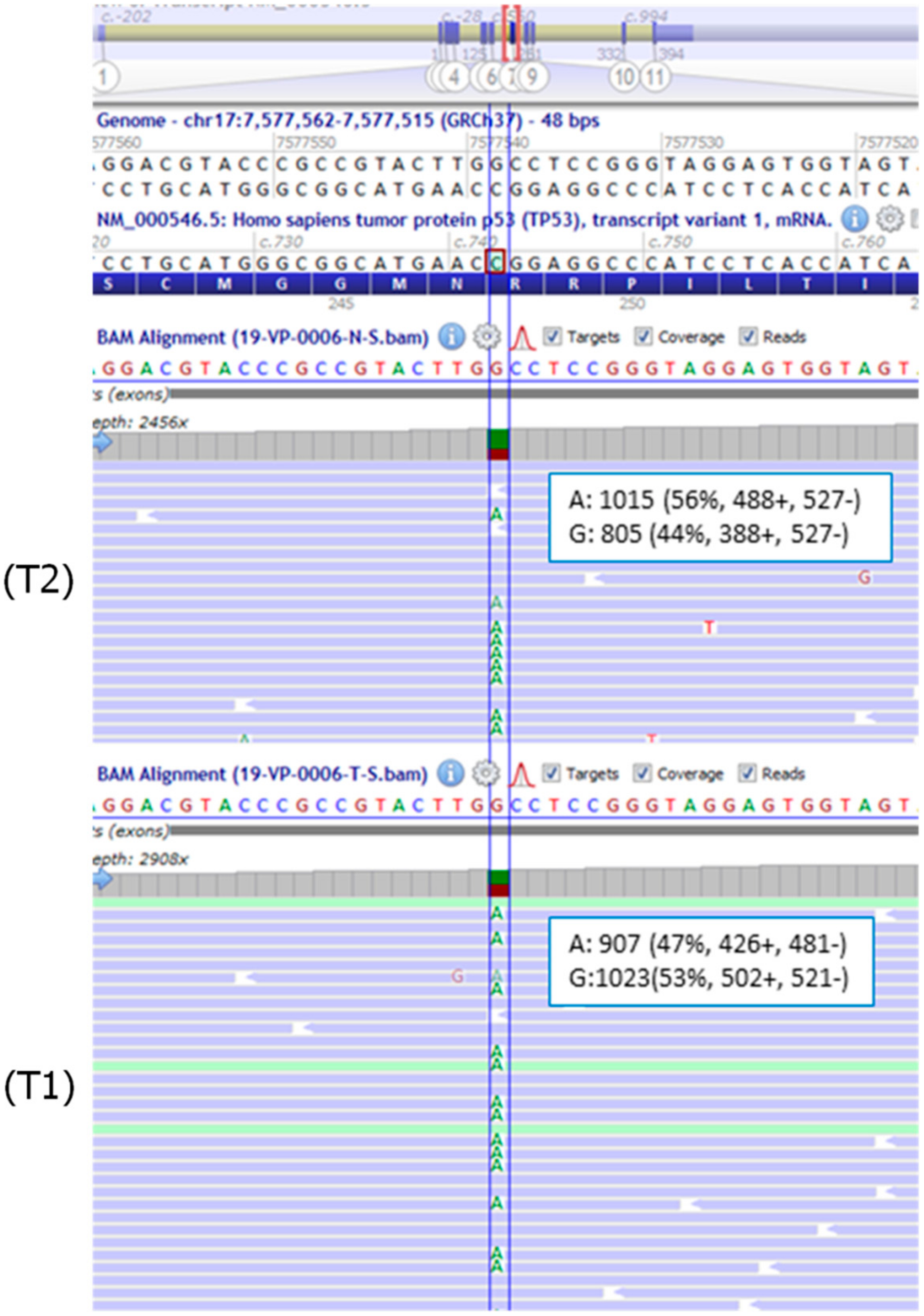
2.2. Patient 2
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Samples
4.3. Immunohistochemistry
4.4. STRs Segregation Analysis
4.5. Sanger Sequencing
4.6. Multiplex Ligation-Dependent Probe Amplification (MLPA)
4.7. Next Generation Sequencing (NGS)
4.8. Quantitative Polymerase Chain Reaction (qPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.-M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugiéres, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical Characteristics of Families With p53 Germline Mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Renaux-Petel, M.; Charbonnier, F.; Théry, J.-C.; Fermey, P.; Lienard, G.; Bou, J.; Coutant, S.; Vezain, M.; Kasper, E.; Fourneaux, S.; et al. Contribution of de novo and mosaic TP53 mutations to Li-Fraumeni syndrome. J. Med. Genet. 2017, 55, 173–180. [Google Scholar] [CrossRef]
- Coffee, B.; Cox, H.C.; Kidd, J.; Sizemore, S.; Brown, K.; Manley, S.; Mancini-DiNardo, D. Detection of somatic variants in peripheral blood lymphocytes using a next generation sequencing multigene pan cancer panel. Cancer Genet. 2017, 211, 5–8. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Chao, E.C.; Nehoray, B.; Van Tongeren, L.R.; LaDuca, H.; Blazer, K.R.; Slavin, T.; Facmg, D.A.B.M.D.; Pesaran, T.; Rybak, C.; et al. Somatic TP53 variants frequently confound germ-line testing results. Genet. Med. 2017, 20, 809–816. [Google Scholar] [CrossRef]
- Batalini, F.; Peacock, E.G.; Stobie, L.; Robertson, A.; Garber, J.E.; Weitzel, J.N.; Tung, N. Li-Fraumeni syndrome: Not a straightforward diagnosis anymore—The interpretation of pathogenic variants of low allele frequency and the differences between germline PVs, mosaicism, and clonal hematopoiesis. Breast Cancer Res. 2019, 21, 107–110. [Google Scholar] [CrossRef]
- Coffee, B.; Cox, H.C.; Bernhisel, R.; Manley, S.; Bowles, K.; Roa, B.B.; Mancini-DiNardo, D. A substantial proportion of apparently heterozygous TP53 pathogenic variants detected with a next-generation sequencing hereditary pan-cancer panel are acquired somatically. Hum. Mutat. 2019, 41, 203–211. [Google Scholar] [CrossRef]
- Mester, J.L.; Jackson, S.A.; Postula, K.; Stettner, A.; Solomon, S.; Bissonnette, J.; Murphy, P.D.; Klein, R.T.; Hruska, K.S. Apparently Heterozygous TP53 Pathogenic Variants May Be Blood Limited in Patients Undergoing Hereditary Cancer Panel Testing. J. Mol. Diagn. 2020, 22, 396–404. [Google Scholar] [CrossRef]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.H.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef]
- Tabori, U.; Baskin, B.; Shago, M.; Alon, N.; Taylor, M.; Ray, P.N.; Bouffet, E.; Malkin, D.; Hawkins, C.E. Universal Poor Survival in Children with Medulloblastoma Harboring Somatic TP53 Mutations. J. Clin. Oncol. 2010, 28, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugières, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Murnyák, B.; Hortobágyi, T. Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget 2016, 7, 64910–64920. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.B.; Almutairi, F.; Haffner, M.C.; Rajoria, G.; Liu, Z.; Klimek, S.; Zoino, R.; Yousefi, K.; Sharma, R.; De Marzo, A.M.; et al. Analytic, Preanalytic, and Clinical Validation of p53 IHC for Detection of TP53 Missense Mutation in Prostate Cancer. Clin. Cancer Res. 2017, 23, 4693–4703. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; Van De Vorst, M.; Van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef]
- Jin, Z.-B.; Wu, J.; Huang, X.-F.; Feng, C.-Y.; Cai, X.-B.; Mao, J.-Y.; Xiang, L.; Wu, K.-C.; Xiao, X.; Kloss, B.A.; et al. Trio-based exome sequencing arrests de novo mutations in early-onset high myopia. Proc. Natl. Acad. Sci. USA 2017, 114, 4219–4224. [Google Scholar] [CrossRef]
- Bishop, M.R.; Perez, K.K.D.; Sun, M.; Ho, S.; Chopra, P.; Mukhopadhyay, N.; Hetmanski, J.B.; Taub, M.A.; Moreno-Uribe, L.M.; Valencia-Ramirez, L.C.; et al. Genome-wide Enrichment of De Novo Coding Mutations in Orofacial Cleft Trios. Am. J. Hum. Genet. 2020, 107, 124–136. [Google Scholar] [CrossRef]
- Franaszczyk, M.; Truszkowska, G.; Chmielewski, P.; Rydzanicz, M.; Kosinska, J.; Rywik, T.; Biernacka, A.; Spiewak, M.; Kostrzewa, G.; Stepien-Wojno, M.; et al. Analysis of De Novo Mutations in Sporadic Cardiomyopathies Emphasizes Their Clinical Relevance and Points to Novel Candidate Genes. J. Clin. Med. 2020, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- Howrigan, D.P.; Rose, S.A.; Samocha, K.E.; Fromer, M.; Cerrato, F.; Chen, W.J.; Churchhouse, C.; Chambert, K.; Chandler, S.D.; Daly, M.; et al. Exome sequencing in schizophrenia-affected parent–offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci. 2020, 23, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Gajecka, M. Unrevealed mosaicism in the next-generation sequencing era. Mol. Genet. Genom. 2015, 291, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Ye, A.Y.; Dou, Y.; Yang, X.; Wang, S.; Huang, A.Y.; Wei, L. A model for postzygotic mosaicisms quantifies the allele fraction drift, mutation rate, and contribution to de novo mutations. Genome Res. 2018, 28, 943–951. [Google Scholar] [CrossRef]
- Kool, M.; Jones, D.T.W.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.B.; Zavadil, J.; Olivier, M. TP53Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Calhoun, S.; Daggett, V. Structural Effects of the L145Q, V157F, and R282W Cancer-Associated Mutations in the p53 DNA-Binding Core Domain. Biochemistry 2011, 50, 5345–5353. [Google Scholar] [CrossRef]
- Mizuarai, S.; Yamanaka, K.; Kotani, H. Mutant p53 Induces the GEF-H1 Oncogene, a Guanine Nucleotide Exchange Factor-H1 for RhoA, Resulting in Accelerated Cell Proliferation in Tumor Cells. Cancer Res. 2006, 66, 6319–6326. [Google Scholar] [CrossRef]
- Prochazkova, K.; Pavlikova, K.; Minarik, M.; Sumerauer, D.; Kodet, R.; Sedlacek, Z. SomaticTP53mutation mosaicism in a patient with Li-Fraumeni syndrome. Am. J. Med. Genet. Part A 2009, 149, 206–211. [Google Scholar] [CrossRef]
- Behjati, S.; Maschietto, M.; Williams, R.D.; Side, L.; Hubank, M.; West, R.; Pearson, K.; Sebire, N.J.; Tarpey, P.; Futreal, A.; et al. A Pathogenic Mosaic TP53 Mutation in Two Germ Layers Detected by Next Generation Sequencing. PLoS ONE 2014, 9, e96531. [Google Scholar] [CrossRef]
- Trubicka, J.; Filipek, I.; Iwanowski, P.; Rydzanicz, M.; Grajkowska, W.; Piekutowska-Abramczuk, D.; Chrzanowska, K.H.; Karkucińska-Więckowska, A.; Iwanicka-Pronicka, K.; Pronicki, M.; et al. Constitutional mosaicism of a de novo TP53 mutation in a patient with bilateral choroid plexus carcinoma. Cancer Genet. 2017, 217, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Perfumo, C.; Bisio, A.; Ciribilli, Y.; Menichini, P.; Russo, D.; Umbach, D.M.; Resnick, M.A.; Inga, A.; Fronza, G. Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol. Cancer Res. 2011, 9, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Zerdoumi, Y.; Aury-Landas, J.; Bonaïti-Pellié, C.; Derambure, C.; Sesboüé, R.; Renaux-Petel, M.; Frebourg, T.; Bougeard, G.; Flaman, J.-M. Drastic Effect of GermlineTP53Missense Mutations in Li-Fraumeni Patients. Hum. Mutat. 2013, 34, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef]
- Amadou, A.; Achatz, M.I.; Hainaut, P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers. Curr. Opin. Oncol. 2018, 30, 23–29. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, 941–947. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Allan Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Storer, N.Y.; Zon, L.I. Zebrafish Models of p53 Functions. Cold Spring Harb. Perspect. Biol. 2010, 2, 1123. [Google Scholar] [CrossRef]
- Kleihues, P.; Schäuble, B.; Hausen, A.Z.; Estève, J.; Ohgaki, H. Tumors associated with p53 germline mutations: A synopsis of 91 families. Am. J. Pathol. 1997, 150, 1–13. [Google Scholar]
- Orr, B.A.; Clay, M.R.; Pinto, E.M.; Kesserwan, C. An update on the central nervous system manifestations of Li–Fraumeni syndrome. Acta Neuropathol. 2019, 139, 669–687. [Google Scholar] [CrossRef]
- Northcott, P.A.; Dubuc, A.M.; Pfister, S.; Taylor, M. Molecular subgroups of medulloblastoma. Expert Rev. Neurother. 2012, 12, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.W.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Meeteren, A.S.-V.; Van Vuurden, D.G.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Langer, A.; Klaar, S.; Eyfjord, J.; Lidereau, R.; Bièche, I.; Varley, J.; Bignon, Y.; Winqvist, R.; Jukkola-Vuorinen, A.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.; Chin, Y.M.; Nakamura, Y.; Low, S.-K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Gender | Tumor (Age) | TP53 Variant | Allele Fraction in Blood | Allele Fraction in Other Tissues | LOH |
|---|---|---|---|---|---|---|
| Patient 2 | M | MB (9) | c.742=/C>T p.(Arg248=/Trp) | 26% | 26% (BS); 42–77% (T) | yes |
| Prochazkova et al., 2008 [29] | F | ACA (1); OS (5) | c.844=/C>T p.(Arg282=/Trp) | 33% * | 33% * (U); 50% * (BS) | N/A |
| Behjati et al., 2014 [30] | M | MFB (0, 7); sarcoma (0, 9); NB (1, 3) | c.743=/G>A p.(Arg248=/Gln) | 14% | 86–97% (T) | yes |
| Trubicka et al., 2017 [31] | M | CPC (1, 5) | c.742=/C>T p.(Arg248=/Trp) | 13% | 33% (BS); 15% (U); 87.5% (T) | yes |
| Renaux-Petel et al., 2018 [4] | M | ACC (0, 3) | c.722=/C>T p.(Ser241=/Phe) | 17% | NA | yes |
| F | ACC (0, 7) | c.548=/C>A p.(Ser183=/ *) | 17% | NA | yes | |
| F | ACC (1) | c.75-10_81=/dup p.(Glu28=/Cysfs * 22) | 4% | NA | N/A | |
| M | CPC (2) | c.742=/C>T p.(Arg248=/Trp) | 14% | NA | N/A | |
| M | Atypical CPP (0, 5) | c.818=/G>A p.(Arg273=/His) | 19% | NA | N/A | |
| F | BBC (27, 34) | c.1024=/C>T p.(Arg342=/ *) | 17% | NA | N/A | |
| F | OS (12); BBC (35, 35) | c.375+1=/G>A p.(=/?) | 7% | NA | yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzollini, J.; Schiavello, E.; Buttarelli, F.R.; Clerici, C.A.; Tizzoni, L.; Vecchi, G.D.; Capra, F.; Pisati, F.; Biassoni, V.; Runza, L.; et al. Pre- and Post-Zygotic TP53 De Novo Mutations in SHH-Medulloblastoma. Cancers 2020, 12, 2503. https://doi.org/10.3390/cancers12092503
Azzollini J, Schiavello E, Buttarelli FR, Clerici CA, Tizzoni L, Vecchi GD, Capra F, Pisati F, Biassoni V, Runza L, et al. Pre- and Post-Zygotic TP53 De Novo Mutations in SHH-Medulloblastoma. Cancers. 2020; 12(9):2503. https://doi.org/10.3390/cancers12092503
Chicago/Turabian StyleAzzollini, Jacopo, Elisabetta Schiavello, Francesca Romana Buttarelli, Carlo Alfredo Clerici, Laura Tizzoni, Giovanna De Vecchi, Fabio Capra, Federica Pisati, Veronica Biassoni, Letterio Runza, and et al. 2020. "Pre- and Post-Zygotic TP53 De Novo Mutations in SHH-Medulloblastoma" Cancers 12, no. 9: 2503. https://doi.org/10.3390/cancers12092503
APA StyleAzzollini, J., Schiavello, E., Buttarelli, F. R., Clerici, C. A., Tizzoni, L., Vecchi, G. D., Capra, F., Pisati, F., Biassoni, V., Runza, L., Carrabba, G., Giangaspero, F., Massimino, M., Pensotti, V., & Manoukian, S. (2020). Pre- and Post-Zygotic TP53 De Novo Mutations in SHH-Medulloblastoma. Cancers, 12(9), 2503. https://doi.org/10.3390/cancers12092503