Systems Biology Approach Identifies Prognostic Signatures of Poor Overall Survival and Guides the Prioritization of Novel BET-CHK1 Combination Therapy for Osteosarcoma

 , , , , , , ,
, , , , , , , 
Abstract
1. Introduction
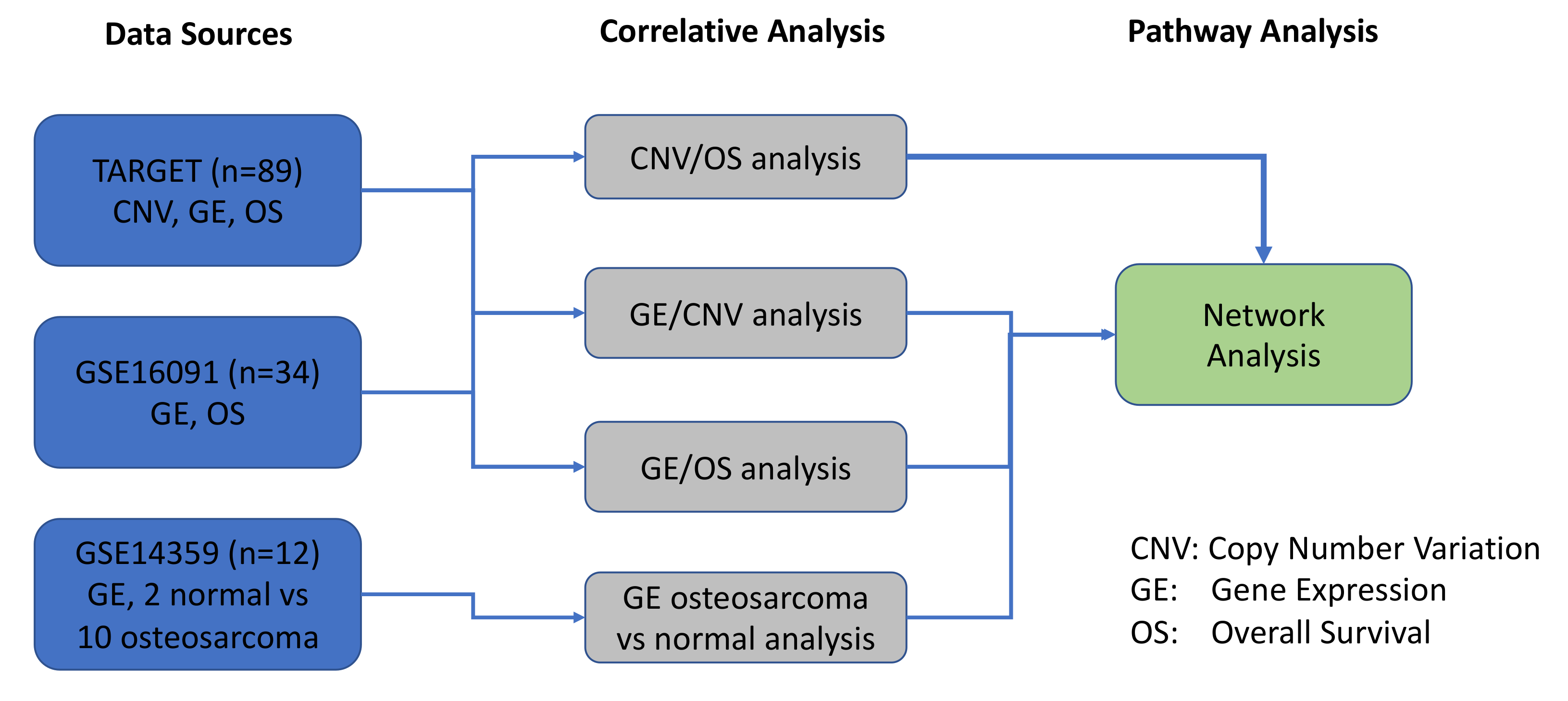
2. Results
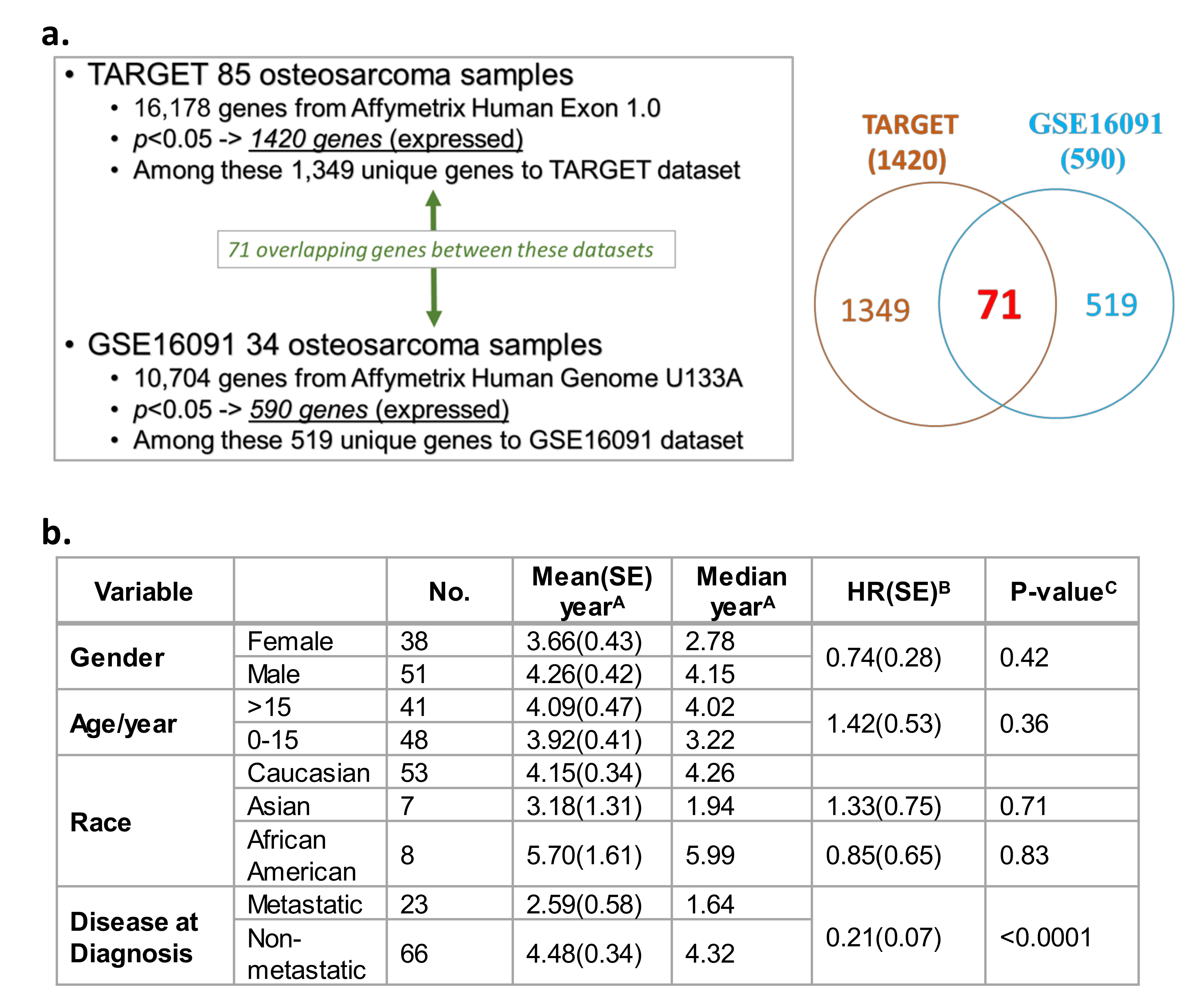
2.1. Significant Correlation between Gene Expression Profiles and Survival in Two Independent OS Datasets
2.2. CNVs Significantly Correlated with the Overall Survival in OS Patients
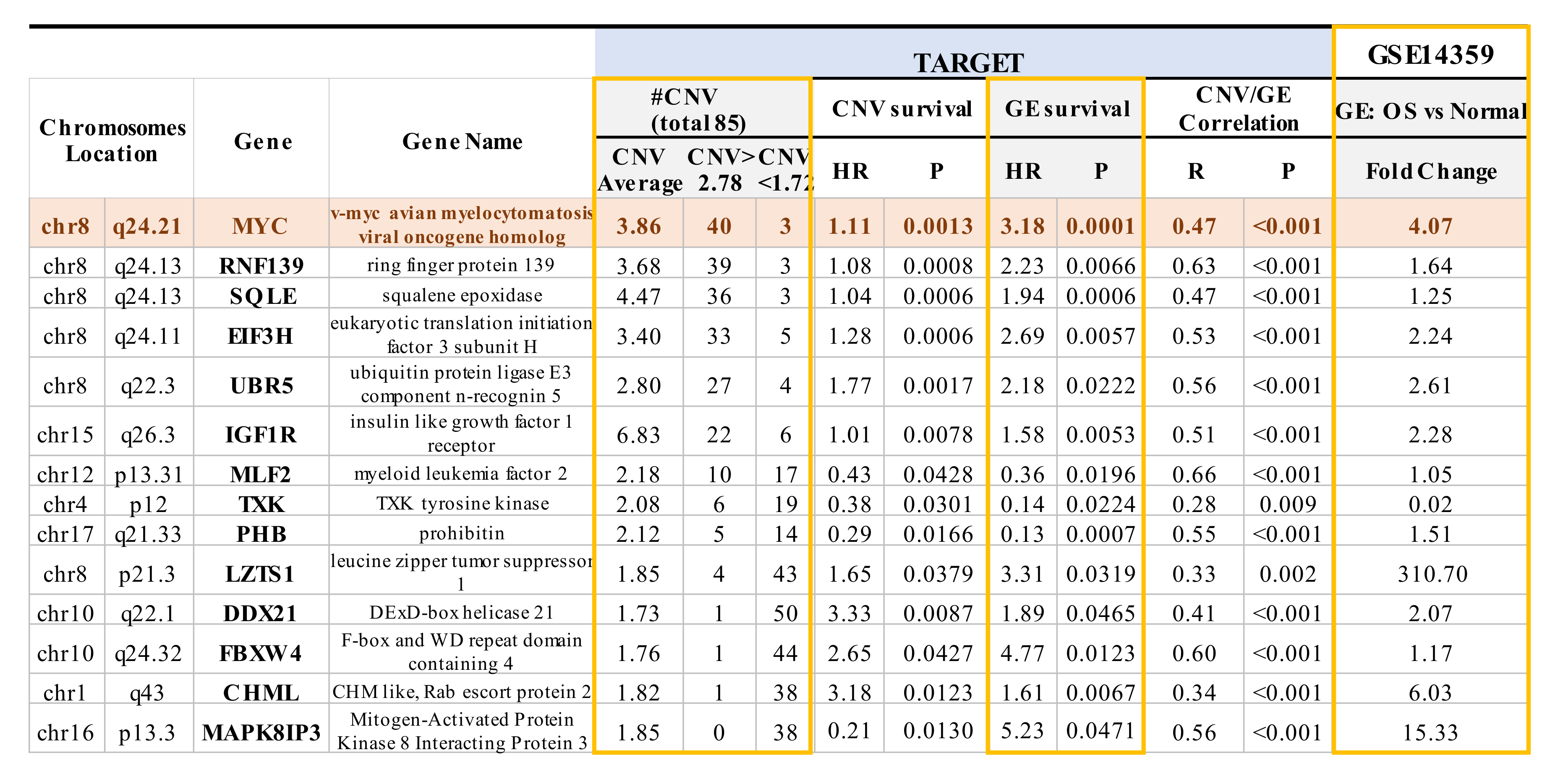
2.3. Mapping of CNVs to Their Corresponding Genes that are Associated with Overall Survival in OS Patients
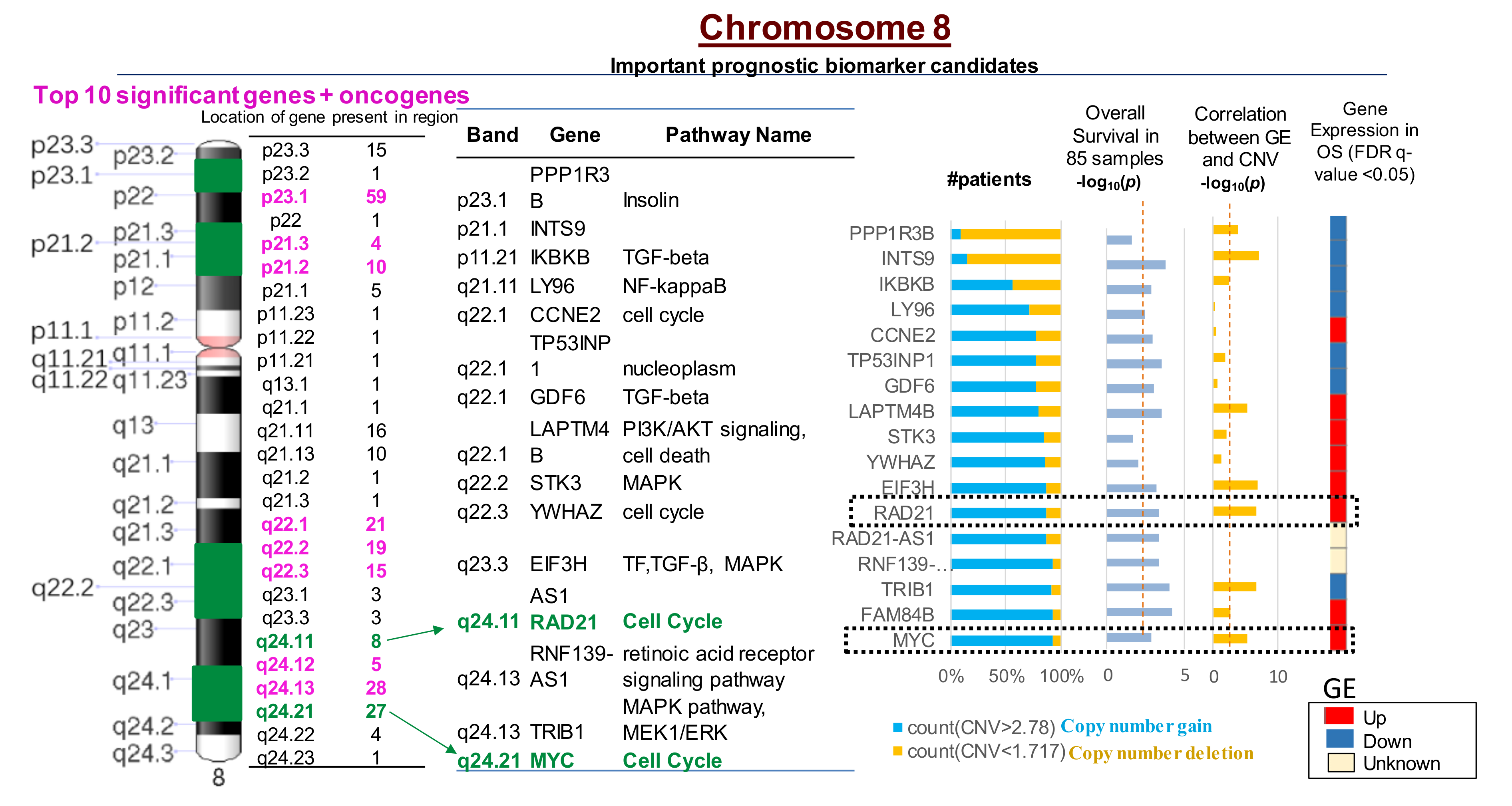
2.4. Multiple CNVs Associated with Increased Gene Expression and Poor Overall Survival in OS Are Present on Chromosome 8
2.5. Ingenuity Pathway Analysis of Genes with CNVs Significantly Associated with Overall Survival in OS Patients
2.6. Prioritization of the MYC-RAD21+ Risk Signature for Investigations in Pre-Clinical Models of Pediatric and AYA OS
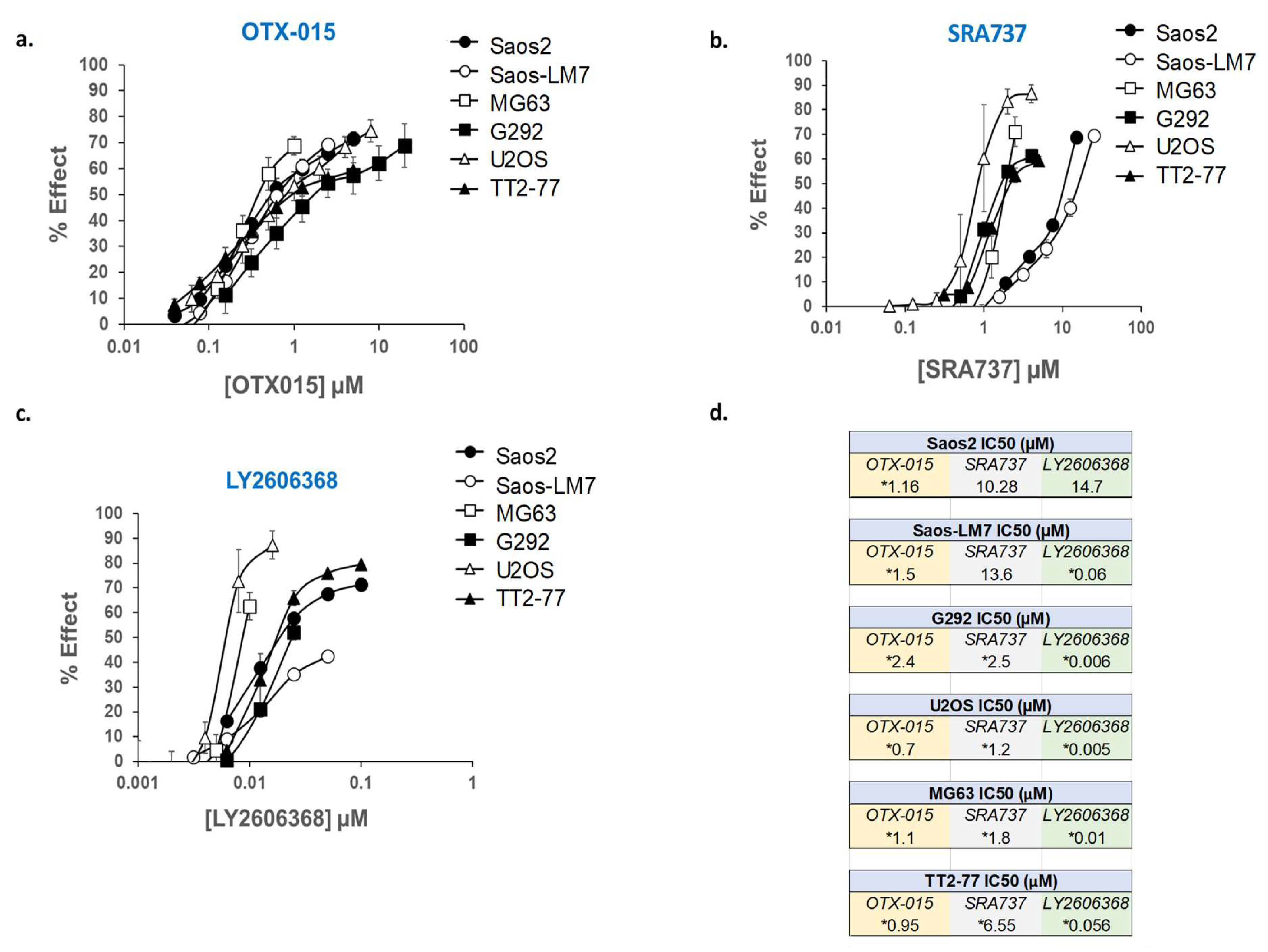
2.7. In Vitro Assessment of Cell Growth in MYC-RAD21+ Pediatric OS Lines Following the Pharmacological Inhibition of BET Proteins and CHK1
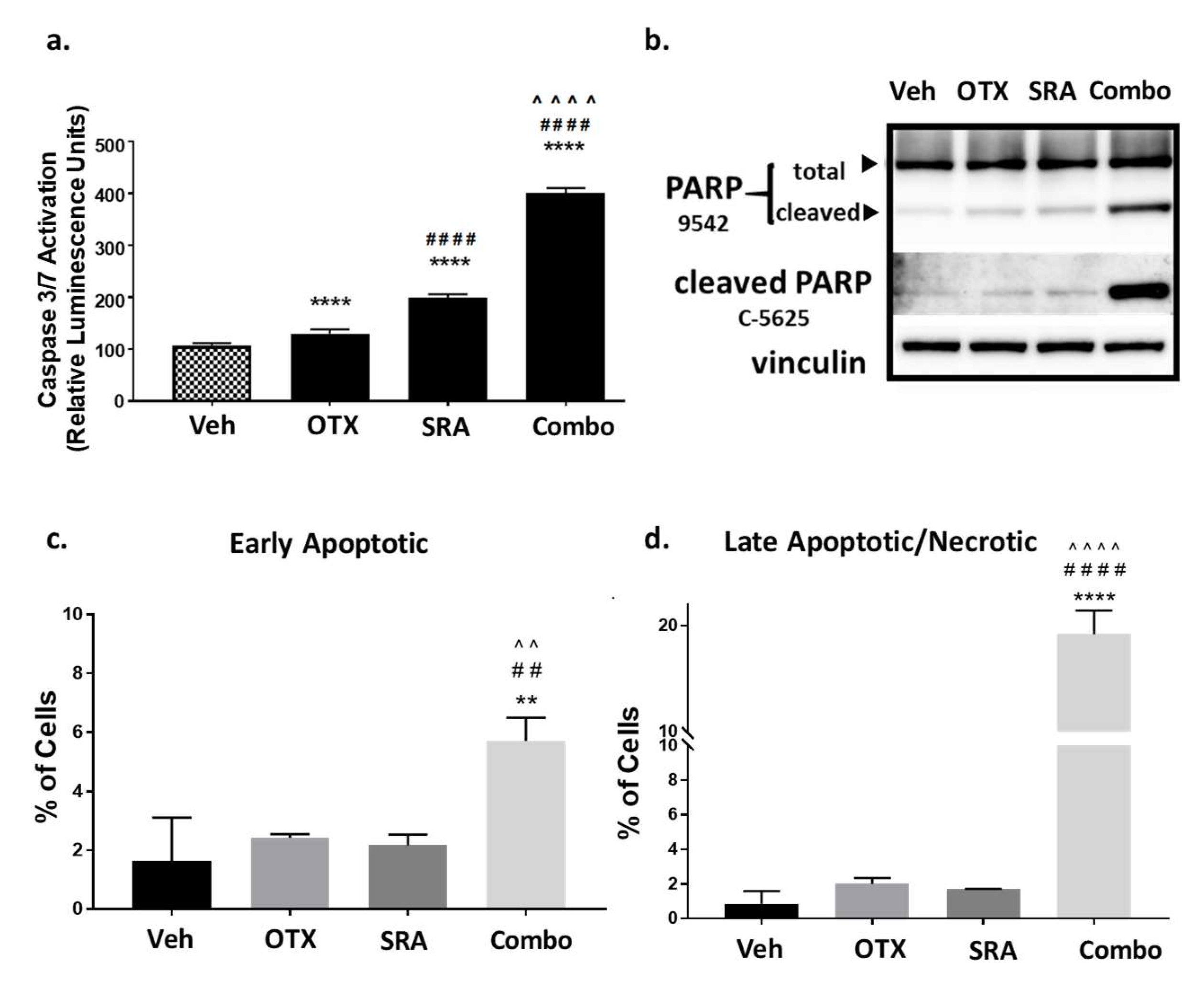
2.8. In Vitro Dual-Inhibition of BET and CHK1 Signifcantly Induces Apoptosis in Saos2 Cells
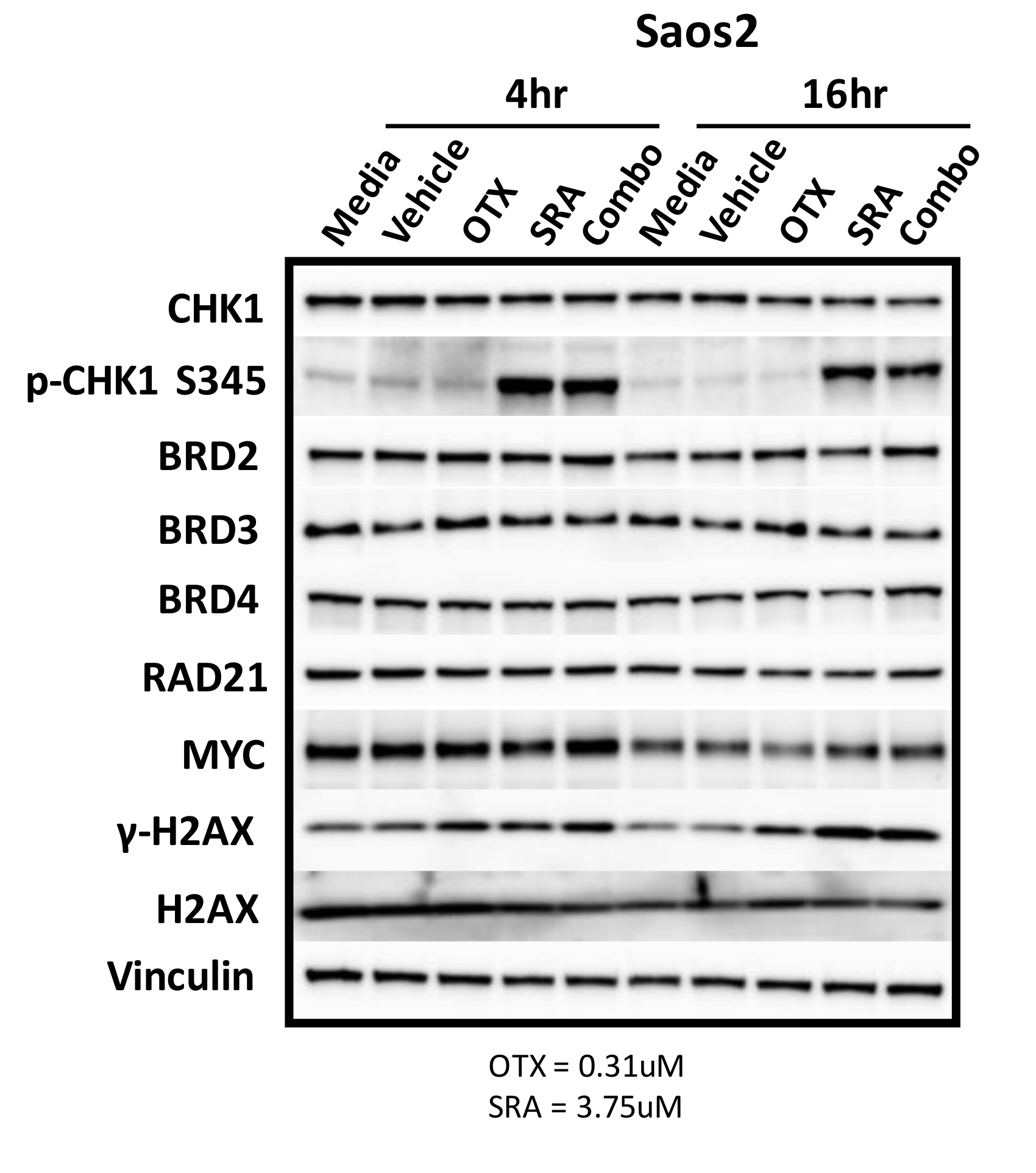
2.9. BET Inhibition does not Decrease MYC Protein Expression but does Inhibit OS Cell-Growth by BRD4-Dependent Mechanisms
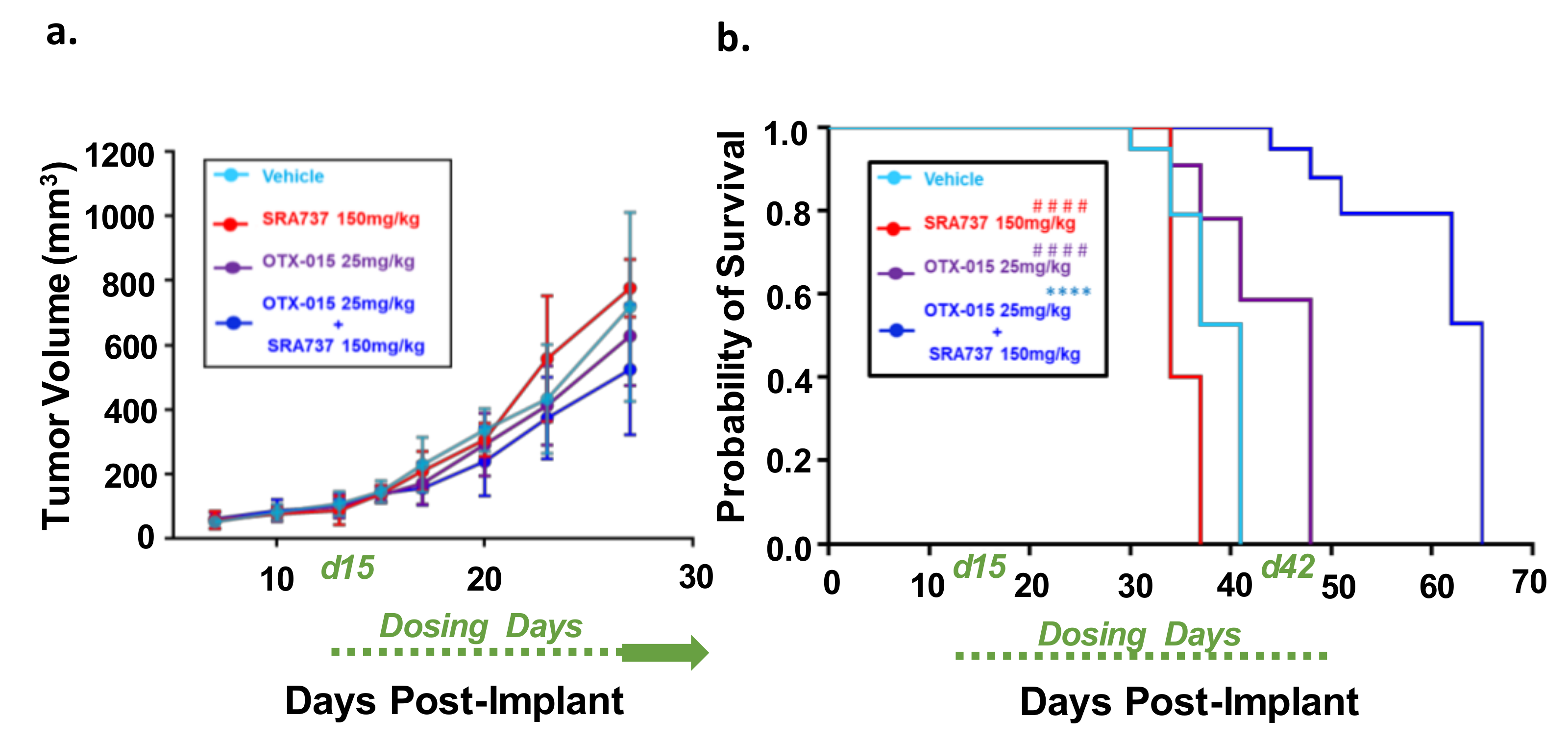
2.10. Dual-Inhibition of BET and CHK1 in a Saos2 Cell Line-Derived Xenograft (CDX) Model Increases the Probability of Survival over Time
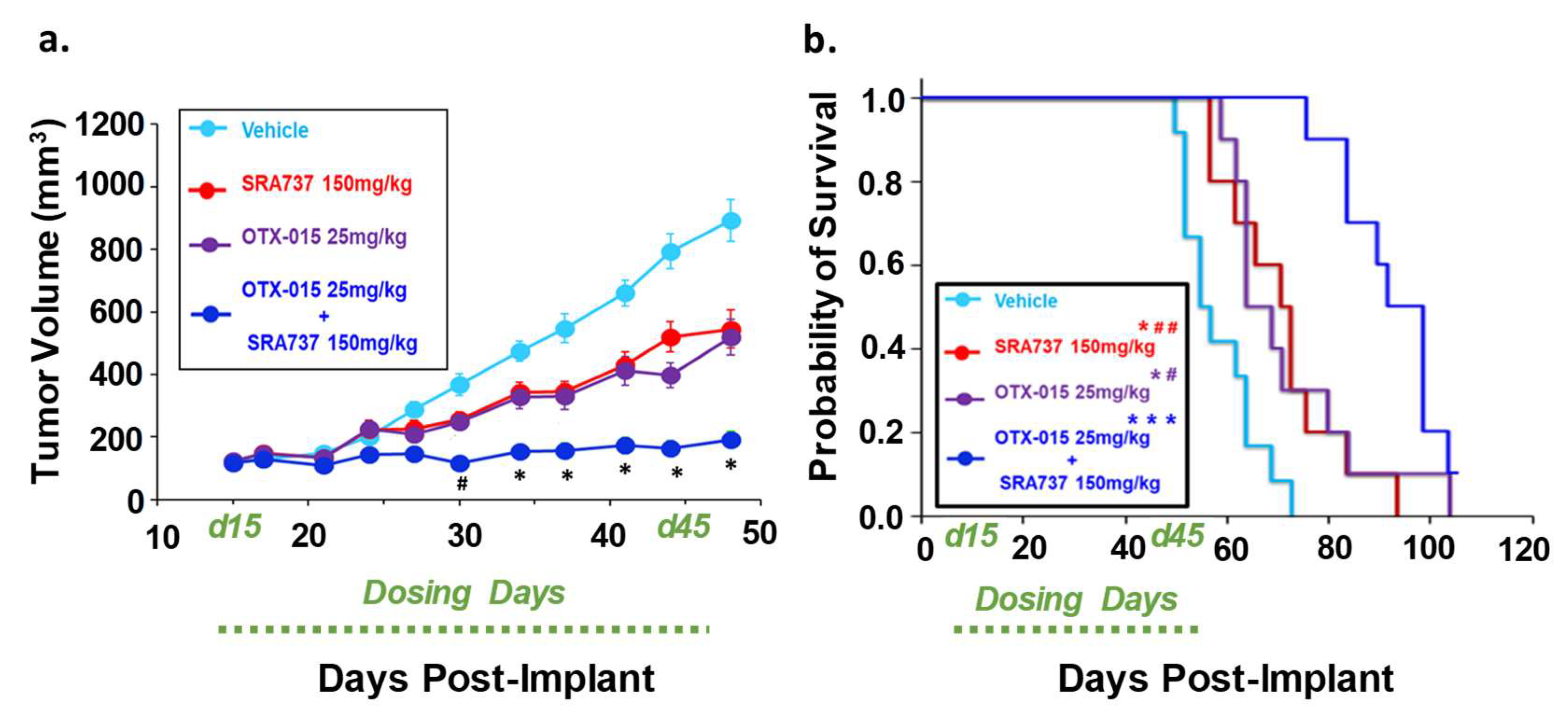
2.11. Dual-Inhibition of BET and CHK1 in a PDX Model of Relapsed OS Signficantly Arrested Tumor Growth During the Dosing Period and Increased the Probability of Survival over Time
3. Discussion
4. Materials and Methods
4.1. Osteosarcoma Genomics Datasets for a Systems Biology Approach
4.2. Gene Annotations for a Systems Biology Approach
4.3. Correlative Analysis between Gene Expression and Patient Overall Survival
4.4. Correlative Analysis between CNV and Patient Overall Survival
4.5. Correlative Analysis between CNV and Gene Expression
4.6. Differential Expression Calculation
4.7. Pathway Analysis (Gene Enrichment Analysis and Network Analysis)
4.8. Cell Lines
4.9. Compounds
4.10. Cell Proliferation Assay
4.11. In Vitro Analysis of Apoptosis by Activated Caspase-3/7
4.12. Apoptosis Flow Cytometry
4.13. Transient Knockdown of BRD4 with siRNA
4.14. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG)
4.15. Development of an Saos2 CDX Model of OS
4.16. Development of TT2-77 PDX from a Pediatric Patient with Relapsed OS
4.17. Development of TT2-77 Xenoline from PDX
4.18. Western Blot Analysis
4.19. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benjamin, R.S.; Patel, S.R. Pediatric and Adult Osteosarcoma: Comparisons and Contrasts in Presentation and Therapy. In Pediatric and Adolescent Osteosarcoma; Jaffe, N., Bruland, O.S., Bielack, S., Eds.; Springer US: Boston, MA, USA, 2010; pp. 355–363. [Google Scholar] [CrossRef]
- Reed, D.R.; Hayashi, M.; Wagner, L.; Binitie, O.; Steppan, D.A.; Brohl, A.S.; Shinohara, E.T.; Bridge, J.A.; Loeb, D.M.; Borinstein, S.C.; et al. Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer 2017, 123, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Taran, S.J.; Taran, R.; Malipatil, N.B. Pediatric Osteosarcoma: An Updated Review. Indian J. Med. Paediatr. Oncol. Off. J. Indian Soc. Med. Paediatr. Oncol. 2017, 38, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Saraf, A.J.; Fenger, J.M.; Roberts, R.D. Osteosarcoma: Accelerating Progress Makes for a Hopeful Future. Front. Oncol. 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.J.; Khanna, C. Osteosarcoma Genetics and Epigenetics: Emerging Biology and Candidate Therapies. Crit. Rev. Oncog. 2015, 20, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Ji, T.; Guo, W. Anti-angiogenesis target therapy for advanced osteosarcoma (Review). Oncol. Rep. 2017, 38, 625–636. [Google Scholar] [CrossRef]
- Greenberg, R.G.; Gamel, B.; Bloom, D.; Bradley, J.; Jafri, H.S.; Hinton, D.; Nambiar, S.; Wheeler, C.; Tiernan, R.; Smith, P.B.; et al. Parents’ perceived obstacles to pediatric clinical trial participation: Findings from the clinical trials transformation initiative. Contemp. Clin. Trials Commun. 2018, 9, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, F.; Baud’huin, M.; Rodriguez Calleja, L.; Jacques, C.; Berreur, M.; Redini, F.; Lecanda, F.; Bradner, J.E.; Heymann, D.; Ory, B. Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat. Commun. 2014, 5, 3511. [Google Scholar] [CrossRef]
- Squire, J.A.; Pei, J.; Marrano, P.; Beheshti, B.; Bayani, J.; Lim, G.; Moldovan, L.; Zielenska, M. High-resolution mapping of amplifications and deletions in pediatric osteosarcoma by use of CGH analysis of cDNA microarrays. Genes Chromosomes Cancer 2003, 38, 215–225. [Google Scholar] [CrossRef]
- Kuijjer, M.L.; Rydbeck, H.; Kresse, S.H.; Buddingh, E.P.; Lid, A.B.; Roelofs, H.; Burger, H.; Myklebost, O.; Hogendoorn, P.C.; Meza-Zepeda, L.A.; et al. Identification of osteosarcoma driver genes by integrative analysis of copy number and gene expression data. Genes Chromosomes Cancer 2012, 51, 696–706. [Google Scholar] [CrossRef]
- Cheng, L.; Pandya, P.H.; Liu, E.; Chandra, P.; Wang, L.; Murray, M.E.; Carter, J.; Ferguson, M.; Saadatzadeh, M.R.; Bijangi-Visheshsaraei, K.; et al. Integration of genomic copy number variations and chemotherapy-response biomarkers in pediatric sarcoma. BMC Med. Genom. 2019, 12, 23. [Google Scholar] [CrossRef]
- Smida, J.; Baumhoer, D.; Rosemann, M.; Walch, A.; Bielack, S.; Poremba, C.; Remberger, K.; Korsching, E.; Scheurlen, W.; Dierkes, C.; et al. Genomic Alterations and Allelic Imbalances Are Strong Prognostic Predictors in Osteosarcoma. Clin. Cancer Res. 2010, 16, 4256–4267. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.C.; Harris, C.P.; Lu, X.Y.; Perlaky, L.; Gogineni, S.; Chintagumpala, M.; Hicks, J.; Johnson, M.E.; Davino, N.A.; Huvos, A.G.; et al. Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes Cancer 2004, 39, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Forus, A.; Weghuis, D.O.; Smeets, D.; Fodstad, Ø.; Myklebost, O.; van Kessel, A.G. Comparative genomic hybridization analysis of human sarcomas: II. Identification of novel amplicons at 6p and 17p in osteosarcomas. Genes Chromosomes Cancer 1995, 14, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Kager, L.; Fink, F.M.; Gadner, H.; Ambros, P.F. Chromosomal regions involved in the pathogenesis of osteosarcomas. Genes Chromosomes Cancer 2000, 28, 329–336. [Google Scholar] [CrossRef]
- Man, T.K.; Lu, X.Y.; Jaeweon, K.; Perlaky, L.; Harris, C.P.; Shah, S.; Ladanyi, M.; Gorlick, R.; Lau, C.C.; Rao, P.H. Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer 2004, 4, 45. [Google Scholar] [CrossRef]
- Martin, J.W.; Yoshimoto, M.; Ludkovski, O.; Thorner, P.S.; Zielenska, M.; Squire, J.A.; Nuin, P.A. Analysis of segmental duplications, mouse genome synteny and recurrent cancer-associated amplicons in human chromosome 6p21-p12. Cytogenet. Genome Res. 2010, 128, 199–213. [Google Scholar] [CrossRef]
- Entz-Werle, N.; Lavaux, T.; Metzger, N.; Stoetzel, C.; Lasthaus, C.; Marec, P.; Kalifa, C.; Brugieres, L.; Pacquement, H.; Schmitt, C.; et al. Involvement of MET/TWIST/APC combination or the potential role of ossification factors in pediatric high-grade osteosarcoma oncogenesis. Neoplasia 2007, 9, 678–688. [Google Scholar] [CrossRef]
- Zielenska, M.; Bayani, J.; Pandita, A.; Toledo, S.; Marrano, P.; Andrade, J.; Petrilli, A.; Thorner, P.; Sorensen, P.; Squire, J.A. Comparative genomic hybridization analysis identifies gains of 1p35 ~ p36 and chromosome 19 in osteosarcoma. Cancer Genet. Cytogenet. 2001, 130, 14–21. [Google Scholar] [CrossRef]
- Pompetti, F.; Rizzo, P.; Simon, R.M.; Freidlin, B.; Mew, D.J.; Pass, H.I.; Picci, P.; Levine, A.S.; Carbone, M. Oncogene alterations in primary, recurrent, and metastatic human bone tumors. J. Cell. Biochem. 1996, 63, 37–50. [Google Scholar] [CrossRef]
- Ladanyi, M.; Park, C.K.; Lewis, R.; Jhanwar, S.C.; Healey, J.H.; Huvos, A.G. Sporadic amplification of the MYC gene in human osteosarcomas. Diagn. Mol. Pathol. Am. J. Surg. Pathol. Part B 1993, 2, 163–167. [Google Scholar] [CrossRef]
- Yotov, W.V.; Hamel, H.; Rivard, G.E.; Champagne, M.A.; Russo, P.A.; Leclerc, J.M.; Bernstein, M.L.; Levy, E. Amplifications of DNA primase 1 (PRIM1) in human osteosarcoma. Genes Chromosomes Cancer 1999, 26, 62–69. [Google Scholar] [CrossRef]
- Lonardo, F.; Ueda, T.; Huvos, A.G.; Healey, J.; Ladanyi, M. p53 and MDM2 alterations in osteosarcomas: Correlation with clinicopathologic features and proliferative rate. Cancer 1997, 79, 1541–1547. [Google Scholar] [CrossRef]
- Tarkkanen, M.; Karhu, R.; Kallioniemi, A.; Elomaa, I.; Kivioja, A.H.; Nevalainen, J.; Bohling, T.; Karaharju, E.; Hyytinen, E.; Knuutila, S.; et al. Gains and losses of DNA sequences in osteosarcomas by comparative genomic hybridization. Cancer Res. 1995, 55, 1334–1338. [Google Scholar] [PubMed]
- Henriksen, J.; Aagesen, T.H.; Maelandsmo, G.M.; Lothe, R.A.; Myklebost, O.; Forus, A. Amplification and overexpression of COPS3 in osteosarcomas potentially target TP53 for proteasome-mediated degradation. Oncogene 2003, 22, 5358–5361. [Google Scholar] [CrossRef][Green Version]
- Yan, T.; Wunder, J.S.; Gokgoz, N.; Gill, M.; Eskandarian, S.; Parkes, R.K.; Bull, S.B.; Bell, R.S.; Andrulis, I.L. COPS3 amplification and clinical outcome in osteosarcoma. Cancer 2007, 109, 1870–1876. [Google Scholar] [CrossRef]
- Atiye, J.; Wolf, M.; Kaur, S.; Monni, O.; Bohling, T.; Kivioja, A.; Tas, E.; Serra, M.; Tarkkanen, M.; Knuutila, S. Gene amplifications in osteosarcoma-CGH microarray analysis. Genes Chromosomes Cancer 2005, 42, 158–163. [Google Scholar] [CrossRef]
- Nielsen, G.P.; Burns, K.L.; Rosenberg, A.E.; Louis, D.N. CDKN2A gene deletions and loss of p16 expression occur in osteosarcomas that lack RB alterations. Am. J. Pathol. 1998, 153, 159–163. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Sekine, K.; Hinohara, S.; Namiki, T.; Nobori, T.; Kaneko, Y. Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet. Cytogenet. 2000, 120, 91–98. [Google Scholar] [CrossRef]
- Miller, C.W.; Aslo, A.; Campbell, M.J.; Kawamata, N.; Lampkin, B.C.; Koeffler, H.P. Alterations of the p15, p16,and p18 genes in osteosarcoma. Cancer Genet. Cytogenet. 1996, 86, 136–142. [Google Scholar] [CrossRef]
- Patino-Garcia, A.; Pineiro, E.S.; Diez, M.Z.; Iturriagagoitia, L.G.; Klussmann, F.A.; Ariznabarreta, L.S. Genetic and epigenetic alterations of the cell cycle regulators and tumor suppressor genes in pediatric osteosarcomas. J. Pediatric Hematol. Oncol. 2003, 25, 362–367. [Google Scholar] [CrossRef]
- Mendoza, S.; David, H.; Gaylord, G.M.; Miller, C.W. Allelic loss at 10q26 in osteosarcoma in the region of the BUB3 and FGFR2 genes. Cancer Genet. Cytogenet. 2005, 158, 142–147. [Google Scholar] [CrossRef]
- Yan, G.N.; Lv, Y.F.; Guo, Q.N. Advances in osteosarcoma stem cell research and opportunities for novel therapeutic targets. Cancer Lett. 2016, 370, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.W.; Squire, J.A.; Zielenska, M. The genetics of osteosarcoma. Sarcoma 2012, 2012, 627254. [Google Scholar] [CrossRef] [PubMed]
- Varshney, J.; Scott, M.C.; Largaespada, D.A.; Subramanian, S. Understanding the Osteosarcoma Pathobiology: A Comparative Oncology Approach. Vet. Sci. 2016, 3, 3. [Google Scholar] [CrossRef]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.J.; Li, Y.; Yu, A.; Shen, J.; Rao, P.H.; Lau, S.S.; Vannucci, M.; Lau, C.C.; Man, T.K. A systems biology approach reveals common metastatic pathways in osteosarcoma. BMC Syst. Biol. 2012, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Zhao, N.; Wang, C.; Kamar, S.; Zhou, Y.; He, Z.; Yang, J.; Sun, B.; Shi, X.; et al. Progress in the chemotherapeutic treatment of osteosarcoma. Oncol. Lett. 2018, 16, 6228–6237. [Google Scholar] [CrossRef]
- Remondini, D. Systems Biology approaches to cancer: Towards new therapeutical strategies and personalized approaches. Mol. Cell. Oncol. 2019, 6, 1561118. [Google Scholar] [CrossRef]
- Chuang, H.Y.; Hofree, M.; Ideker, T. A decade of systems biology. Annu. Rev. Cell Dev. Biol. 2010, 26, 721–744. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Somvanshi, P.R.; Venkatesh, K.V. A conceptual review on systems biology in health and diseases: From biological networks to modern therapeutics. Syst. Synth. Biol. 2014, 8, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Xu, H.; Tuynman, J.; George, J.; Yan, Y.; Li, J.; Ward, R.L.; Mortensen, N.; Hawkins, N.J.; McKay, M.J.; et al. RAD21 cohesin overexpression is a prognostic and predictive marker exacerbating poor prognosis in KRAS mutant colorectal carcinomas. Br. J. Cancer 2014, 110, 1606–1613. [Google Scholar] [CrossRef]
- Yu, L.; Guo, W.; Zhao, S.; Tang, J.; Liu, J. Knockdown of Mad2 induces osteosarcoma cell apoptosis-involved Rad21 cleavage. J. Orthop. Sci. Off. J. Jpn. Orthop. Assoc. 2011, 16, 814–820. [Google Scholar] [CrossRef]
- Rohban, S.; Campaner, S. Myc induced replicative stress response: How to cope with it and exploit it. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 517–524. [Google Scholar] [CrossRef]
- Campaner, S.; Amati, B. Two sides of the Myc-induced DNA damage response: From tumor suppression to tumor maintenance. Cell Div. 2012, 7, 6. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef]
- Gu, L.; Chu, P.; Lingeman, R.; McDaniel, H.; Kechichian, S.; Hickey, R.J.; Liu, Z.; Yuan, Y.C.; Sandoval, J.A.; Fields, G.B.; et al. The Mechanism by Which MYCN Amplification Confers an Enhanced Sensitivity to a PCNA-Derived Cell Permeable Peptide in Neuroblastoma Cells. EBioMedicine 2015, 2, 1923–1931. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef]
- Hills, S.A.; Diffley, J.F.X. DNA Replication and Oncogene-Induced Replicative Stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Spentzos, D.; Hoffman, R.M.; Shi, H.; Duan, Z. Myc is a prognostic biomarker and potential therapeutic target in osteosarcoma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920922055. [Google Scholar] [CrossRef] [PubMed]
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Khoueiry, P.; Ward Gahlawat, A.; Petretich, M.; Michon, A.M.; Simola, D.; Lam, E.; Furlong, E.E.; Benes, V.; Dawson, M.A.; Prinjha, R.K.; et al. BRD4 bimodal binding at promoters and drug-induced displacement at Pol II pause sites associates with I-BET sensitivity. Epigenetics Chromatin 2019, 12, 39. [Google Scholar] [CrossRef]
- Li, W.; Gupta, S.K.; Han, W.; Kundson, R.A.; Nelson, S.; Knutson, D.; Greipp, P.T.; Elsawa, S.F.; Sotomayor, E.M.; Gupta, M. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol. 2019, 12, 73. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Berenguer-Daizé, C.; Astorgues-Xerri, L.; Odore, E.; Cayol, M.; Cvitkovic, E.; Noel, K.; Bekradda, M.; MacKenzie, S.; Rezai, K.; Lokiec, F.; et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 2016, 139, 2047–2055. [Google Scholar] [CrossRef]
- Zhang, J.; Dulak, A.M.; Hattersley, M.M.; Willis, B.S.; Nikkilä, J.; Wang, A.; Lau, A.; Reimer, C.; Zinda, M.; Fawell, S.E.; et al. BRD4 facilitates replication stress-induced DNA damage response. Oncogene 2018, 37, 3763–3777. [Google Scholar] [CrossRef]
- Takashima, Y.; Kikuchi, E.; Kikuchi, J.; Suzuki, M.; Kikuchi, H.; Maeda, M.; Shoji, T.; Furuta, M.; Kinoshita, I.; Dosaka-Akita, H.; et al. Bromodomain and extraterminal domain inhibition synergizes with WEE1-inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int. J. Cancer. 2020, 146, 1114–1124. [Google Scholar] [CrossRef]
- Rohban, S.; Cerutti, A.; Morelli, M.J.; d’Adda di Fagagna, F.; Campaner, S. The cohesin complex prevents Myc-induced replication stress. Cell Death Dis. 2017, 8, e2956. [Google Scholar] [CrossRef]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Plummer, E.R.; Kristeleit, R.S.; Cojocaru, E.; Haris, N.M.; Carter, L.; Jones, R.H.; Blagden, S.P.; Evans, T.R.J.; Arkenau, H.T.; Sarker, D.; et al. A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. J. Clin. Oncol. 2019, 37, 3094. [Google Scholar] [CrossRef]
- Wicaksono, B.D. Investigating the Predictors of Sensitivity to the Chk1 Inhibitor PF-00477736 in Melanoma. Master’s Thesis, The University of Melbourne, Melbourne, Australia, 2013. [Google Scholar]
- Dorsett, D.; Ström, L. The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr. Biol. 2012, 22, R240–R250. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Guan, Y.; Qin, W.; Zhai, X.; Yu, B.; Liu, H. Targeting Brd4 for cancer therapy: Inhibitors and degraders. Medchemcomm 2018, 9, 1779–1802. [Google Scholar] [CrossRef] [PubMed]
- Tarkkanen, M.; Elomaa, I.; Blomqvist, C.; Kivioja, A.H.; Kellokumpu-Lehtinen, P.; Böhling, T.; Valle, J.; Knuutila, S. DNA sequence copy number increase at 8q: A potential new prognostic marker in high-grade osteosarcoma. Int. J. Cancer 1999, 84, 114–121. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, W.; Ma, X.; Hao, Y.; Liu, G.; Hu, X.; Shang, H.; Wu, P.; Zhao, Z.; Liu, W. Logistic regression analysis for the identification of the metastasis-associated signaling pathways of osteosarcoma. Int. J. Mol. Med. 2018, 41, 1233–1244. [Google Scholar] [CrossRef]
- Takeba, Y.; Nagafuchi, H.; Takeno, M.; Kashiwakura, J.I.; Suzuki, N. Txk, a Member of Nonreceptor Tyrosine Kinase of Tec Family, Acts as a Th1 Cell-Specific Transcription Factor and Regulates IFN-γ Gene Transcription. J. Immunol. 2002, 168, 2365–2370. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, T. miR-214 promotes the proliferation and invasion of osteosarcoma cells through direct suppression of LZTS1. Biochem. Biophys. Res. Commun. 2014, 449, 190–195. [Google Scholar] [CrossRef]
- Yao, P.; Wang, Z.B.; Ding, Y.Y.; Ma, J.M.; Hong, T.; Pan, S.N.; Zhang, J. Regulatory network of differentially expressed genes in metastatic osteosarcoma. Mol. Med. Rep. 2015, 11, 2104–2110. [Google Scholar] [CrossRef]
- Cabeza-Arvelaiz, Y.; Sepulveda, J.L.; Lebovitz, R.M.; Thompson, T.C.; Chinault, A.C. Functional identification of LZTS1 as a candidate prostate tumor suppressor gene on human chromosome 8p22. Oncogene 2001, 20, 4169–4179. [Google Scholar] [CrossRef][Green Version]
- Myhre, S.; Lingjærde, O.C.; Hennessy, B.T.; Aure, M.R.; Carey, M.S.; Alsner, J.; Tramm, T.; Overgaard, J.; Mills, G.B.; Børresen-Dale, A.L.; et al. Influence of DNA copy number and mRNA levels on the expression of breast cancer related proteins. Mol. Oncol. 2013, 7, 704–718. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Gaudio, E.; Tarantelli, C.; Ponzoni, M.; Odore, E.; Rezai, K.; Bernasconi, E.; Cascione, L.; Rinaldi, A.; Stathis, A.; Riveiro, E.; et al. Bromodomain inhibitor OTX015 (MK-8628) combined with targeted agents shows strong in vivo antitumor activity in lymphoma. Oncotarget 2016, 7, 58142–58147. [Google Scholar] [CrossRef] [PubMed]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef] [PubMed]
- Najafova, Z.; Tirado-Magallanes, R.; Subramaniam, M.; Hossan, T.; Schmidt, G.; Nagarajan, S.; Baumgart, S.J.; Mishra, V.K.; Bedi, U.; Hesse, E.; et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2016, 45, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Engelke, C.G.; Parsels, L.A.; Qian, Y.; Zhang, Q.; Karnak, D.; Robertson, J.R.; Tanska, D.M.; Wei, D.; Davis, M.A.; Parsels, J.D.; et al. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4412–4421. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Boklan, J. Little patients, losing patience: Pediatric cancer drug development. Mol. Cancer Ther. 2006, 5, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Chandra, P.; Wang, L.; Pollok, K.; Pandya, P.; Murray, M.; Carter, J.; Ferguson, M.; Reza, M.; Mark, M.; et al. Abstract 1281: Genomic structure variation in large screening for pediatric sarcoma therapy. Cancer Res. 2018, 78, 1281. [Google Scholar] [CrossRef]
- Moriarity, B.S.; Otto, G.M.; Rahrmann, E.P.; Rathe, S.K.; Wolf, N.K.; Weg, M.T.; Manlove, L.A.; LaRue, R.S.; Temiz, N.A.; Molyneux, S.D.; et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat. Genet. 2015, 47, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.; Utset-Ward, T.J.; Reed, D.R.; Lynch, C.C. Re-calculating! Navigating through the osteosarcoma treatment roadblock. Pharmacol. Res. 2017, 117, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Iranmanesh, S.M.; Guo, N.L. Integrated DNA Copy Number and Gene Expression Regulatory Network Analysis of Non-small Cell Lung Cancer Metastasis. Cancer Inform. 2014, 13, 13–23. [Google Scholar] [CrossRef]
- Ren, L.; Chen, L.; Wu, W.; Garribba, L.; Tian, H.; Liu, Z.; Vogel, I.; Li, C.; Hickson, I.D.; Liu, Y. Potential biomarkers of DNA replication stress in cancer. Oncotarget 2017, 8, 36996–37008. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Viktorsson, K.; Lewensohn, R.; Zhivotovsky, B. Systems biology approaches to develop innovative strategies for lung cancer therapy. Cell Death Dis. 2014, 5, e1260. [Google Scholar] [CrossRef]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Tomaszewski, J.M.; Vazquez, M.; Verschoor, S.; et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef]
- He, H.; Ni, J.; Huang, J.U.N. Molecular mechanisms of chemoresistance in osteosarcoma (Review). Oncol. Lett. 2014, 7, 1352–1362. [Google Scholar] [CrossRef]
- Gajduskova, P.; Snijders, A.M.; Kwek, S.; Roydasgupta, R.; Fridlyand, J.; Tokuyasu, T.; Pinkel, D.; Albertson, D.G. Genome position and gene amplification. Genome Biol. 2007, 8, R120. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, R.; Zhuang, Z.; Ren, J.; Wang, Z.; Liu, Y.; Wu, J.; Jiang, S.; Wang, K. A risk signature-based on metastasis-associated genes to predict survival of patients with osteosarcoma. J. Cell. Biochem. 2020, 121, 3479–3490. [Google Scholar] [CrossRef] [PubMed]
- Pongas, G.; Kim, M.K.; Min, D.J.; House, C.D.; Jordan, E.; Caplen, N.; Chakka, S.; Ohiri, J.; Kruhlak, M.J.; Annunziata, C.M. BRD4 facilitates DNA damage response and represses CBX5/Heterochromatin protein 1 (HP1). Oncotarget 2017, 8, 51402–51415. [Google Scholar] [CrossRef] [PubMed]
- Da-Rè, C.; Halazonetis, T.D. DNA replication stress as an Achilles’ heel of cancer. Oncotarget 2015, 6, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Fritsche-Guenther, R.; Noske, A.; Ungethum, U.; Kuban, R.J.; Schlag, P.M.; Tunn, P.U.; Karle, J.; Krenn, V.; Dietel, M.; Sers, C. De novo expression of EphA2 in osteosarcoma modulates activation of the mitogenic signalling pathway. Histopathology 2010, 57, 836–850. [Google Scholar] [CrossRef]
- Bruford, E.A.; Lush, M.J.; Wright, M.W.; Sneddon, T.P.; Povey, S.; Birney, E. The HGNC Database in 2008: A resource for the human genome. Nucleic Acids Res. 2008, 36, D445–D448. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Gatto, L.; Christoforou, A. Using R and Bioconductor for proteomics data analysis. Biochim. Biophys. Acta 2014, 1844, 42–51. [Google Scholar] [CrossRef]
- Cox, D.R. Regression Models and Life-Tables. J. R. Stat. Soc. Ser. B Methodol. 1972, 34, 187–220. [Google Scholar] [CrossRef]
- Bradburn, M.J.; Clark, T.G.; Love, S.B.; Altman, D.G. Survival analysis part II: Multivariate data analysis—an introduction to concepts and methods. Br. J. Cancer 2003, 89, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Tonsing-Carter, E.; Bailey, B.J.; Saadatzadeh, M.R.; Ding, J.; Wang, H.; Sinn, A.L.; Peterman, K.M.; Spragins, T.K.; Silver, J.M.; Sprouse, A.A.; et al. Potentiation of carboplatin-mediated DNA damage by the Mdm2 modulator Nutlin-3a in a humanized orthotopic breast-to-lung metastatic model. Mol. Cancer Ther. 2015, 14, 2850–2863. [Google Scholar] [CrossRef]
- Mattar, M.; McCarthy, C.R.; Kulick, A.R.; Qeriqi, B.; Guzman, S.; de Stanchina, E. Establishing and Maintaining an Extensive Library of Patient-Derived Xenograft Models. Front. Oncol. 2018, 8, 19. [Google Scholar] [CrossRef]
- Liu, Q.; Yin, X.; Languino, L.R.; Altieri, D.C. Evaluation of drug combination effect using a Bliss independence dose-response surface model. Stat. Biopharm. Res. 2018, 10, 112–122. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Total Gene # | Significant Gene # | Significant Gene % | CNV Gains # | CNV Loss# | p-Value | Enrichment Odds Ratio | −log p |
|---|---|---|---|---|---|---|---|---|
| 1 | 2652 | 220 | 8.30 | 58 | 205 | 0.997 | 0.800 | 0.003 |
| 2 | 1728 | 56 | 3.24 | 16 | 56 | 1.000 | 0.287 | 0.000 |
| 3 | 1477 | 60 | 4.06 | 18 | 60 | 1.000 | 0.368 | 0.000 |
| 4 | 1011 | 88 | 8.70 | 47 | 82 | 0.813 | 0.856 | 0.207 |
| 5 | 1216 | 26 | 2.14 | 9 | 26 | 1.000 | 0.189 | 0.000 |
| 6 | 1374 | 21 | 1.53 | 15 | 21 | 1.000 | 0.133 | 0.000 |
| 7 | 1269 | 10 | 0.79 | 6 | 10 | 1.000 | 0.068 | 0.000 |
| 8 | 962 | 261 | 27.13 | 188 | 160 | 1.05 × 10−72 | 3.617 | 165.736 |
| 9 | 1063 | 132 | 12.42 | 41 | 132 | 7.75 × 10−03 | 1.294 | 4.860 |
| 10 | 1061 | 367 | 34.59 | 8 | 367 | 1.44 × 10−163 | 5.375 | 374.960 |
| 11 | 1609 | 428 | 26.60 | 129 | 428 | 3.13 × 10−116 | 3.705 | 265.958 |
| 12 | 1326 | 127 | 9.58 | 116 | 127 | 0.344 | 0.954 | 1.067 |
| 13 | 601 | 6 | 1.00 | 2 | 6 | 1.000 | 0.089 | 0.000 |
| 14 | 876 | 12 | 1.37 | 11 | 2 | 1.000 | 0.121 | 0.000 |
| 15 | 931 | 193 | 20.73 | 81 | 191 | 1.33 × 10−28 | 2.467 | 64.185 |
| 16 | 1086 | 280 | 25.78 | 7 | 278 | 4.18 × 10−70 | 3.386 | 159.750 |
| 17 | 1498 | 207 | 13.82 | 70 | 205 | 4.02 × 10−7 | 1.485 | 14.728 |
| 18 | 407 | 12 | 2.95 | 2 | 12 | 1.000 | 0.271 | 0.000 |
| 19 | 1725 | 108 | 6.26 | 77 | 108 | 1.000 | 0.586 | 0.000 |
| 20 | 749 | 10 | 1.34 | 6 | 10 | 1.000 | 0.119 | 0.000 |
| 21 | 341 | 1 | 0.29 | 1 | 1 | 1.000 | 0.026 | 0.000 |
| 22 | 587 | 37 | 6.30 | 35 | 37 | 0.997 | 0.602 | 0.003 |
| X | 1118 | 8 | 0.72 | 7 | 8 | 1.000 | 0.062 | 0.000 |
| Y | 106 | 0 | 0.00 | 0 | 0 | 0.999 | 0.000 | 0.001 |
| (a) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Saos2: Mean+SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| SRA737 (µM) | *0.04 | *0.08 | *0.16 | *0.31 | *0.63 | *1.25 | 2.5 | 5 | |
| *0.94 | 3 ± 0 | 11 ± 1 | 13 ± 1 | 16 ± 0 | 15 ± 0 | 16 ± 0 | 15 ± 0 | 15 ± 1 | |
| *1.88 | 9 ± 1 | 16 ± 1 | 20 ± 1 | 20 ± 1 | 19 ± 0 | 20 ± 1 | 17 ± 0 | 15 ± 0 | |
| *3.75 | 10 ± 0 | 17 ± 0 | 19 ± 1 | 21 ± 0 | 19 ± 1 | 17 ± 1 | 14 ± 0 | 13 ± 0 | |
| 7.5 | 7 ± 0 | 12 ± 1 | 15 ± 1 | 16 ± 0 | 15 ± 1 | 13 ± 1 | 12 ± 0 | 9 ± 1 | |
| 15 | −3 ± 1 | −1 ± 0 | 0 ± 1 | 3 ± 0 | 3 ± 0 | 2 ± 1 | 1 ± 1 | −1 ± 0 | |
| Saos-LM7: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| SRA737 (µM) | *0.02 | *0.04 | *0.08 | *0.16 | *0.31 | *0.63 | *1.25 | 2.5 | |
| *1.56 | 6 ± 1 | 3 ± 2 | 4 ± 2 | 7 ± 2 | 9 ± 2 | 10 ± 2 | 8 ± 1 | 7 ± 1 | |
| *3.13 | 9 ± 0 | 7 ± 1 | 8 ± 3 | 7 ± 2 | 9 ± 2 | 12 ± 1 | 8 ± 1 | 6 ± 1 | |
| 6.25 | 6 ± 2 | 5 ± 2 | 7 ± 1 | 6 ± 1 | 4 ± 1 | 4 ± 2 | 4 ± 1 | 2 ± 1 | |
| 12.5 | 2 ± 1 | 2 ± 1 | 4 ± 0 | 4 ± 1 | 4 ± 0 | 3 ± 1 | 3 ± 1 | 0 ± 1 | |
| 25 | 2 ± 0 | 2 ± 1 | 0 ± 3 | 5 ± 1 | 4 ± 1 | 3 ± 1 | 0 ± 1 | −2 ± 1 | |
| MG63: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| SRA737 (µM) | *0.02 | *0.04 | *0.08 | *0.16 | *0.31 | *0.63 | *1.25 | 2.5 | |
| *0.16 | −7 ± 1 | −7 ± 1 | −6 ± 1 | −6 ± 0 | −2 ± 0 | 0 ± 1 | 1 ± 0 | −1 ± 0 | |
| *0.31 | −9 ± 0 | −10 ± 1 | −7 ± 1 | −6 ± 0 | −4 ± 0 | 0 ± 1 | 1 ± 1 | 0 ± 0 | |
| *0.63 | −7 ± 1 | −8 ± 1 | −6 ± 1 | −6 ± 0 | −1 ± 0 | 3 ± 1 | 6 ± 0 | 4 ± 1 | |
| *1.25 | −6 ± 0 | −11 ± 1 | −14 ± 2 | −14 ± 3 | −9 ± 3 | 0 ± 2 | 3 ± 3 | 2 ± 1 | |
| *2.5 | −6 ± 1 | −17 ± 2 | −30 ± 2 | −36 ± 1 | −30 ± 1 | −18 ± 1 | −14 ± 1 | −13 ± 1 | |
| G292: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| SRA737 (µM) | *0.13 | *0.25 | *0.5 | *1 | *2 | *4 | *8 | 16 | |
| *0.25 | −7 ± 0 | −10 ± 0 | −7 ± 0 | −9 ± 1 | −4 ± 0 | −5 ± 2 | −3 ± 1 | 0 ± 1 | |
| *0.5 | −5 ± 2 | −8 ± 1 | −7 ± 1 | −8 ± 1 | −3 ± 1 | −2 ± 1 | 0 ± 1 | 2 ± 2 | |
| *1 | −13 ± 3 | −18 ± 2 | −17 ± 2 | −17 ± 2 | −11 ± 1 | −10 ± 2 | −7 ± 2 | −3 ± 2 | |
| *2 | −16 ± 2 | −22 ± 1 | −24 ± 2 | −22 ± 2 | −19 ± 2 | −16 ± 2 | −13 ± 1 | −8 ± 1 | |
| *4 | −16 ± 2 | −22 ± 2 | −25 ± 1 | −22 ± 2 | −19 ± 2 | −16 ± 2 | −14 ± 2 | −9 ± 2 | |
| U2OS: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| SRA737 (µM) | *0.02 | *0.05 | *0.09 | *0.18 | *0.38 | *0.75 | *1.5 | 3 | |
| *0.09 | 1 ± 1 | −1 ± 2 | 0 ± 1 | 1 ± 1 | 2 ± 1 | 0 ± 0 | 1 ± 0 | 1 ± 1 | |
| *0.19 | 0 ± 2 | −3 ± 2 | 0 ± 2 | 3 ± 0 | 2 ± 1 | 2 ± 0 | 3 ± 1 | 2 ± 1 | |
| *0.38 | −8 ± 2 | −11 ± 1 | −10 ± 1 | −7 ± 3 | −6 ± 3 | −5 ± 3 | −4 ± 3 | −3 ± 3 | |
| *0.75 | −8 ± 3 | −9 ± 3 | −10 ± 4 | −10 ± 2 | −10 ± 2 | −10 ± 1 | −9 ± 0 | −9 ± 1 | |
| *1.5 | 2+1 | −4 ± 1 | −6 ± 1 | −8 ± 1 | −8 ± 1 | −9 ± 1 | −9 ± 1 | −8 ± 0 | |
| TT2-77 Xenoline: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| SRA737 (µM) | *0.04 | *0.08 | *0.16 | *0.31 | *0.63 | *1.25 | 2.5 | 5 | |
| *0.31 | −7 ± 1 | −7 ± 1 | −9 ± 1 | −7 ± 1 | −8 ± 1 | −5 ± 1 | −4 ± 1 | −3 ± 1 | |
| *0.63 | −7 ± 0 | −7 ± 1 | −9 ± 1 | −8 ± 1 | −8 ± 1 | −5 ± 1 | −4 ± 1 | −4 ± 1 | |
| *1.25 | −9 ± 1 | −8 ± 0 | −11 ± 1 | −10 ± 1 | −10 ± 1 | −6 ± 1 | −6 ± 1 | −5 ± 1 | |
| *2.5 | −17 ± 2 | −20 ± 4 | −23 ± 4 | −21 ± 4 | −19 ± 4 | −16 ± 3 | −16 ± 3 | −16 ± 3 | |
| *5 | −17 ± 0 | −21 ± 1 | −25 ± 2 | −25 ± 2 | −24 ± 3 | −21 ± 3 | −21 ± 3 | −21 ± 3 | |
| (b) | |||||||||
| Saos2: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| LY2606368 (µM) | *0.01 | *0.02 | *0.03 | *0.06 | *0.13 | *0.25 | *0.5 | *1 | |
| *0.00625 | 1 ± 1 | 2 ± 1 | 8 ± 1 | 9 ± 2 | 16 ± 1 | 19 ± 1 | 21 ± 2 | 18 ± 1 | |
| *0.0125 | 4 ± 1 | 6 ± 2 | 8 ± 1 | 13 ± 2 | 17 ± 0 | 18 ± 0 | 17 ± 0 | 15 ± 1 | |
| *0.025 | 1 ± 1 | 4 ± 0 | 6 ± 1 | 9 ± 1 | 11 ± 0 | 10 ± 0 | 10 ± 0 | 6 ± 1 | |
| *0.05 | 2 ± 0 | 2 ± 1 | 4 ± 1 | 6 ± 0 | 6 ± 1 | 6 ± 0 | 5 ± 0 | 3 ± 1 | |
| *0.1 | 0 ± 1 | 1 ± 1 | 2 ± 1 | 4 ± 1 | 4 ± 0 | 4 ± 0 | 2 ± 0 | 0 ± 1 | |
| Saos-LM7: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| LY2606368 (µM) | *0.01 | *0.02 | *0.03 | *0.06 | *0.13 | *0.25 | *0.5 | *1 | |
| *0.00313 | 3 ± 1 | 2 ± 0 | −1 ± 1 | −2 ± 1 | −1 ± 1 | −3 ± 1 | −1 ± 0 | 0 ± 1 | |
| *0.00625 | 4 ± 0 | 3 ± 1 | 1 ± 1 | 2 ± 0 | 0 ± 3 | 2 ± 1 | 2 ± 1 | 1 ± 1 | |
| *0.0125 | 7 ± 1 | 6 ± 1 | 4 ± 1 | 6 ± 2 | 8 ± 1 | 8 ± 1 | 7 ± 1 | 4 ± 1 | |
| *0.025 | 4 ± 1 | 3 ± 2 | 3 ± 1 | 6 ± 1 | 7 ± 1 | 9 ± 1 | 7 ± 2 | 4 ± 2 | |
| *0.05 | 14 ± 9 | 14 ± 9 | 13 ± 7 | 14 ± 9 | 16 ± 9 | 16 ± 7 | 12left6 | 8 ± 5 | |
| MG63: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| LY2606368 (µM) | *0.01 | *0.02 | *0.03 | *0.06 | *0.13 | *0.25 | *0.5 | *1 | |
| *0.00063 | 2 ± 1 | 2 ± 0 | 1 ± 1 | 2 ± 1 | −1 ± 1 | 0 ± 1 | 0 ± 2 | 2 ± 1 | |
| *0.00125 | 2 ± 1 | 2 ± 2 | 1 ± 1 | 2 ± 0 | 2 ± 1 | 1 ± 0 | 0 ± 0 | 0 ± 1 | |
| *0.0025 | 3 ± 2 | 3 ± 3 | 5 ± 3 | 2 ± 2 | 2 ± 1 | 4 ± 1 | 1 ± 1 | 2 ± 1 | |
| *0.005 | 5 ± 2 | 4 ± 2 | 2 ± 2 | 1 ± 2 | 1 ± 1 | 0 ± 1 | 1 ± 2 | 2 ± 1 | |
| *0.01 | −4 ± 0 | −7 ± 1 | −17 ± 1 | −29 ± 2 | −31 ± 1 | −21 ± 2 | −13 ± 1 | −10 ± 1 | |
| G292: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| LY2606368 (µM) | *0.16 | *0.31 | *0.63 | *1.25 | 2.5 | 5 | 10 | 20 | |
| *0.25 | 1 ± 2 | −1 ± 1 | 1 ± 1 | 1 ± 1 | −1 ± 0 | 0 ± 1 | 2 ± 1 | 2 ± 1 | |
| *0.5 | 1 ± 3 | −3 ± 2 | −1 ± 2 | 1 ± 1 | −1 ± 1 | 1 ± 2 | 3 ± 1 | 5 ± 2 | |
| 1 | −5 ± 4 | −4 ± 3 | −3 ± 2 | 0 ± 2 | 0 ± 1 | 2 ± 1 | 4 ± 1 | 8 ± 2 | |
| 2 | −11 ± 3 | −13 ± 2 | −10 ± 2 | −7 ± 2 | −5 ± 1 | −1 ± 2 | 2 ± 1 | 5 ± 1 | |
| 4 | −22 ± 3 | −24 ± 3 | −19 ± 1 | −16 ± 1 | −13 ± 0 | −9 ± 1 | −6 ± 1 | −1 ± 0 | |
| U2OS: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| LY2606368 (µM) | *0.06 | *0.13 | *0.25 | *0.5 | *1 | *2 | 4 | 8 | |
| *0.00156 | 2 ± 5 | 3 ± 5 | 2 ± 2 | −1 ± 2 | −1 ± 2 | 1 ± 3 | 1 ± 4 | 2 ± 3 | |
| *0.003 | 1 ± 2 | 5 ± 3 | 3 ± 3 | 0 ± 1 | 1 ± 2 | 3 ± 2 | 2 ± 3 | 4 ± 3 | |
| *0.006 | −3 ± 2 | 1 ± 1 | −1 ± 4 | −4 ± 4 | −3 ± 2 | 0 ± 2 | 1 ± 1 | 4 ± 1 | |
| *0.0125 | −10 ± 1 | −13 ± 2 | −15 ± 1 | −18 ± 2 | −17 ± 1 | −14 ± 2 | −9 ± 2 | −6 ± 2 | |
| *0.025 | −3 ± 0 | −5 ± 1 | −8 ± 2 | −10 ± 2 | −11 ± 2 | −10 ± 2 | −8 ± 2 | −7 ± 1 | |
| TT2-77 Xenoline: Mean ± SEM Observed–Expected (% Effect) | |||||||||
| OTX-015 (µM) | |||||||||
| LY2606368 (µM) | *0.04 | *0.08 | *0.16 | *0.31 | *0.63 | *1.25 | 2.5 | 5 | |
| *0.00625 | −4 ± 1 | −7 ± 1 | −8 ± 2 | −9 ± 1 | −6 ± 1 | −5 ± 0 | −3 ± 1 | −3 ± 1 | |
| *0.0125 | −14 ± 1 | −18 ± 2 | −22 ± 2 | −21 ± 1 | −18 ± 1 | −15 ± 1 | −14 ± 2 | −13 ± 2 | |
| *0.025 | −15 ± 1 | −21 ± 1 | −28 ± 1 | −30 ± 1 | −28 ± 1 | −26 ± 1 | −25 ± 1 | −24 ± 1 | |
| *0.05 | −7 ± 1 | −13 ± 0 | −18 ± 0 | −24 ± 1 | −25 ± 1 | −26 ± 1 | −26 ± 1 | −25 ± 1 | |
| *0.1 | −5 ± 1 | −9 ± 1 | −14 ± 2 | −20 ± 1 | −22 ± 1 | −24 ± 0 | −26 ± 1 | −26 ± 1 | |
| Key | |||||||||
| <−10 | Antagonism | ||||||||
| −10 to 10 | Additive | ||||||||
| 10 to 20 | Synergistic | ||||||||
| 20 to >30 | Markedly Synergistic | ||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandya, P.H.; Cheng, L.; Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Tang, S.; Sinn, A.L.; Trowbridge, M.A.; Coy, K.L.; Bailey, B.J.; Young, C.N.; et al. Systems Biology Approach Identifies Prognostic Signatures of Poor Overall Survival and Guides the Prioritization of Novel BET-CHK1 Combination Therapy for Osteosarcoma. Cancers 2020, 12, 2426. https://doi.org/10.3390/cancers12092426
Pandya PH, Cheng L, Saadatzadeh MR, Bijangi-Vishehsaraei K, Tang S, Sinn AL, Trowbridge MA, Coy KL, Bailey BJ, Young CN, et al. Systems Biology Approach Identifies Prognostic Signatures of Poor Overall Survival and Guides the Prioritization of Novel BET-CHK1 Combination Therapy for Osteosarcoma. Cancers. 2020; 12(9):2426. https://doi.org/10.3390/cancers12092426
Chicago/Turabian StylePandya, Pankita H., Lijun Cheng, M. Reza Saadatzadeh, Khadijeh Bijangi-Vishehsaraei, Shan Tang, Anthony L. Sinn, Melissa A. Trowbridge, Kathryn L. Coy, Barbara J. Bailey, Courtney N. Young, and et al. 2020. "Systems Biology Approach Identifies Prognostic Signatures of Poor Overall Survival and Guides the Prioritization of Novel BET-CHK1 Combination Therapy for Osteosarcoma" Cancers 12, no. 9: 2426. https://doi.org/10.3390/cancers12092426
APA StylePandya, P. H., Cheng, L., Saadatzadeh, M. R., Bijangi-Vishehsaraei, K., Tang, S., Sinn, A. L., Trowbridge, M. A., Coy, K. L., Bailey, B. J., Young, C. N., Ding, J., Dobrota, E. A., Dyer, S., Elmi, A., Thompson, Q., Barghi, F., Shultz, J., Albright, E. A., Shannon, H. E., ... Pollok, K. E. (2020). Systems Biology Approach Identifies Prognostic Signatures of Poor Overall Survival and Guides the Prioritization of Novel BET-CHK1 Combination Therapy for Osteosarcoma. Cancers, 12(9), 2426. https://doi.org/10.3390/cancers12092426