Anti-Glucosylsphingosine Autoimmunity, JAK2V617F-Dependent Interleukin-1β and JAK2V617F-Independent Cytokines in Myeloproliferative Neoplasms

 , , and
, , and 
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Description of Patients
2.2. Levels of Inflammation-Linked Cytokines in MPN Patients
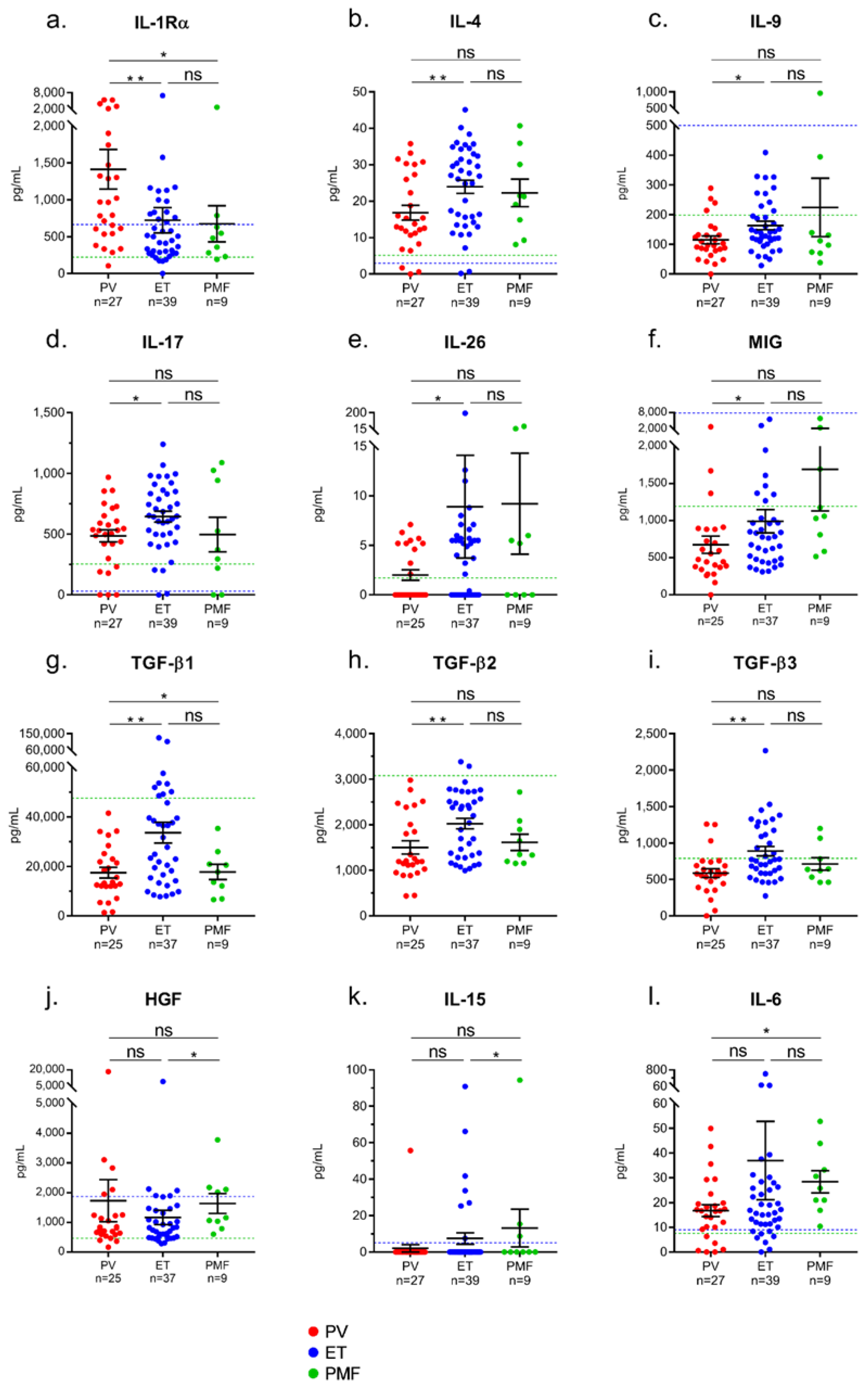
2.3. Differences in Inflammation-Linked Cytokine Levels According to MPN Phenotype
2.4. Correlations between Cytokine Levels and Blood Cell Counts in MPN Patients
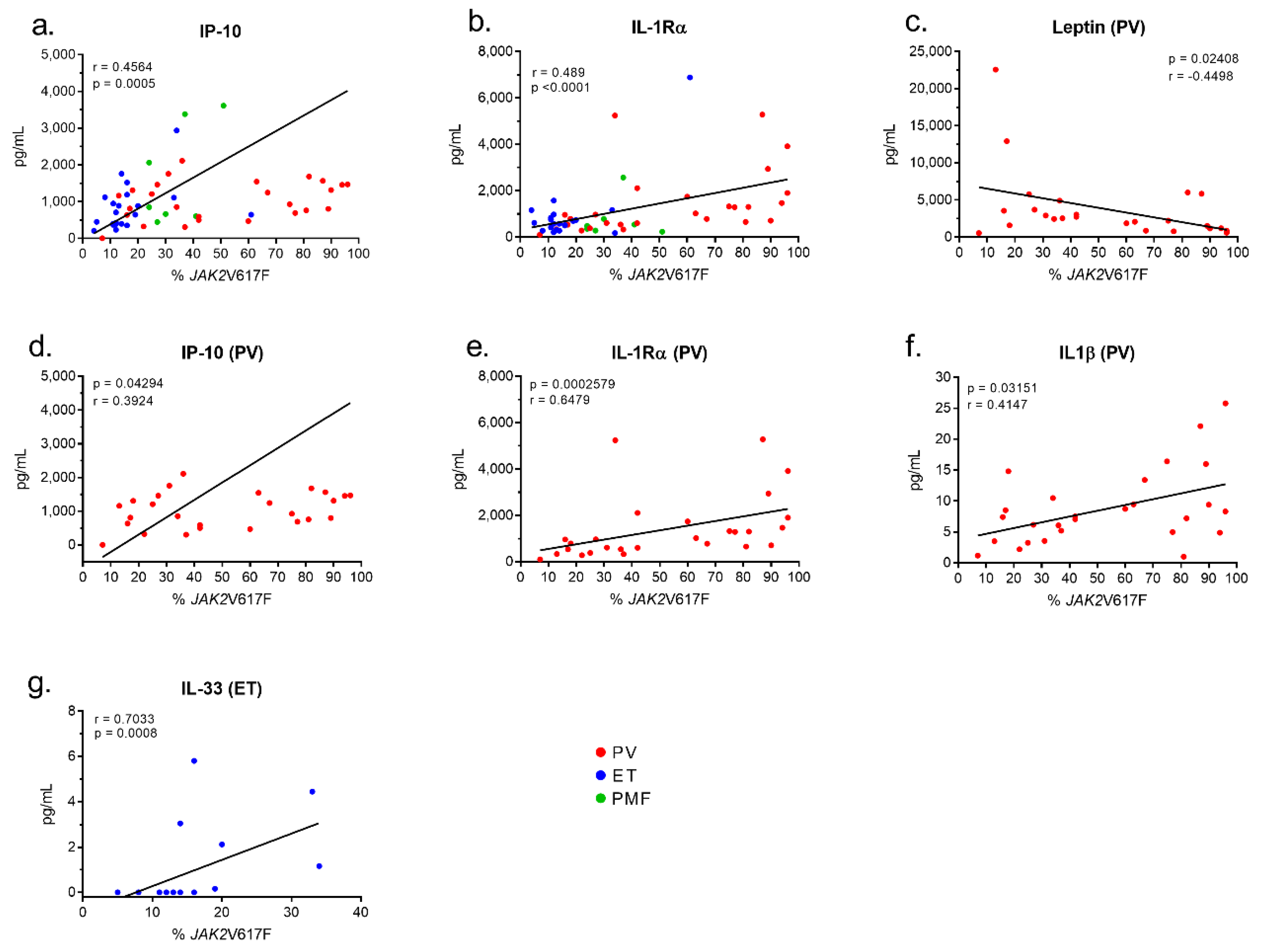
2.5. Inflammation-Linked Cytokines or Receptors Linked to the JAK2V617F Mutation
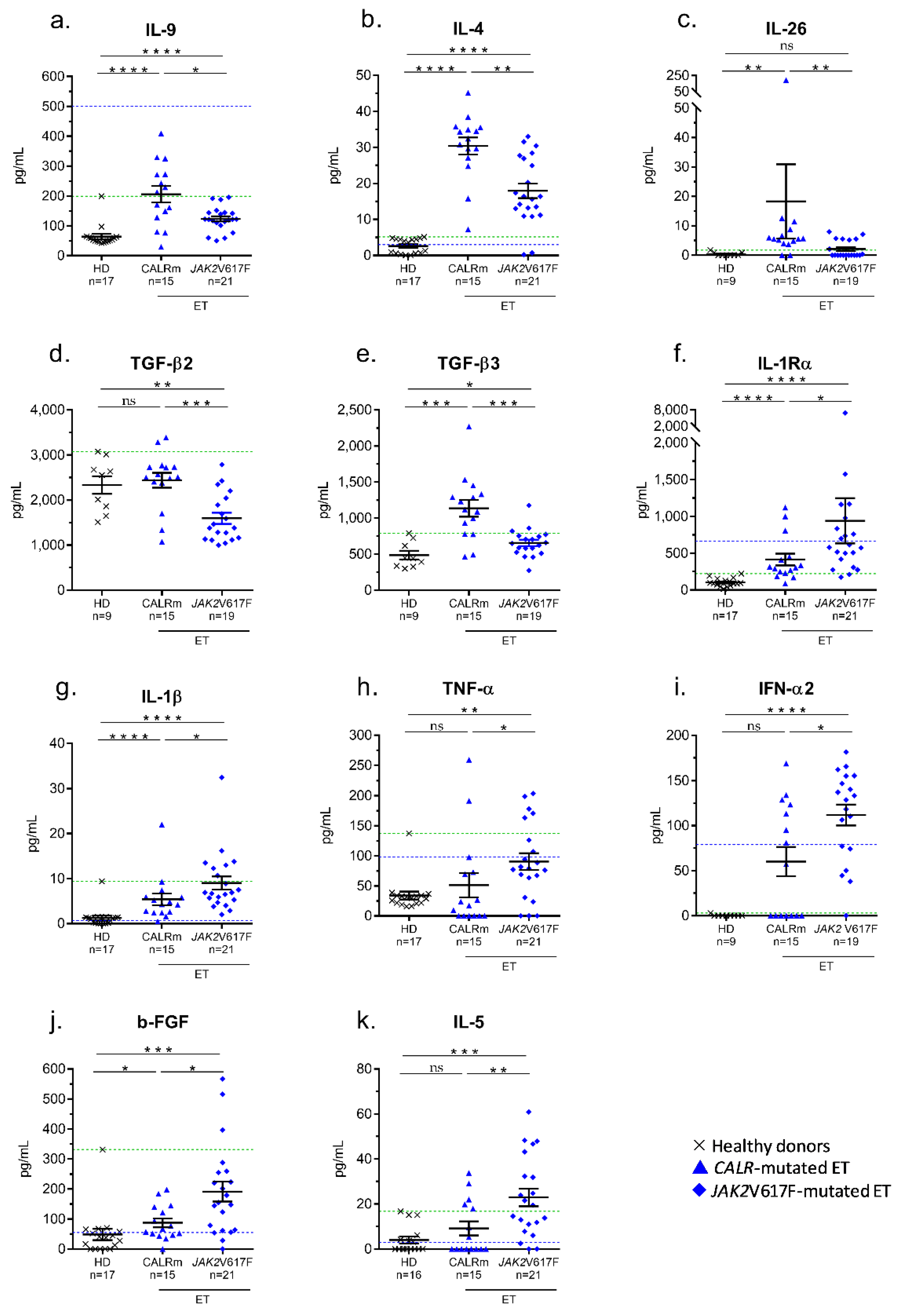
2.6. Inflammation-Linked Cytokines Associated with CALR Exon 9 Mutated-ET
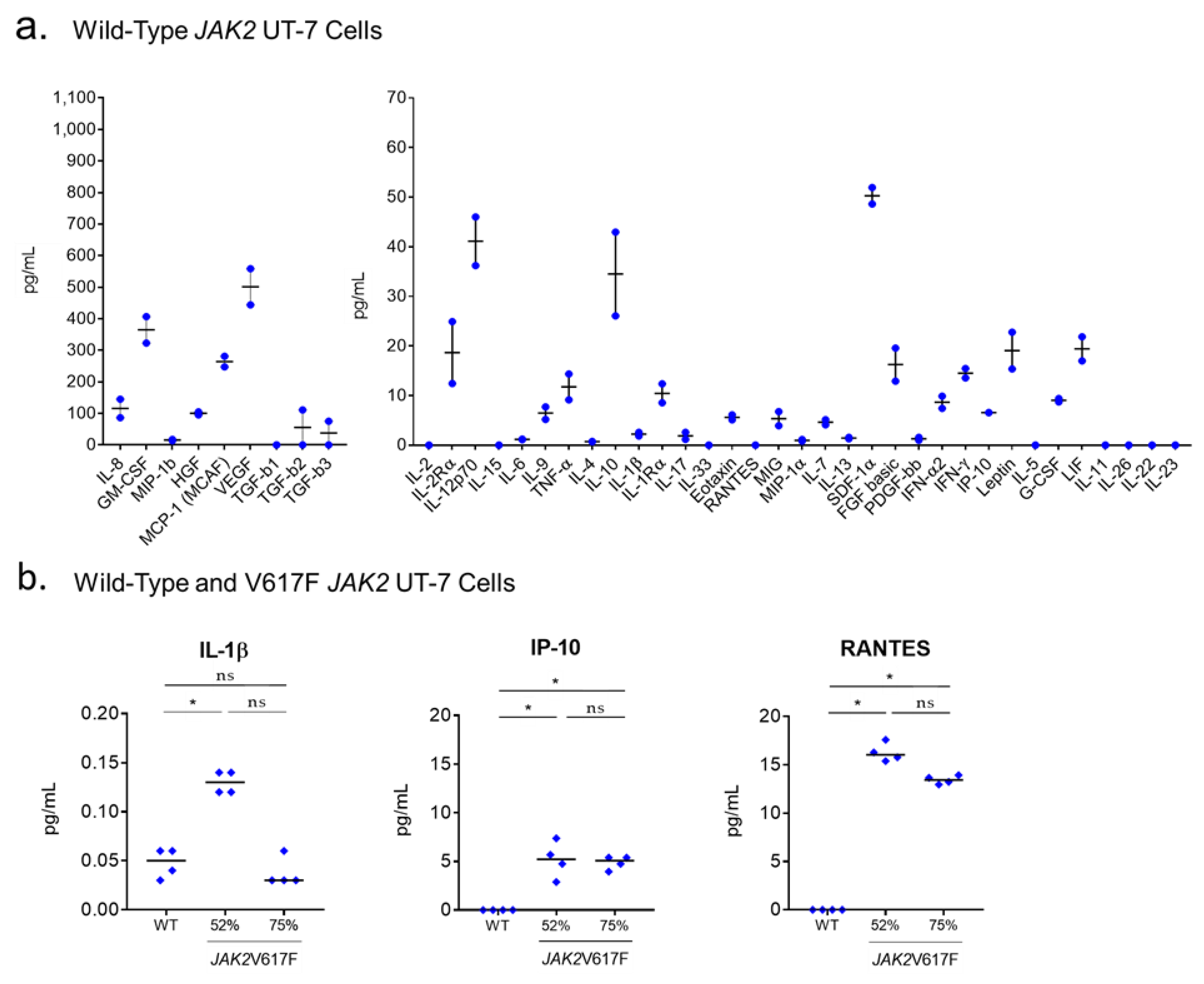
2.7. Production of Cytokines by UT-7 Cells Expressing the JAK2V617F or CALR Mutants
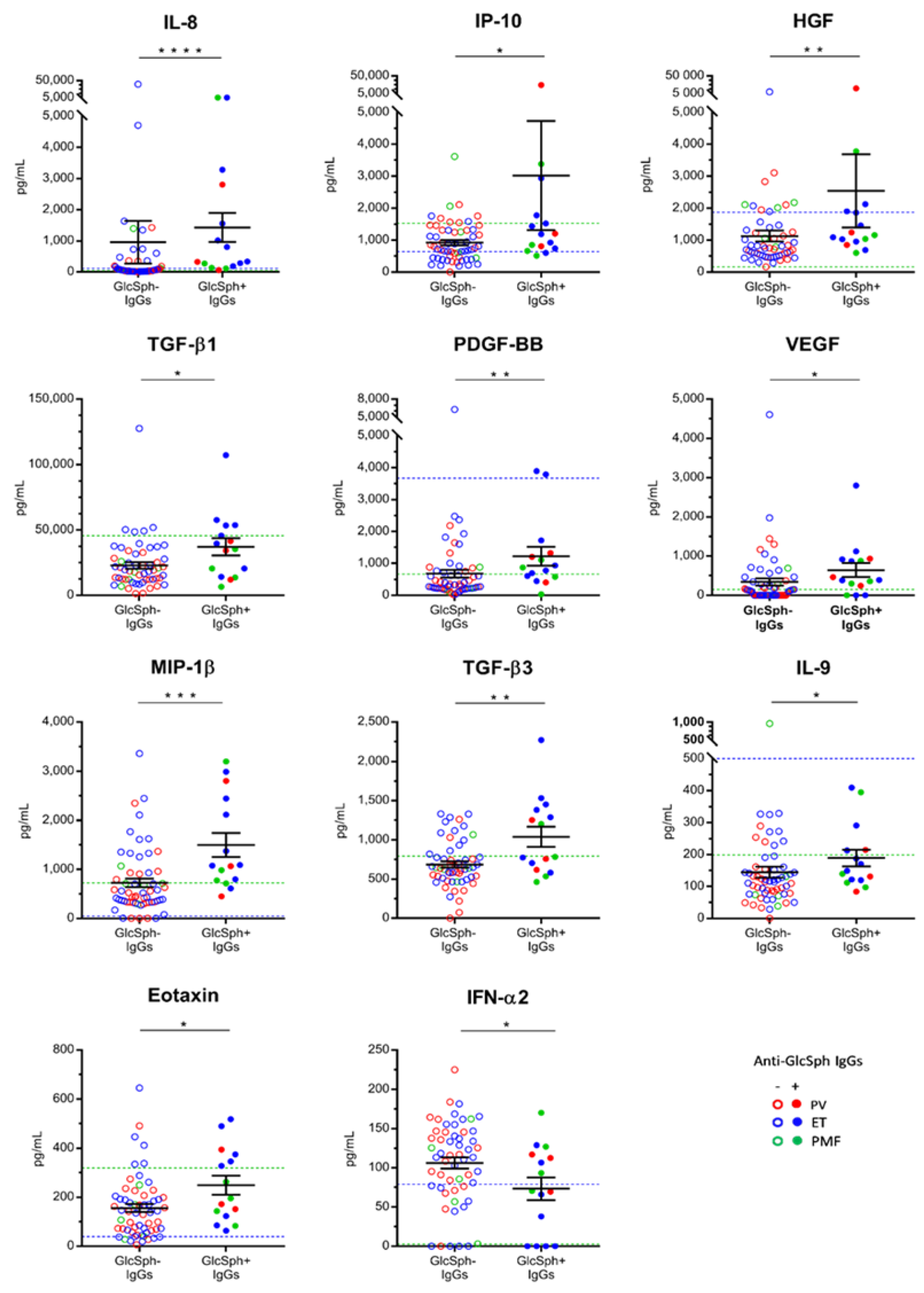
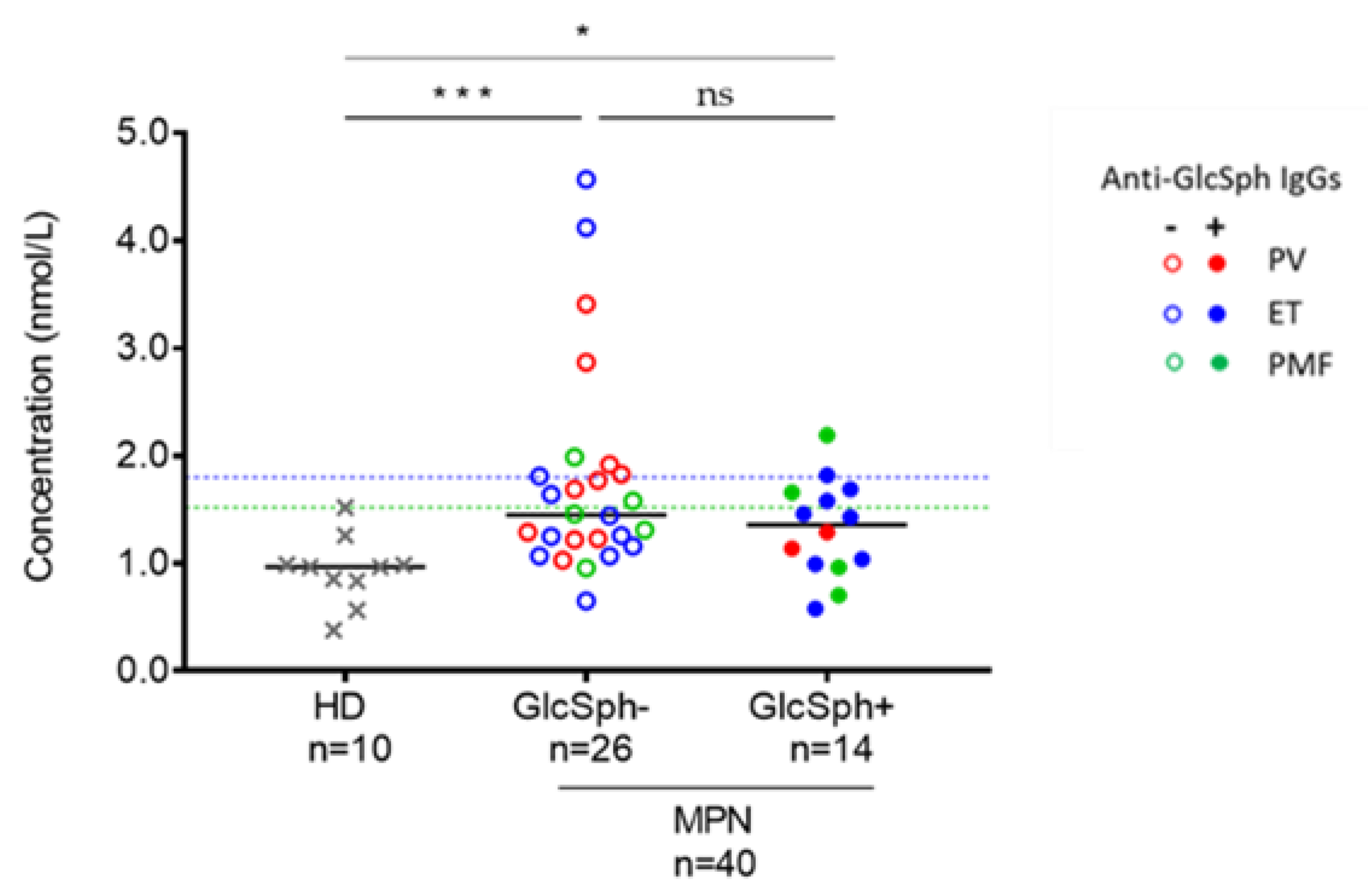
2.8. Auto-Immunity Against Glucosylsphingosine (GlcSph) in MPN Patients
2.8.1. Detection of GlcSph-Reactive IgGs in the Serum of MPN Patients
2.8.2. Characteristics of MPN Patients with GlcSph-Reactive IgGs
2.8.3. Analysis of GlcSph Levels in the Serum of MPN Patients
3. Discussions
4. Materials and Methods
4.1. Patients
4.2. UT-7 Cells
4.3. Generation of JAK2V617F+ UT-7 Cells Using the CRISPR/CAS-9 Technology
4.4. Quantification of JAK2V617F in UT-7 Clones
4.5. Quantification of Inflammation-Linked Cytokines
4.6. Detection of GlcSph-Reactive Igs
4.7. GlcSph Quantification
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Klampfl, T.; Them, N.C.C.; Berg, T.; Vladimer, G.I.; Bagienski, K.; Milanesi, C.; Casetti, I.C.; Sant’Antonio, E.; Ferretti, V.V.; Schischlik, F.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with non-mutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Staerk, J.; Constantinescu, S.N. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. JAK-STAT 2012, 1, 184–190. [Google Scholar] [CrossRef]
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of myeloproliferative neoplasms. Exp. Hematol. 2015, 43, 599–608. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef]
- Lambert, J.R.; Everington, T.; Linch, D.C.; Gale, R.E. In essential thrombocythemia, multiple JAK2-V617F clones are present in most mutant-positive patients: A new disease paradigm. Blood 2009, 114, 3018–3023. [Google Scholar] [CrossRef]
- Schaub, F.X.; Jäger, R.; Looser, R.; Hao-Shen, H.; Hermouet, S.; Girodon, F.; Tichelli, A.; Gisslinger, H.; Kralovics, R.; Skoda, R.C. Clonal analysis of deletions on chromosome 20q and JAK2-V617F in MPD suggests that del20q acts independently and is not one of the predisposing mutations for JAK2-V617F. Blood 2009, 113, 2022–2027. [Google Scholar] [CrossRef]
- Hermouet, S. Pathogenesis of myeloproliferative neoplasms: More than mutations. Exp. Hematol. 2015, 43, 993–994. [Google Scholar] [CrossRef] [PubMed]
- Hermouet, S.; Bigot-Corbel, E.; Gardie, B. Pathogenesis of Myeloproliferative Neoplasms: Role and Mechanisms of Chronic Inflammation. Mediat. Inflamm. 2015, 2015, 145293. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.L.; Dueck, A.C.; Scherber, R.M.; Mesa, R.A. Impact of Inflammation on Myeloproliferative Neoplasm Symptom Development. Mediat. Inflamm. 2015, 2015, 284706. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Perspectives on the impact of JAK-inhibitor therapy upon inflammation-mediated comorbidities in myelofibrosis and related neoplasms. Expert Rev. Hematol. 2014, 7, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Kantarjian, H.; Kadia, T.; Cortes, J.; Borthakur, G.; Newberry, K.; Garcia-Manero, G.; Ravandi, F.; Jabbour, E.; Dellasala, S.; et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2014, 15, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Greenfield, G.; McPherson, S.; Mills, K.I.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16, 360. [Google Scholar] [CrossRef]
- Li, B.; Rampal, R.K.; Xiao, Z. Targeted therapies for myeloproliferative neoplasms. Biomark. Res. 2019, 7, 15. [Google Scholar] [CrossRef]
- Hobbs, G.; Rozelle, S.; Mullally, A. The Development and Use of Janus Kinase 2 Inhibitors for the Treatment of Myeloproliferative Neoplasms. Hematol. Clin. 2017, 31, 613–626. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Cassinat, B.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Menot, M.-L.; Massonnet, G.; Dutel, J.-L.; Ghomari, K.; et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon-2a. Blood 2006, 108, 2037–2040. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Chomienne, C.; Fenaux, P. Interferon-α therapy in bcr-abl-negative myeloproliferative neoplasms. Leukemia 2008, 22, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.; McLornan, D.P.; Curto-Garcia, N.; O’Sullivan, J.; Alimam, S.; Keohane, C.; Woodley, C.; Francis, Y.; Kordasti, S.Y.; Radia, D.H.; et al. Essential thrombocythaemia treated with recombinant interferon: ‘real world’ United Kingdom referral centre experience. Br. J. Haematol. 2019, 186, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.K.; Andersen, M.; Knudsen, T.A.; Sajid, Z.; Gudmand-Hoeyer, J.; Dam, M.J.B.; Skov, V.; Kjaer, L.; Ellervik, C.; Larsen, T.S.; et al. Data-driven analysis of JAK2V617F kinetics during interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms. Cancer Med. 2020, 9, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.J.; Bug, G.; Aulitzky, E.W.; Huber, C.; Peschel, C. Inhibition of interleukin-11 by interferon-alpha in human bone marrow stromal cells. Exp. Hematol. 1996, 24, 863–867. [Google Scholar] [PubMed]
- Radaeva, S.; Jaruga, B.; Hong, F.; Kim, W.; Fan, S.; Cai, H.; Strom, S.C.; Liu, Y.; El-Assal, O.; Gao, B. Interferon-α activates multiple STAT signals and down-regulates c-Met in primary human hepatocytes. Gastroenterology 2002, 122, 1020–1034. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Hasselbalch, H.C. Minimal residual disease or cure in MPNs? Rationales and perspectives on combination therapy with interferon-alpha2 and ruxolitinib. Expert Rev. Hematol. 2017, 94, 1–12. [Google Scholar] [CrossRef]
- Mikkelsen, S.U.; Kjær, L.; Bjørn, M.E.; Knudsen, T.A.; Sørensen, A.L.; Andersen, C.B.L.; Bjerrum, O.W.; Brochmann, N.; El Fassi, D.; Kruse, T.A.; et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018, 7, 3571–3581. [Google Scholar] [CrossRef]
- Czech, J.; Cordua, S.; Weinbergerova, B.; Baumeister, J.; Crepcia, A.; Han, L.; Maié, T.; Costa, I.G.; Denecke, B.; Maurer, A.; et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia 2019, 33, 995–1010. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Landgren, O.; Samuelsson, J.; Bjorkholm, M.; Goldin, L.R. Autoimmunity and the risk of auto-immune neoplasms. Haematologica 2010, 7, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.M.; Bak, M.; Sørensen, A.L.; Zwisler, A.-D.; Ellervik, C.; Larsen, M.K.; Hasselbalch, H.C.; Tolstrup, J.S. Smoking is associated with increased risk of myeloproliferative neoplasms: A general population-based cohort study. Cancer Med. 2018, 7, 5796–5802. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, N.A.; Kjaergaard, A.D.; Pedersen, K.M.; Sørensen, A.L.; Bak, M.; Larsen, M.K.; Nordestgaard, B.G.; Bojesen, S.E.; Çolak, Y.; Skov, V.; et al. Smoking, blood cells and myeloproliferative neoplasms: Meta-analysis and Mendelian randomization of 2·3 million people. Br. J. Haematol. 2020, 189, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Branagan, A.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef]
- Nair, S.; Sng, J.; Boddupalli, C.S.; Seckinger, A.; Chesi, M.; Fulciniti, M.; Zhang, L.; Rauniyar, N.; López, M.; Neparidze, N.; et al. Antigen-mediated regulation in monoclonal gammopathies and myeloma. JCI Insight 2018, 3, e98259. [Google Scholar] [CrossRef]
- Nair, S.; Bar, N.; Xu, M.L.; Dhodapkar, M.; Mistry, P. Glucosylsphingosine but not Saposin C, is the target antigen in Gaucher disease-associated gammopathy. Mol. Genet. Metab. 2020, 129, 286–291. [Google Scholar] [CrossRef]
- Bosseboeuf, A.; Seillier, C.; Mennesson, N.; Allain-Maillet, S.; Fourny, M.; Tallet, A.; Piver, E.; Lehours, P.; Mégraud, F.; Berthelot, L.; et al. Analysis of the Targets and Glycosylation of Monoclonal IgAs From MGUS and Myeloma Patients. Front. Immunol. 2020, 11, 854. [Google Scholar] [CrossRef]
- Bosseboeuf, A.; Mennesson, N.; Allain-Maillet, S.; Tallet, A.; Piver, E.; Decaux, O.; Moreau, C.; Moreau, P.; Lehours, P.; Megraud, F.; et al. Characteristics of MGUS and Multiple Myeloma According to the Target of Monoclonal Immunoglobulins, Glucosylsphingosine, or Epstein-Barr Virus EBNA-1. Cancers 2020, 12, 1254. [Google Scholar] [CrossRef]
- Ayto, R.; Hughes, D. Gaucher Disease and Myeloma. Crit. Rev. Oncog. 2013, 18, 247–268. [Google Scholar] [CrossRef]
- Mistry, P.; Taddei, T.H.; Dahl, S.V.; Rosenbloom, B.E. Gaucher disease and malignancy: A model for cancer pathogenesis in an inborn error of metabolism. Crit. Rev. Oncog. 2013, 18, 235–246. [Google Scholar] [CrossRef]
- Linari, S.; Castaman, G. Hematological manifestations and complications of Gaucher disease. Expert Rev. Hematol. 2015, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; McKay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Wątek, M.; Piktel, E.; Wollny, T.; Durnaś, B.; Fiedoruk, K.; Lech-Marańda, E.; Bucki, R. Defective Sphingolipids Metabolism and Tumor Associated Macrophages as the Possible Links Between Gaucher Disease and Blood Cancer Development. Int. J. Mol. Sci. 2019, 20, 843. [Google Scholar] [CrossRef] [PubMed]
- Lippert, E.; Boissinot, M.; Kralovics, R.; Girodon, F.; Dobo, I.; Praloran, V.; Boiret-Dupré, N.; Skoda, R.C.; Hermouet, S. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood 2006, 108, 1865–1867. [Google Scholar] [CrossRef]
- Bosseboeuf, A.; Allain, S.; Mennesson, N.; Tallet, A.; Rossi, C.; Garderet, L.; Caillot, D.; Moreau, P.; Piver, E.; Girodon, F.; et al. Pro-inflammatory state in MGUS and Myeloma is characterized by low sialylation of pathogen-specific and other monoclonal and polyclonal immunoglobulin G. Front. Immunol. 2017, 8, 1347. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Pietra, D.; Della Porta, M.G.; Boveri, E.; Pascutto, C.; Vanelli, L.; Arcaini, L.; Burcheri, S.; Malcovati, L.; et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood 2006, 107, 3676–3682. [Google Scholar] [CrossRef]
- Rotunno, G.; Mannarelli, C.; Guglielmelli, P.; Pacilli, A.; Pancrazzi, A.; Pieri, L.; Fanelli, T.; Bosi, A.; Vannucchi, A.M.; on behalf of the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014, 123, 1552–1555. [Google Scholar] [CrossRef]
- Lippert, E.; Girodon, F.; Hammond, E.; Carillo, S.; Richard, C.; Fehse, B.; Hermans, M.; James, I.; Jelinek, J.; Marzac, C.; et al. Concordance of Assays Designed for the Quantitation of JAK2 1849G>T (V617F): A Multi-Centre Study. Blood 2007, 110, 2529. [Google Scholar] [CrossRef]
- Jovanovic, J.V.; Ivey, A.; Vannucchi, A.M.; Lippert, E.; Oppliger Leibundgut, E.; Cassinat, B.; Pallisgaard, N.; Maroc, N.; Hermouet, S.; Nickless, G.; et al. Establishing Optimal Quantitative-Polymerase Chain Reaction Assays for Routine Diagnosis and Tracking Minimal Residual Disease in JAK2-V617F Associated Myeloid Neoplasms: A Joint European LeukemiaNet/MPN&MPNr-EuroNet (COST Action BM0902) Study. Leukemia 2013, 27, 2032–2039. [Google Scholar]
- Mansier, O.; Prouzet-Mauléon, V.; Jégou, G.; Barroso, K.; Raymundo, D.P.; Chauveau, A.; Dumas, P.-Y.; Lagarde, V.; Turcq, B.; Pasquet, J.M.; et al. The Expression of Myeloproliferative Neoplasm-Associated Calreticulin Variants Depends on the Functionality of ER-Associated Degradation. Cancers 2019, 11, 1921. [Google Scholar] [CrossRef]
- Duo, C.-C.; Gong, F.-Y.; He, X.-Y.; Li, Y.-M.; Wang, J.; Zhang, J.; Gao, X.-M. Soluble Calreticulin Induces Tumor Necrosis Factor-α (TNF-α) and Interleukin (IL)-6 Production by Macrophages through Mitogen-Activated Protein Kinase (MAPK) and NFκB Signaling Pathways. Int. J. Mol. Sci. 2014, 15, 2916–2928. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma cytokines in polycythemia vera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am. J. Hematol. 2012, 87, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene 2010, 30, 990–1001. [Google Scholar] [CrossRef]
- Nooij, F.; Van Der Sluijs-Gelling, A.; Der Zijde, C.J.-V.; Van Tol, M.; Haas, H.; Radl, J. Immunoblotting techniques for the detection of low level homogeneous immunoglobulin components in serum. J. Immunol. Methods 1990, 134, 273–281. [Google Scholar] [CrossRef]
- Braun, W.; Abraham, R. Modified diffusion blotting for rapid and efficient protein transfer with PhastSystem. Electrophoresis 1989, 10, 249–253. [Google Scholar] [CrossRef]
- Pettazzoni, M.; Froissart, R.; Pagan, C.; Vanier, M.T.; Ruet, S.; Latour, P.; Guffon, N.; Fouilhoux, A.; Germain, M.P.; Levade, T.; et al. LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: A novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease. PLoS ONE 2017, 12, e0181700. [Google Scholar] [CrossRef]
- Xiao, M.; Wang, Y.; Tao, C.; Wang, Z.; Yang, J.; Chen, Z.; Zou, Z.; Li, M.; Liu, A.; Jia, C.; et al. Osteoblasts support megakaryopoiesis through production of interleukin-9. Blood 2017, 129, 3196–3209. [Google Scholar] [CrossRef]
- Feng, Y.; Yu, M.; Zhu, F.; Zhang, S.; Ding, P.; Wang, M. IL-9 Promotes the Development of Deep Venous Thrombosis by Facilitating Platelet Function. Thromb. Haemost. 2018, 118, 1885–1894. [Google Scholar] [CrossRef]
- Ding, P.; Zhang, S.; Yu, M.; Feng, Y.; Long, Q.; Yang, H.; Li, J.; Wang, M. IL-17A promotes the formation of deep vein thrombosis in a mouse model. Int. Immunopharmacol. 2018, 57, 132–138. [Google Scholar] [CrossRef]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Mondet, J.; Hussein, K.; Mossuz, P. Circulating Cytokine Levels as Markers of Inflammation in Philadelphia Negative Myeloproliferative Neoplasms: Diagnostic and Prognostic Interest. Mediat. Inflamm. 2015, 2015, 670580. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; Latocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Vilaine, M.; Hermouet, S. The hepatocyte growth factor (HGF)/c-MET axis: A neglected target in the treatment of chronic myeloproliferative neoplasms? Cancers 2014, 6, 1631–1669. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.; Hansen, N.; Hao-Shen, H.; Dirnhofer, S.; Tata, N.R.; Skoda, R.C. IL-1β Secreted from Mutant Cells Carrying JAK2-V617Ffavors Early Clonal Expansion and Promotes MPN Disease Initiation and Progression. Blood 2019, 134 (Suppl. 1), 307. [Google Scholar] [CrossRef]
- Øbro, N.F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.S.; Rao, T.N.; Karow, A.; Riedel, L.M.; Harris, O.B.; Baxter, E.J.; et al. Longitudinal Cytokine Profiling Identifies GRO-α and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. HemaSphere 2020, 4, e371. [Google Scholar] [CrossRef]
- Olcaydu, D.; Harutyunyan, A.S.; Jäger, R.; Berg, T.; Gisslinger, B.; Pabinger, I.; Gisslinger, H.; Kralovics, R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat. Genet. 2009, 41, 450–454. [Google Scholar] [CrossRef]
- Hermouet, S.; Vilaine, M. The JAK2 46/1 haplotype: A marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica 2011, 96, 1575–1579. [Google Scholar] [CrossRef]
- Jager, R.; Harutyunyan, A.S.; Rumi, E.; Pietra, D.; Berg, T.; Olcaydu, D.; Houlston, R.S.; Cazzola, M.; Kralovics, R. Common germline variation at the TERT locus contributes to familial clustering of myeloproliferative neoplasms. Am. J. Hematol. 2014, 89, 1107–1110. [Google Scholar] [CrossRef]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef]
- Saliba, J.; Saint-Martin, C.; Di Stefano, A.; Lenglet, G.; Marty, C.; Keren, B.; Pasquier, F.; Della Valle, V.; Secardin, L.; Leroy, G.; et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat. Genet. 2015, 47, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar] [PubMed]
- Dinarello, C.A.; Van Der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Arriero, M.D.M.; Villatoro, A. Interleukin-1β as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017, 31, 306–317. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients | PV | ET | PMF | |
|---|---|---|---|---|
| Number | 75 | 27 | 39 | 9 |
| Sex, M/F (male%) | 38/37 (50.7%) | 16/11 (59.3%) | 15/24 (41.0%) | 7/2 (77.8%) |
| Age (Yr) | ||||
| Median | 67.0 | 69.0 | 73.0 | 62.0 |
| (Range) | (33–95) | (33–93) | (37–95) | (50–86) |
| %JAK2V617F | ||||
| Median | 16.0 | 42.0 | 5.0 | 27.0 |
| (Range) | (0–96) | (7–96) | (0–61 *) | (0–51) |
| Blood Counts | ||||
| Hematocrit (L/L) | ||||
| Median | 46.3 | 53.4 | 44.1 | 36.2 |
| (Range) | (25.1–70.0) | (43.1 **–70.0) | (35.3–63.3 ***) | (25.1–41.0) |
| Hemoglobin (g/dL) | ||||
| Median | 14.8 | 17.4 | 14.4 | 11.0 |
| (Range) | (7.9–22.4) | (13.8 **–22.4) | (10.4–20.0 ***) | (7.9–13.0) |
| Leukocytes (×109/L) | ||||
| Median | 8.7 | 9.7 | 8.3 | 11.3 |
| (Range) | (2.8–41.0) | (2.8–37.8) | (4.5–41.0) | (5.0–27.5) |
| Platelets (×109/L) | ||||
| Median | 527.5 | 377 | 644 | 165 |
| (Range) | (47–2300) | (89–851) | (191–2300) | (47–586) |
| Molecules (pg/mL) | HD n = 17 | All MPNs n = 75 | PV n = 27 | ET n = 39 | PMF n = 9 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Med. | (Range) | Med. | (Range) | Med. | Range | Med. | (Range) | Med. | (Range) | |
| Markers of poor prognosis in MPNs | ||||||||||
| sIL-2Rα | 69.8 | (0.0–162) | 295.9 | (30.3–1438) | 287.2 | (39.5–1129) | 275.6 | (30.3–1438) | 382.6 | (205–601) |
| IL-8 | 15.4 | (3.0–43.1) | 80.2 | (2.4–40,808) | 54.1 | (2.4–2804) | 85.5 | (14.4–40,808) | 120.4 | (13.8–5093) |
| IL-12p70 | 13.4 | (1.4–277) | 78.0 | (0.0–789) | 76.3 | (0.0–378) | 86.2 | (0.0–789) | 81.8 | (0.0–303) |
| IL-15 | 0.0 | (0.0–0.0) | 0.0 | (0.0–94.4) | 0.0 | (0.0–55.7) | 0.0 | (0.0–90.9) | 0.0 | (0.0–94.4) |
| GM-CSF | 0.0 | (0.0–2.11) | 0.0 | (0.0–382) | 0.0 | (0.0–156) | 0.0 | (0.0–266) | 0.0 | (0.0–382) |
| MIP-1β | 310.3 | (1.34–727) | 612.7 | (0.0–3362) | 448.1 | (0.0–2802) | 701.6 | (1.7–3362) | 714.2 | (292–3199) |
| Anti-inflammation | ||||||||||
| IL-4 | 3.6 | (0.0–5.1) | 20.2 | (0.0–45.1) | 13.8 | (0.0–35.8) | 26.9 | (0.2–45.1) | 21.3 | (8.1–40.7) |
| IL-10 | 0.0 | (0.0–166) | 15.8 | (0.0–152) | 24.8 | (0.0–103) | 1.5 | (0.0–152) | 22.6 | (0.0–86.3) |
| HGF | 302.6 | (125–471) | 837.4 | (166–18,303) | 815.0 | (165–18,303) | 815.6 | (286–9302) | 1158 | (600–3778) |
| Pro-inflammation | ||||||||||
| TNF-α | 29.7 | (25.7–38.7) | 78.9 | (0.0–294) | 88.4 | (0.0–294) | 69.2 | (0.0–259) | 105.0 | (0.0–191) |
| IL-1β | 1.0 | (0.0–9.4) | 6.2 | (0.5–42.5) | 7.4 | (1.0–25.8) | 5.8 | (0.5–42.5) | 4.8 | (1.3–19.2) |
| IL-1Rα | 96.9 | (13.5–222) | 605.0 | (87.6–6884) | 966.3 | (105.2–5284) | 500.8 | (87.6–6884) | 480.3 | (193.2–2570) |
| IL-2 | 0.0 | (0.0–3.42) | 0.0 | (0.0–152) | 0.0 | (0.0–59.9) | 0.0 | (0.0–115) | 0.0 | (0.0–152) |
| IL-6 | 0.8 | (0.0–7.6) | 16.9 | (0.0–623) | 16.7 | (0.0–49.9) | 15.9 | (0.0–623) | 25.8 | (10.4–52.8) |
| IL-7 | 0.0 | (0.0–10.6) | 31.9 | (0.0–190) | 30.2 | (0.0–89.7) | 33.7 | (0.0–190) | 33.9 | (5.0–69.7) |
| IL-9 | 53.2 | (42.6–198.7) | 122.0 | (0.0–962) | 96.9 | (0.0–289) | 141.6 | (29.0–409) | 112.0 | (38.5–962) |
| IL-11 | 0.0 | (0.0–0.0) | 0.0 | (0.0–0.1) | 0.0 | (0.0–0.1) | 0.0 | (0.0–0.0) | 0.0 | (0.0–0.0) |
| IL-13 | 1.2 | (0.0–6.49) | 5.7 | (0.0–83.6) | 16.1 | (0.0–42.3) | 1.1 | (0.0–65.0) | 0.0 | (0.0–83.6) |
| IL-17 | 124.1 | (0.0–255) | 575.0 | (0.0–1240) | 529.0 | (0.0–968) | 648.2 | (0.0–1240) | 371.3 | (0.0–1089) |
| IL-22 | 0.0 | (0.0–0.0) | 0.0 | (0.0–0.0) | 0.0 | (0.0–0.0) | 0.0 | (0.0–0.0) | 0.0 | (0.0–0.0) |
| IL-23 | 0.0 | (0.0–2.8) | 0.0 | (0.0–6.2) | 0.0 | (0.0–0.0) | 0.0 | (0.0–6.2) | 0.0 | (0.0–0.0) |
| IL-26 | 0.0 | (0.0–1.71) | 3.2 | (0.0–195) | 0.0 | (0.0–7.1) | 4.9 | (0.0–195) | 5.2 | (0.0–46.4) |
| IL-33 | 0.27 | (0.0–3.46) | 0.0 | (0.0–11.1) | 0.0 | (0.0–11.1) | 0.0 | (0.0–5.8) | 0.0 | (0.0–7.6) |
| IFN-α2 | 0.0 | (0.0–2.9) | 110.5 | (0.0–225) | 115.9 | (0.0–225) | 106.6 | (0.0–182) | 93.3 | (3.1–170) |
| IFN-γ | 17.2 | (2.0–34.7) | 143.8 | (0.0–689) | 156.5 | (0.0–690) | 147.0 | (0.0–509) | 129.3 | (0.0–498) |
| IP-10 | 626.2 | (298–1528) | 847.6 | (0.0–26,760) | 1161 | (0.0–26,760) | 721.4 | (189–2935) | 847.6 | (442–3613) |
| Eotaxin | 111.5 | (44.2–319) | 146.5 | (6.1–646) | 141.7 | (6.1–491) | 169.3 | (19.4–646) | 143.4 | (28.0–2623) |
| FGF basic | 38.5 | (0.0–332) | 121.2 | (0.0–567) | 128.6 | (0.0–405) | 121.2 | (0.0–567) | 111.3 | (0.0–332) |
| Leptin | 2206 | (584–10,200) | 2713 | (463–36,254) | 2449 | (552–22,567) | 2.84 | (2840–2816) | 1781 | (733–4489) |
| MCP-1 | 0.0 | (0.0–123) | 0.0 | (0.0–1524) | 0.0 | (0.0–197) | 0.0 | (0.0–1524) | 0.0 | (0.0–1211) |
| MIG | 325.3 | (211–1193) | 699.7 | (0.0–5878) | 477.3 | (0.0–2820) | 777.8 | (312–5605) | 1059 | (516–5878) |
| MIP-1α | 5.0 | (1.0–615) | 13.4 | (0.0–848) | 13.7 | (0.0–848) | 13.2 | (0.0–620) | 10.1 | (0.0–18.2) |
| PDGF-BB | 464 | (34.1–666) | 418.2 | (25.8–6263) | 350.3 | (25.8–2181) | 603.7 | (41.4–6263) | 232.1 | (34.9–1107) |
| RANTES | 13,592 | (3108–25,751) | 12,342 | (724–66,241) | 11,132 | (724–48,946) | 14,943 | (3372–66,241) | 18,805 | (2451–33,031) |
| SDF-1α | 750 | (610–970) | 1093 | (0.0–1769) | 1093 | (0.0–1602) | 1106 | (540–1769) | 1,03 | (360–1416) |
| VEGF | 0.0 | (0.0–144) | 147.6 | (0.0–4605) | 124.3 | (0.0–1443) | 174.7 | (0.0–4605) | 280.0 | (0.0–880) |
| Other hematopoietic growth factors | ||||||||||
| IL-5 | 0.0 | (0.0–16.8) | 13.8 | (0.0–84.6) | 17.8 | (0.0–84.6) | 13.0 | (0.0–60.9) | 18.6 | (0.0–49.3) |
| G-CSF | 12.2 | (0.0–16.8) | 123.1 | (0.0–265) | 123.1 | (0.0–265) | 122.8 | (0.0–212) | 156.6 | (38.7–218) |
| LIF | 0.0 | (0.0–4.52) | 41.5 | (0.0–299) | 56.1 | (0.0–292) | 27.6 | (0.0–299) | 6.4 | (0.0–275) |
| TGF-β1 | 16,765 | (4928–47,560) | 20,530 | (1355–127,571) | 13,639 | (1355–41,498) | 27,802 | (7782–127,571) | 17,741 | (6566–35,311) |
| TGF-β2 | 2554 | (1512–3078) | 1647 | (439–3386) | 1195 | (439–2983) | 2203 | (996–3386) | 1351 | (1154–2721) |
| TGF-β3 | 441.6 | (300–790) | 659.6 | (0.0–2268) | 577.2 | (0.0–1260) | 772.7 | (275–2268) | 638.2 | (462–1201) |
| Molecules | Dg | Neutrophils | Monocytes | Hematocrit | Platelets | Lymphocytes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | r | p Value | n | r | p Value | n | r | p Value | n | r | p Value | n | r | p Value | ||
| IL-1Rα | PV | 24 | 0.800 | <0.0001 | - | - | - | - | - | - | - | - | ||||
| IL-1β | PV | 24 | 0.496 | 0.0137 | - | - | - | - | - | - | - | - | ||||
| IP-10 | PV | 24 | 0.476 | 0.0186 | 25 | 0.576 | 0.0026 | - | - | - | - | - | - | |||
| IL-8 | PMF | - | - | 9 | −0.786 | 0.0279 | - | - | - | - | - | - | ||||
| MIP-1α | PMF | - | - | - | - | - | - | - | - | 9 | −0.810 | 0.0218 | ||||
| MIP-1β | PV | - | - | - | - | - | - | - | - | 24 | 0.440 | 0.0314 | ||||
| PMF | - | - | 9 | −0.738 | 0.0458 | - | - | - | - | 9 | −0.762 | 0.0368 | ||||
| Leptin | PV | 15 M | −0.600 | 0.0223 | - | - | - | - | - | - | - | - | ||||
| PMF | - | - | 8 | −0.786 | 0.0279 | - | - | - | - | - | - | |||||
| IL-2Rα | ET | - | - | - | - | - | - | 38 | 0.613 | 0.0052 | - | - | ||||
| SDF-1α | PV | - | - | - | - | - | - | 24 | 0.605 | 0.0017 | - | - | ||||
| ET | - | - | - | - | - | - | 38 | 0.520 | 0.0012 | - | - | |||||
| IL-2 | ET | - | - | - | - | 38 | −0.519 | 0.0009 | - | - | - | - | ||||
| IL-4 | ET | - | - | - | - | 38 | −0.464 | 0.0034 | - | - | 38 | 0.439 | 0.0065 | |||
| IL-7 | PV | - | - | - | - | - | - | 24 | 0.525 | 0.0070 | - | - | ||||
| IL-9 | PV | - | - | - | - | - | - | - | - | 24 | 0.412 | 0.0454 | ||||
| ET | - | - | - | - | - | - | 38 | 0.420 | 0.0087 | 38 | 0.451 | 0.0050 | ||||
| IL-17 | PV | - | - | - | - | - | - | 24 | 0.508 | 0.0095 | - | - | ||||
| ET | - | - | - | - | - | - | 38 | 0.468 | 0.0030 | - | - | |||||
| IL-26 | ET | - | - | - | - | 38 | −0.464 | 0.0043 | - | - | - | - | ||||
| IL-33 | PV | - | - | - | - | - | - | - | - | 24 | 0.499 | 0.0154 | ||||
| HGF | PV | 24 | 0.501 | 0.0149 | - | - | - | - | - | - | - | - | ||||
| MIG | PV | 24 | 0.462 | 0.0266 | - | - | - | - | 24 | 0.450 | 0.0275 | - | - | |||
| TGF-β1 | ET | - | - | - | - | - | - | 38 | −0.489 | 0.0024 | - | - | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allain-Maillet, S.; Bosseboeuf, A.; Mennesson, N.; Bostoën, M.; Dufeu, L.; Choi, E.H.; Cleyrat, C.; Mansier, O.; Lippert, E.; Le Bris, Y.; et al. Anti-Glucosylsphingosine Autoimmunity, JAK2V617F-Dependent Interleukin-1β and JAK2V617F-Independent Cytokines in Myeloproliferative Neoplasms. Cancers 2020, 12, 2446. https://doi.org/10.3390/cancers12092446
Allain-Maillet S, Bosseboeuf A, Mennesson N, Bostoën M, Dufeu L, Choi EH, Cleyrat C, Mansier O, Lippert E, Le Bris Y, et al. Anti-Glucosylsphingosine Autoimmunity, JAK2V617F-Dependent Interleukin-1β and JAK2V617F-Independent Cytokines in Myeloproliferative Neoplasms. Cancers. 2020; 12(9):2446. https://doi.org/10.3390/cancers12092446
Chicago/Turabian StyleAllain-Maillet, Sophie, Adrien Bosseboeuf, Nicolas Mennesson, Mégane Bostoën, Laura Dufeu, Eun Ho Choi, Cédric Cleyrat, Olivier Mansier, Eric Lippert, Yannick Le Bris, and et al. 2020. "Anti-Glucosylsphingosine Autoimmunity, JAK2V617F-Dependent Interleukin-1β and JAK2V617F-Independent Cytokines in Myeloproliferative Neoplasms" Cancers 12, no. 9: 2446. https://doi.org/10.3390/cancers12092446
APA StyleAllain-Maillet, S., Bosseboeuf, A., Mennesson, N., Bostoën, M., Dufeu, L., Choi, E. H., Cleyrat, C., Mansier, O., Lippert, E., Le Bris, Y., Gombert, J.-M., Girodon, F., Pettazzoni, M., Bigot-Corbel, E., & Hermouet, S. (2020). Anti-Glucosylsphingosine Autoimmunity, JAK2V617F-Dependent Interleukin-1β and JAK2V617F-Independent Cytokines in Myeloproliferative Neoplasms. Cancers, 12(9), 2446. https://doi.org/10.3390/cancers12092446