Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Clinical Features
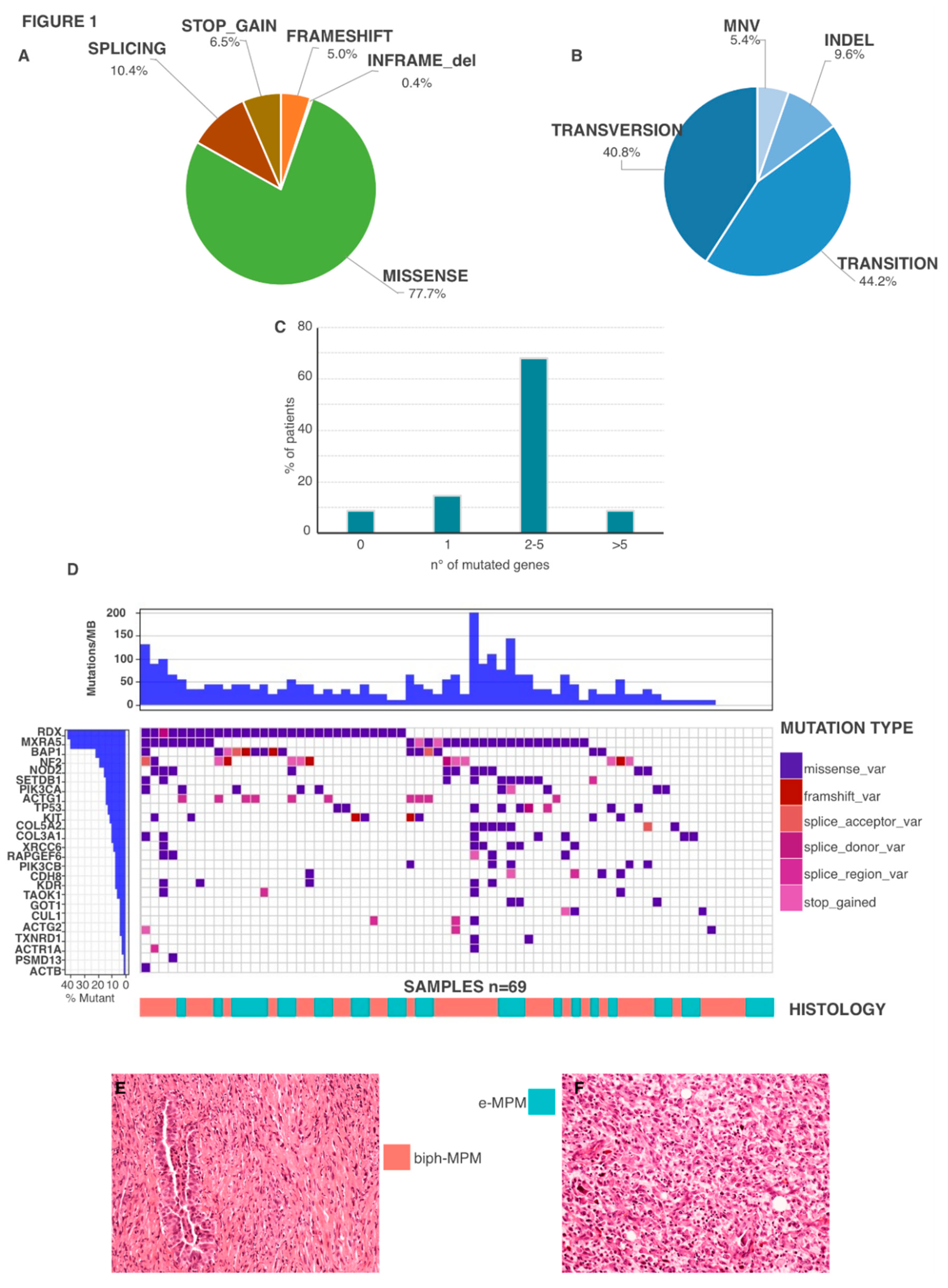
2.2. Genetic Features
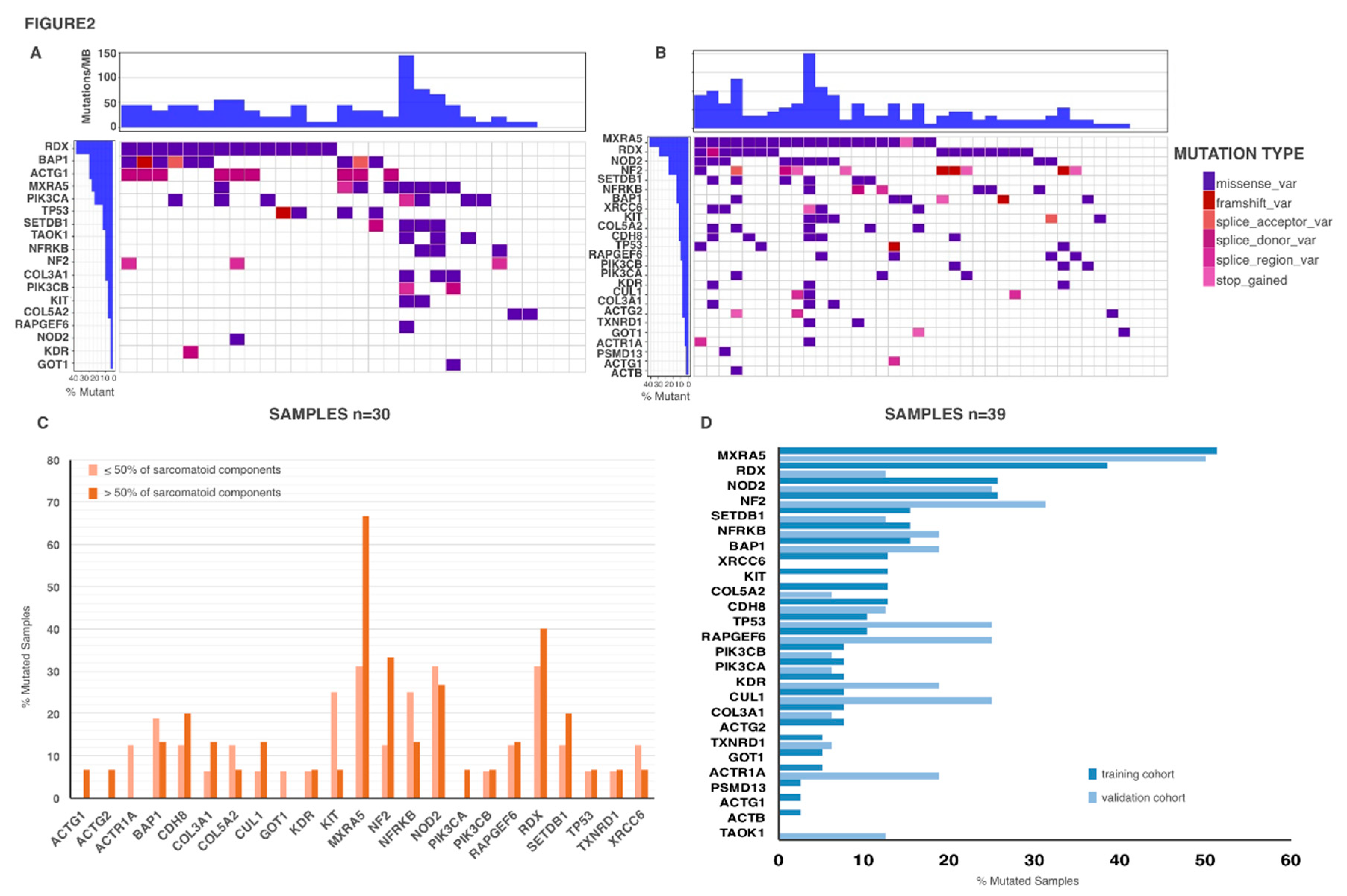
2.3. Genetic Mutations in Biph-MPM
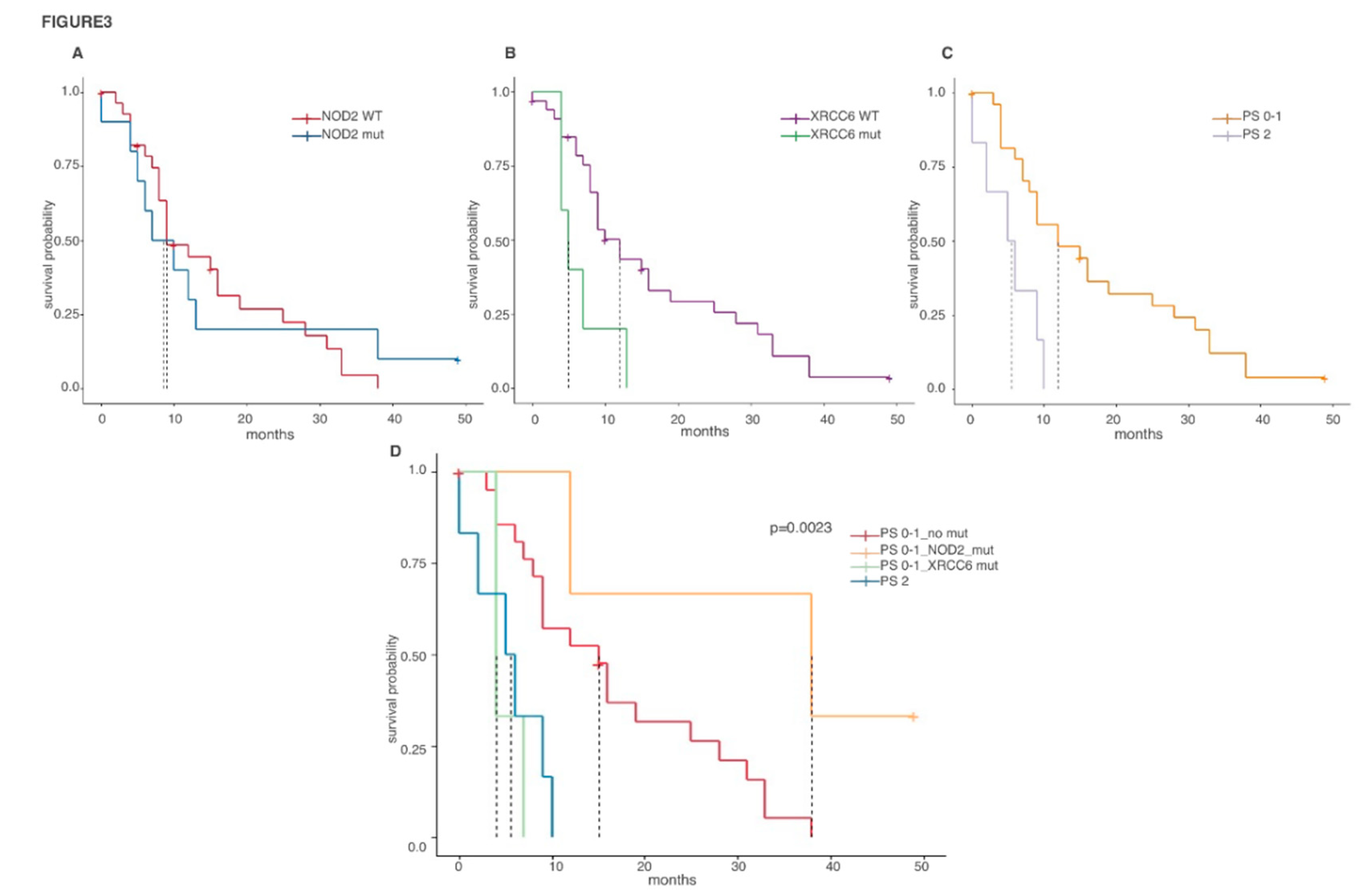
2.4. Prognostic Value of Biph-MPM-Associated Genetic Mutations
3. Discussion
4. Materials and Methods
4.1. Patients Selection
4.2. Next Generation Sequencing
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef]
- Bianchi, C.; Bianchi, T. Malignant mesothelioma: Global incidence and relationship with asbestos. Ind. Health 2007, 45, 379–387. [Google Scholar] [CrossRef]
- Huang, S.X.; Jaurand, M.C.; Kamp, D.W.; Whysner, J.; Hei, T.K. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J. Toxicol. Environ. Health B Crit. Rev. 2011, 14, 179–245. [Google Scholar] [CrossRef]
- Frank, A.L.; Joshi, T.K. The global spread of asbestos. Ann. Glob. Health 2014, 80, 257–262. [Google Scholar] [CrossRef]
- Bueno, R. Multimodality treatments in the management of malignant pleural mesothelioma: An update. Hematol. Oncol. Clin. N. Am. 2005, 19, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Christoph, D.C.; Eberhardt, W.E. Systemic treatment of malignant pleural mesothelioma: New agents in clinical trials raise hope of relevant improvements. Curr. Opin. Oncol. 2014, 26, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Galateau-Salle, F.; Churg, A.; Roggli, V.; Travis, W.D.; World Health Organization Committee for Tumors of the Pleura. The 2015 World Health Organization classification of tumors of the pleura: Advances since the 2004 classification. J. Thorac. Oncol. 2016, 11, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to the 2015 World Health Organization classification of tumors of the lung, pleura, thymus, and heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef]
- Galateau Salle, F.; Le Stang, N.; Nicholson, A.G.; Pissaloux, D.; Churg, A.; Klebe, S.; Roggli, V.L.; Tazelaar, H.D.; Vignaud, J.M.; Attanoos, R.; et al. New insights on diagnostic reproducibility of biphasic mesotheliomas: A multi-institutional evaluation by the international mesothelioma panel from the MESOPATH reference center. J. Thorac. Oncol. 2018, 13, 1189–1203. [Google Scholar] [CrossRef]
- Balduyck, B.; Trousse, D.; Nakas, A.; Martin-Ucar, A.E.; Edwards, J.; Waller, D.A. Therapeutic surgery for nonepithelioid malignant pleural mesothelioma: Is it really worthwhile? Ann. Thorac. Surg. 2010, 89, 907–911. [Google Scholar] [CrossRef]
- Meyerhoff, R.R.; Yang, C.F.; Speicher, P.J.; Gulack, B.C.; Hartwig, M.G.; D’Amico, T.A.; Harpole, D.H.; Berry, M.F. Impact of mesothelioma histologic subtype on outcomes in the Surveillance, Epidemiology, and End Results database. J. Surg. Res. 2015, 196, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Pass, H.I.; Giroux, D.; Kennedy, C.; Ruffini, E.; Cangir, A.K.; Rice, D.; Asamura, H.; Waller, D.; Edwards, J.; Weder, W.; et al. Supplementary prognostic variables for pleural mesothelioma: A report from the IASLC staging committee. J. Thorac. Oncol. 2014, 9, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Lococo, F.; Torricelli, F.; Lang-Lazdunski, L.; Veronesi, G.; Rena, O.; Paci, M.; Casadio, C.; Piana, S.; Novellis, P.; Di Stefano, T.S.; et al. Survival results in biphasic malignant pleural mesothelioma patients: A multicentric analysis. J. Thorac. Cardiovasc. Surg. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hylebos, M.; Van Camp, G.; van Meerbeeck, J.P.; Op de Beeck, K. The Genetic landscape of malignant pleural mesothelioma: Results from massively parallel sequencing. J. Thorac. Oncol. 2016, 11, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- De Reynies, A.; Jaurand, M.C.; Renier, A.; Couchy, G.; Hysi, I.; Elarouci, N.; Galateau-Salle, F.; Copin, M.C.; Hofman, P.; Cazes, A.; et al. Molecular classification of malignant pleural mesothelioma: Identification of a poor prognosis subgroup linked to the epithelial-to-mesenchymal transition. Clin. Cancer Res. 2014, 20, 1323–1334. [Google Scholar] [CrossRef]
- He, X.; Wang, L.; Riedel, H.; Wang, K.; Yang, Y.; Dinu, C.Z.; Rojanasakul, Y. Mesothelin promotes epithelial-to-mesenchymal transition and tumorigenicity of human lung cancer and mesothelioma cells. Mol. Cancer 2017, 16, 63. [Google Scholar] [CrossRef]
- Iwanami, T.; Uramoto, H.; Nakagawa, M.; Shimokawa, H.; Yamada, S.; Kohno, K.; Tanaka, F. Clinical significance of epithelial-mesenchymal transition-associated markers in malignant pleural mesothelioma. Oncology 2014, 86, 109–116. [Google Scholar] [CrossRef]
- Blum, Y.; Jaurand, M.C.; De Reynies, A.; Jean, D. Unraveling the cellular heterogeneity of malignant pleural mesothelioma through a deconvolution approach. Mol. Cell. Oncol. 2019, 6, 1610322. [Google Scholar] [CrossRef]
- Blum, Y.; Meiller, C.; Quetel, L.; Elarouci, N.; Ayadi, M.; Tashtanbaeva, D.; Armenoult, L.; Montagne, F.; Tranchant, R.; Renier, A.; et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat. Commun. 2019, 10, 1333. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Kim, H.K.; Lee, S.; Mendez, P.; Kim, J.W.; Woodard, G.; Yoon, J.H.; Jen, K.Y.; Fang, L.T.; Jones, K.; et al. Whole exome and targeted deep sequencing identify genome-wide allelic loss and frequent SETDB1 mutations in malignant pleural mesotheliomas. Oncotarget 2016, 7, 8321–8331. [Google Scholar] [CrossRef] [PubMed]
- Sugarbaker, D.J.; Richards, W.G.; Gordon, G.J.; Dong, L.; De Rienzo, A.; Maulik, G.; Glickman, J.N.; Chirieac, L.R.; Hartman, M.L.; Taillon, B.E.; et al. Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 3521–3526. [Google Scholar] [CrossRef]
- Branquinho, D.; Freire, P.; Sofia, C. NOD2 mutations and colorectal cancer—Where do we stand? World J. Gastrointest. Surg. 2016, 8, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Li, G.; Li, K.; Xu, Q.; Pan, Z.; Ding, F.; Vedell, P.; Liu, P.; Cui, P.; Hua, X.; et al. Exome sequencing identifies MXRA5 as a novel cancer gene frequently mutated in non-small cell lung carcinoma from Chinese patients. Carcinogenesis 2012, 33, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Poveda, J.; Sanz, A.B.; Fernandez-Fernandez, B.; Carrasco, S.; Ruiz-Ortega, M.; Cannata-Ortiz, P.; Ortiz, A.; Sanchez-Nino, M.D. MXRA5 is a TGF-beta1-regulated human protein with anti-inflammatory and anti-fibrotic properties. J. Cell. Mol. Med. 2017, 21, 154–164. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, X.; Liu, H.; Xiao, H.; Kwapong, W.R.; Mei, J. Matrix-remodeling associated 5 as a novel tissue biomarker predicts poor prognosis in non-small cell lung cancers. Cancer Biomark. 2015, 15, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Yao, L.; Xu, H.W.; Tang, W.T.; Fu, J.H.; Hu, X.F.; Cui, L.; Xu, X.M. Identification of MXRA5 as a novel biomarker in colorectal cancer. Oncol. Lett. 2013, 5, 544–548. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef]
- Siggers, R.H.; Hackam, D.J. The role of innate immune-stimulated epithelial apoptosis during gastrointestinal inflammatory diseases. Cell. Mol. Life Sci. 2011, 68, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 2018, 173, 972–988.e923. [Google Scholar] [CrossRef] [PubMed]
- Kido, M.; Obata, S.; Tanihara, H.; Rochelle, J.M.; Seldin, M.F.; Taketani, S.; Suzuki, S.T. Molecular properties and chromosomal location of cadherin-8. Genomics 1998, 48, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Shinjo, K.; Kondo, Y.; Shen, L.; Toyota, M.; Suzuki, H.; Gao, W.; An, B.; Fujii, M.; Murakami, H.; et al. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res. 2009, 69, 9073–9082. [Google Scholar] [CrossRef]
- Christensen, B.C.; Godleski, J.J.; Marsit, C.J.; Houseman, E.A.; Lopez-Fagundo, C.Y.; Longacker, J.L.; Bueno, R.; Sugarbaker, D.J.; Nelson, H.H.; Kelsey, K.T. Asbestos exposure predicts cell cycle control gene promoter methylation in pleural mesothelioma. Carcinogenesis 2008, 29, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Compton, C.C. The american joint committee on cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef]
- Berzenji, L.; Van Schil, P.E.; Carp, L. The eighth TNM classification for malignant pleural mesothelioma. Transl. Lung Cancer Res. 2018, 7, 543–549. [Google Scholar] [CrossRef]
- Gandolfi, G.; Ragazzi, M.; de Biase, D.; Visani, M.; Zanetti, E.; Torricelli, F.; Sancisi, V.; Gugnoni, M.; Manzotti, G.; Braglia, L.; et al. Genome-wide profiling identifies the THYT1 signature as a distinctive feature of widely metastatic Papillary Thyroid Carcinomas. Oncotarget 2018, 9, 1813–1825. [Google Scholar] [CrossRef]
- Lococo, F.; Gandolfi, G.; Rossi, G.; Pinto, C.; Rapicetta, C.; Cavazza, A.; Cesario, A.; Galeone, C.; Paci, M.; Ciarrocchi, A. Deep sequencing analysis reveals that KRAS mutation is a marker of poor prognosis in patients with pulmonary sarcomatoid carcinoma. J. Thorac. Oncol. 2016, 11, 1282–1292. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Mutations | Total Number of Mutation | Number of Mutated Patients |
|---|---|---|---|
| ACTB | p.Val45Leu | 1 | 1 |
| ACTG1 | c.984+7_984+9delCGA | 10 | 10 |
| ACTG2 | p.Pro113Ser; p.Gln354Ter; c.255+7G>A | 3 | 3 |
| ACTR1A | p.Val47Ile; c.751-7C>T | 2 | 2 |
| BAP1 | p.Cys39PhefsTer33; p.Gln277Ter; p.Tyr241Ter; p.Asp68Gly; p.Leu180Arg; p.His169Arg; p.Asp74Tyr; c.68-1G>A; p.Glu154Asp; p.Asn229Lys; p.Arg300GlyfsTer6; p.Phe170Val; p.His141Tyr; p.His169Gln; p.His141Arg; c.581-2A>G; p.His141Pro | 17 | 15 |
| CDH8 | p.Pro616Gln; p.Pro214His; p.Asn609Lys; p.Ala174Thr; p.Val636Phe | 5 | 5 |
| COL3A1 | p.Pro49Thr; p.Asn1282Lys; p.Pro135His; p.Pro904His; p.Arg1024Gln; p.Pro89Leu; p.Gly633Val | 7 | 6 |
| COL5A2 | p.Gly489Cys; p.Gly804Asp; p.Pro118Thr; p.Gly570Asp; p.Gly1212Asp; p.Gly819Ser; p.Leu198Phe | 7 | 7 |
| CUL1 | p.Leu92Ile; c.316-8G>T; c.2251-7C>T | 3 | 3 |
| GOT1 | p.Asp64Glu; p.Gln105Ter; p.Arg160His | 3 | 3 |
| KDR | p.Arg1118Gln; p.Asp623Tyr; p.Glu540Ter; p.Ser427Tyr; p.Ser1260Asn; p.Leu383Met; c.162-3C>T | 7 | 4 |
| KIT | p.Pro41Thr; p.Glu198Lys; c.926-1G>T; p.Gln190His; p.Phe522Leu; p.Gly297Glu; p.Leu305Phe | 7 | 7 |
| MXRA5 | p.His2434Gln; p.Leu1313Phe; p.Thr468Ile; p.Thr1507Ile; p.Leu1413Ile; p.Arg1232Ile; p.Pro1112Thr; p.Arg541Trp; p.Glu1540Lys; p.Ala1601Thr; p.Ala1541Thr; p.Arg791Cys; p.Pro1171Ser; p.Asp734Asn; p.Ala557Val; p.IleSerProPro824ValSerProArg; p.Glu230Ter; p.Thr2823Ile; p.Ala623Thr; p.Ala1567Thr; p.Gly733Arg; p.His268Asn; p.Gly1597Glu; p.Pro1516Leu; p.Leu2284Met; p.Trp2277Cys; p.Pro1788Leu; p.Ser1199Phe; p.Gly160Val; p.His2498Asn; p.Ala1187Thr; p.Pro1289Ser; p.Ser1147Phe; p.Ala829Val; p.Ala1512Thr; p.Cys275Phe; p.Asp2230Asn; p.Ser1811Phe; p.Pro1259Leu; p.Ala1979Val; p.Pro1898Arg; p.Leu2279Ile; p.Glu2055Ter | 47 | 28 |
| NF2 | p.Thr180IlefsTer29; p.Glu206Ter; p.Ser288Ter; p.Ser267ArgfsTer27; c.1575-1G>T; p.Ile210Thr; p.Glu167Ter; c.114+2T>A; p.Phe22LeufsTer3; p.Glu182Ter; p.Arg57Ter; p.Tyr217_Val219delinsTer; p.Trp74Ter | 13 | 13 |
| NFRKB | p.Pro927Thr; p.Ala989Thr; p.Arg176Trp; c.891+1G>A; p.Pro764Leu; p.Pro527Leu; c.1460-7C>T; p.Thr1134Asn; p.Ala1033Val; p.Asp737Asn; p.His821Gln | 11 | 9 |
| NOD2 | p.Ala781Val; p.Leu701Phe; p.Asp824Asn; p.Arg391His; p.Val259Ile; p.Arg543His; p.Pro80Ser; p.Ala819Thr; p.Ala266Thr; p.Cys354Tyr; p.Pro528GlnfsTer235 | 12 | 11 |
| PIK3CA | p.Arg524Lys; p.Asn372Lys; p.Leu244Ter; p.Ala1066Thr; p.Glu218Ter; p.Glu418Lys; p.Pro421Ser; p.Thr470Ile | 12 | 10 |
| PIK3CB | p.Glu225Lys; p.Gly28Asp; p.Ser521Asn; p.Arg669Ser; p.Gly971Ter; c.2797-4C>T | 6 | 5 |
| PSMD13 | p.Arg156His | 1 | 1 |
| RAPGEF6 | c.4489+5G>T; p.Ser1440Ile; p.Pro633Ser; p.Met993Ile; p.Arg902Met; p.Asn1565Ser; p.Ala1113Thr; p.Lys926AsnfsTer3; p.Thr285Ile | 9 | 5 |
| RDX | p.Lys448Glu; p.Asn112Lys; p.Pro119Thr; c.959+1G>A; p.Lys3AsnfsTer5; p.Gly286Val | 42 | 29 |
| SETDB1 | p.Gly1010Glu; p.Gly891Ser; p.Glu1011Lys; p.His77GlnfsTer3; p.Pro713Gln; p.Ala560Thr; p.Pro950Ser; c.3130-5T>G; p.Ala23Val; p.Ala833Val; p.Thr66Lys | 13 | 10 |
| TAOK1 | p.Cys215Ser; p.Ser717Cys; p.Glu249Lys; c.750-1G>T | 4 | 3 |
| TP53 | p.Thr55AsnfsTer71; p.Ile195Thr; p.Ala161Thr; p.Val173Ala; p.Arg249Ser; p.Val272Leu; p.Cys242Tyr; p.Met160SerfsTer6; p.Ser241_Gly245del; p.Arg213HisfsTer34; p.Pro359Thr | 11 | 8 |
| TXNRD1 | p.Arg501Ile; p.Ala383Val | 2 | 2 |
| XRCC6 | p.Glu333Ter; p.Gly127Glu; p.Pro532Gln; p.Thr541Ile; p.Pro175Thr | 5 | 5 |
| Epithelioid (n = 30) | Biphasic (n = 39) | Total (n = 69) | p Value | |
|---|---|---|---|---|
| ACTB | 1.000 | |||
| WT | 30 (100.0%) | 38 (97.4%) | 68 (98.6%) | |
| Mutated | 0 (0.0%) | 1 (2.6%) | 1 (1.4%) | |
| ACTG1 | 0.002 | |||
| WT | 21 (70.0%) | 38 (97.4%) | 59 (85.5%) | |
| Mutated | 9 (30.0%) | 1 (2.6%) | 10 (14.5%) | |
| ACTG2 | 0.252 | |||
| WT | 30 (100.0%) | 36 (92.3%) | 66 (95.7%) | |
| Mutated | 0 (0.0%) | 3 (7.7%) | 3 (4.3%) | |
| ACTR1A | 0.501 | |||
| WT | 30 (100.0%) | 37 (94.9%) | 67 (97.1%) | |
| Mutated | 0 (0.0%) | 2 (5.1%) | 2 (2.9%) | |
| BAP1 | 0.238 | |||
| WT | 21 (70.0%) | 33 (84.6%) | 54 (78.3%) | |
| Mutated | 9 (30.0%) | 6 (15.4%) | 15 (21.7%) | |
| CDH8 | 0.064 | |||
| WT | 30 (100.0%) | 34 (87.2%) | 64 (92.8%) | |
| Mutated | 0 (0.0%) | 5 (12.8%) | 5 (7.2%) | |
| COL3A1 | 1.000 | |||
| WT | 27 (90.0%) | 36 (92.3%) | 63 (91.3%) | |
| Mutated | 3 (10.0%) | 3 (7.7%) | 6 (8.7%) | |
| COL5A2 | 0.690 | |||
| WT | 28 (93.3%) | 34 (87.2%) | 62 (89.9%) | |
| Mutated | 2 (6.7%) | 5 (12.8%) | 7 (10.1%) | |
| CUL1 | 0.252 | |||
| WT | 30 (100.0%) | 36 (92.3%) | 66 (95.7%) | |
| Mutated | 0 (0.0%) | 3 (7.7%) | 3 (4.3%) | |
| GOT1 | 1.000 | |||
| WT | 29 (96.7%) | 37 (94.9%) | 66 (95.7%) | |
| Mutated | 1 (3.3%) | 2 (5.1%) | 3 (4.3%) | |
| KDR | 0.627 | |||
| WT | 29 (96.7%) | 36 (92.3%) | 65 (94.2%) | |
| Mutated | 1 (3.3%) | 3 (7.7%) | 4 (5.8%) | |
| KIT | 0.690 | |||
| WT | 28 (93.3%) | 34 (87.2%) | 62 (89.9%) | |
| Mutated | 2 (6.7%) | 5 (12.8%) | 7 (10.1%) | |
| MXRA5 | 0.050 | |||
| WT | 22 (73.3%) | 19 (48.7%) | 41 (59.4%) | |
| Mutated | 8 (26.7%) | 20 (51.3%) | 28 (40.6%) | |
| NF2 | 0.128 | |||
| WT | 27 (90.0%) | 29 (74.4%) | 56 (81.2%) | |
| Mutated | 3 (10.0%) | 10 (25.6%) | 13 (18.8%) | |
| NFRKB | 0.722 | |||
| WT | 27 (90.0%) | 33 (84.6%) | 60 (87.0%) | |
| Mutated | 3 (10.0%) | 6 (15.4%) | 9 (13.0%) | |
| NOD2 | 0.018 | |||
| WT | 29 (96.7%) | 29 (74.4%) | 58 (84.1%) | |
| Mutated | 1 (3.3%) | 10 (25.6%) | 11 (15.9%) | |
| PIK3CA | 0.090 | |||
| WT | 23 (76.7%) | 36 (92.3%) | 59 (85.5%) | |
| Mutated | 7 (23.3%) | 3 (7.7%) | 10 (14.5%) | |
| PIK3CB | 1.000 | |||
| WT | 28 (93.3%) | 36 (92.3%) | 64 (92.8%) | |
| Mutated | 2 (6.7%) | 3 (7.7%) | 5 (7.2%) | |
| PSMD13 | 1.000 | |||
| WT | 30 (100.0%) | 38 (97.4%) | 68 (98.6%) | |
| Mutated | 0 (0.0%) | 1 (2.6%) | 1 (1.4%) | |
| RAPGEF6 | 0.379 | |||
| WT | 29 (96.7%) | 35 (89.7%) | 64 (92.8%) | |
| Mutated | 1 (3.3%) | 4 (10.3%) | 5 (7.2%) | |
| RDX | 0.624 | |||
| WT | 16 (53.3%) | 24 (61.5%) | 40 (58.0%) | |
| Mutated | 14 (46.7%) | 15 (38.5%) | 29 (42.0%) | |
| SETDB1 | 1.000 | |||
| WT | 26 (86.7%) | 33 (84.6%) | 59 (85.5%) | |
| Mutated | 4 (13.3%) | 6 (15.4%) | 10 (14.5%) | |
| TAOK1 | 0.077 | |||
| WT | 27 (90.0%) | 39 (100.0%) | 66 (95.7%) | |
| Mutated | 3 (10.0%) | 0 (0.0%) | 3 (4.3%) | |
| TP53 | 0.720 | |||
| WT | 26 (86.7%) | 35 (89.7%) | 61 (88.4%) | |
| Mutated | 4 (13.3%) | 4 (10.3%) | 8 (11.6%) | |
| TXNRD1 | 0.501 | |||
| WT | 30 (100.0%) | 37 (94.9%) | 67 (97.1%) | |
| Mutated | 0 (0.0%) | 2 (5.1%) | 2 (2.9%) | |
| XRCC6 | 0.064 | |||
| WT | 30 (100.0%) | 34 (87.2%) | 64 (92.8%) | |
| Mutated | 0 (0.0%) | 5 (12.8%) | 5 (7.2%) | |
| mutated genes/patient | 0.331 | |||
| Mean (SD) | 3.282 (2.625) | 2.567 (1.736) | 2.971 (2.294) |
| Univariate Analysis | Multivariate Analysis | |||||
|---|---|---|---|---|---|---|
| HR | C.I. 95% | p Value | HR | C.I. 95% | p Value | |
| MXRA5 | 0.95 | 0.47–1.91 | 0.884 | 1.76 | 0.63–4.92 | 0.279 |
| NOD2 | 0.93 | 0.42–2.05 | 0.852 | 0.21 | 0.06–0.79 | 0.021 |
| CDH8 | 2.2 | 0.74–6.53 | 0.156 | 2.15 | 0.26–17.8 | 0.478 |
| XRCC6 | 1.2 | 0.51–2.30 | 0.022 | 8.16 | 1.16–57.2 | 0.035 |
| Surgery | 0.43 | 0.20–0.95 | 0.036 | 0.85 | 0.28–2.58 | 0.775 |
| Performance Status | 3.89 | 1.44–10.49 | 0.007 | 4.45 | 1.04–19.0 | 0.043 |
| TNM stage 1 vs. TNM stage 2-3-4 | 1.32 | 0.67–2.62 | 0.423 | 1.27 | 0.52–3.13 | 0.602 |
| FEV1% (30–80 vs. >80%) | 0.59 | 0.25–1.37 | 0.223 | 0.35 | 0.11–1.13 | 0.078 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torricelli, F.; Lococo, F.; Di Stefano, T.S.; Lorenzini, E.; Piana, S.; Valli, R.; Rena, O.; Veronesi, G.; Billè, A.; Ciarrocchi, A. Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma. Cancers 2020, 12, 2454. https://doi.org/10.3390/cancers12092454
Torricelli F, Lococo F, Di Stefano TS, Lorenzini E, Piana S, Valli R, Rena O, Veronesi G, Billè A, Ciarrocchi A. Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma. Cancers. 2020; 12(9):2454. https://doi.org/10.3390/cancers12092454
Chicago/Turabian StyleTorricelli, Federica, Filippo Lococo, Teresa Severina Di Stefano, Eugenia Lorenzini, Simonetta Piana, Riccardo Valli, Ottavio Rena, Giulia Veronesi, Andrea Billè, and Alessia Ciarrocchi. 2020. "Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma" Cancers 12, no. 9: 2454. https://doi.org/10.3390/cancers12092454
APA StyleTorricelli, F., Lococo, F., Di Stefano, T. S., Lorenzini, E., Piana, S., Valli, R., Rena, O., Veronesi, G., Billè, A., & Ciarrocchi, A. (2020). Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma. Cancers, 12(9), 2454. https://doi.org/10.3390/cancers12092454