Autocrine Signaling of NRP1 Ligand Galectin-1 Elicits Resistance to BRAF-Targeted Therapy in Melanoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
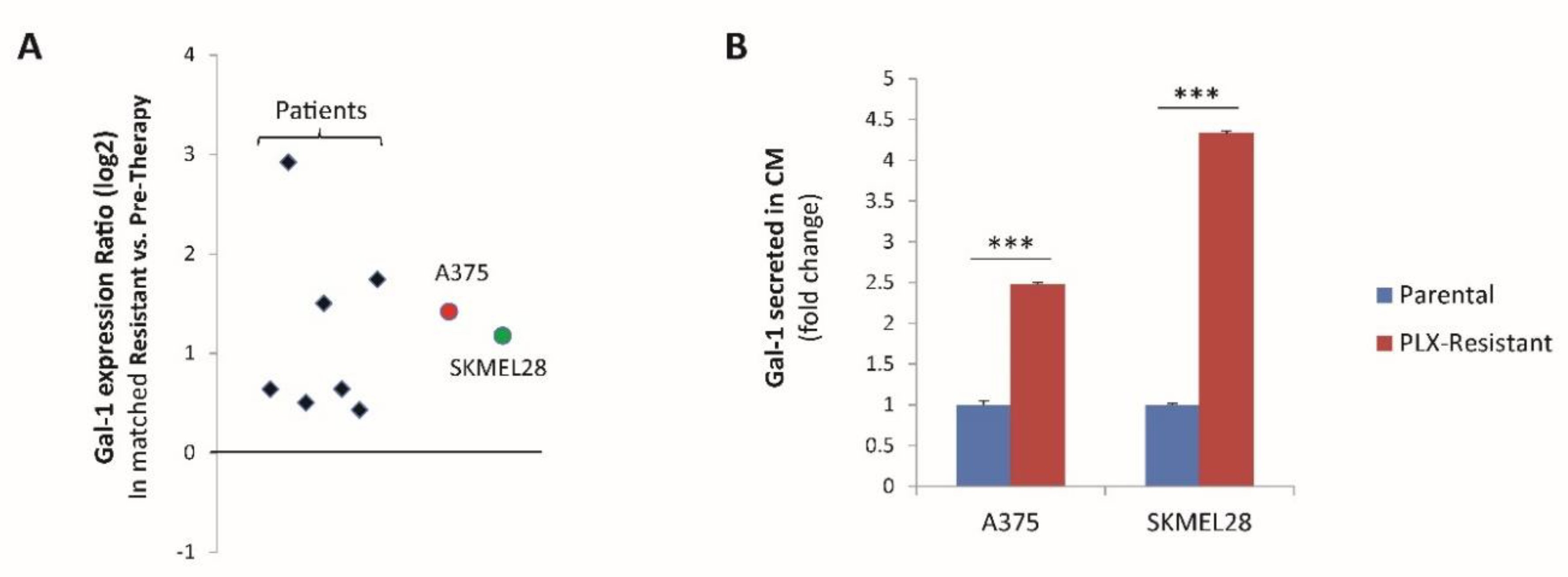
2.1. The Soluble NRP1-Ligand Galectin-1 Is Upregulated in Melanoma Cells Resistant to Targeted Therapy
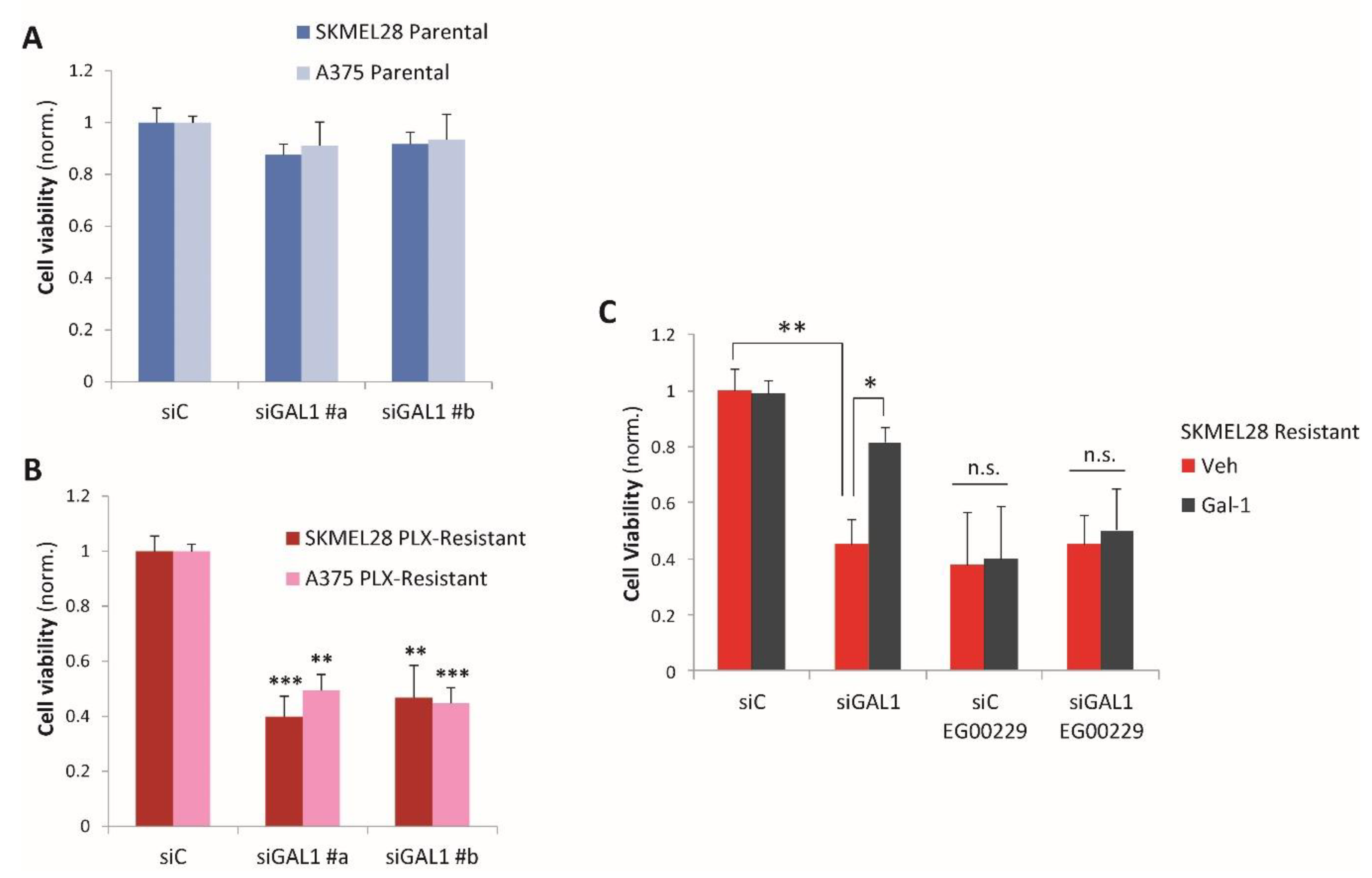
2.2. Galectin-1 Is a Novel Driver of Resistance to BRAF-Targeted Therapy in Melanoma
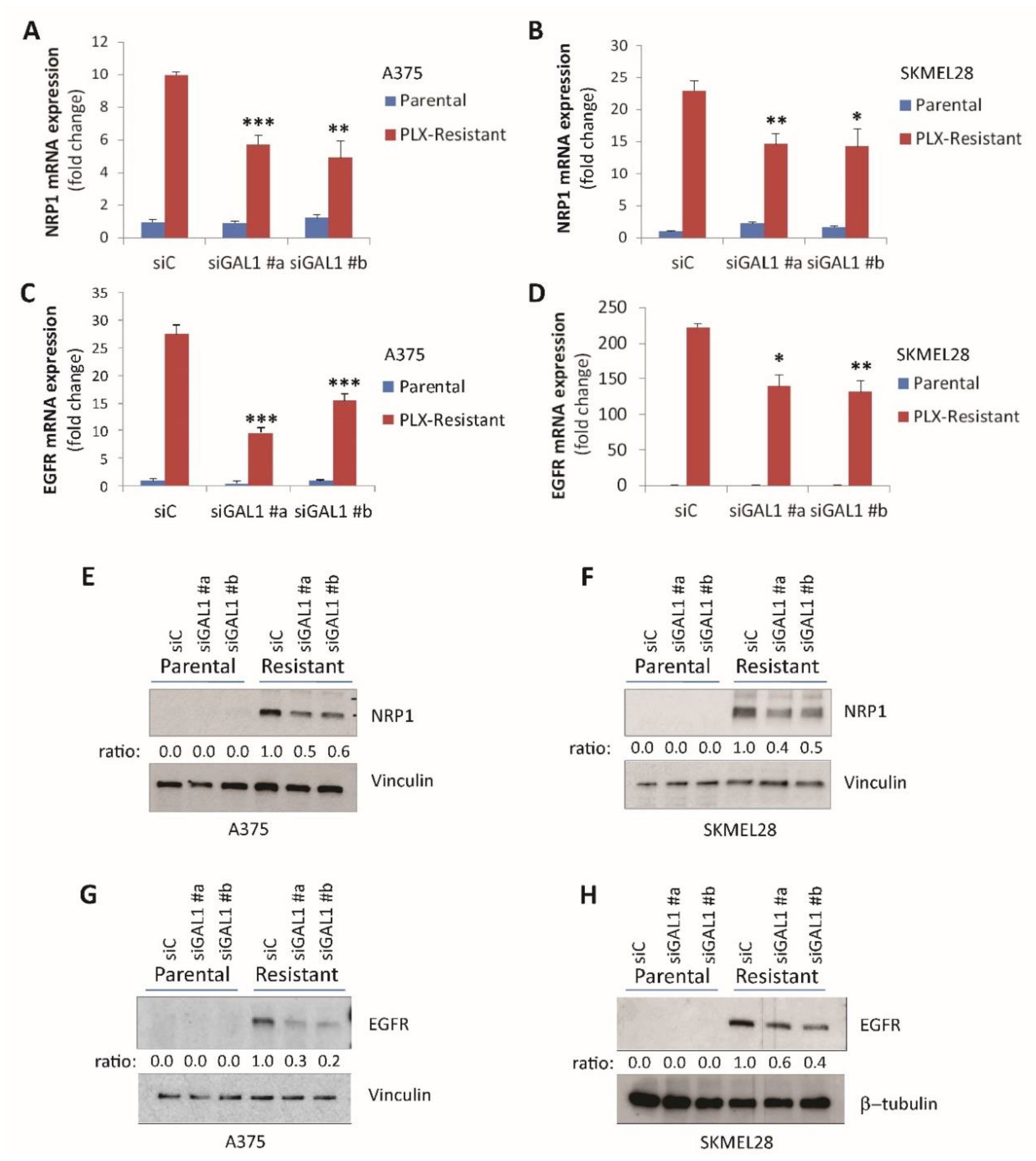
2.3. Galectin-1 Promotes NRP1 and EGFR Upregulation in Drug-Resistant Melanoma Cells
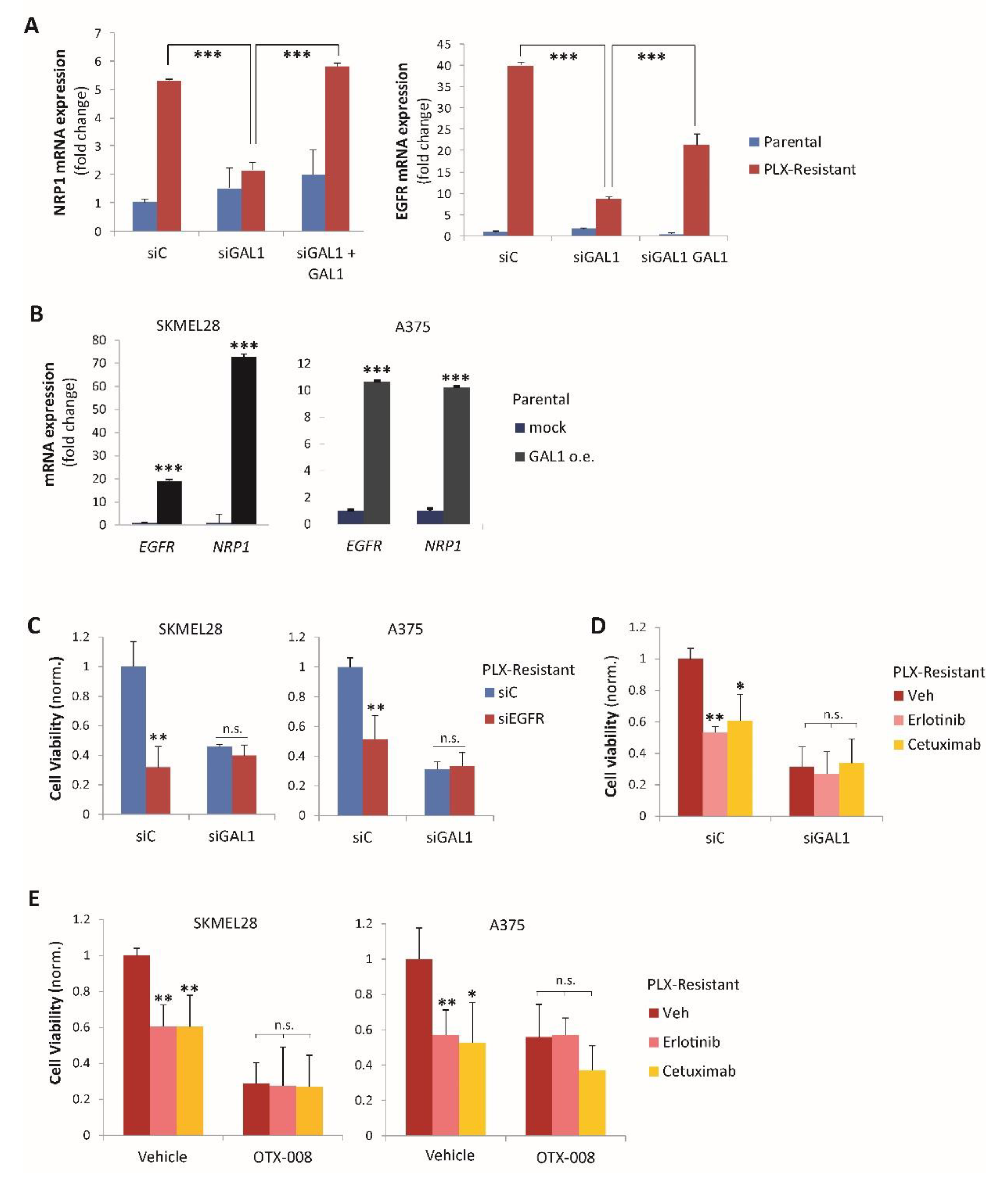
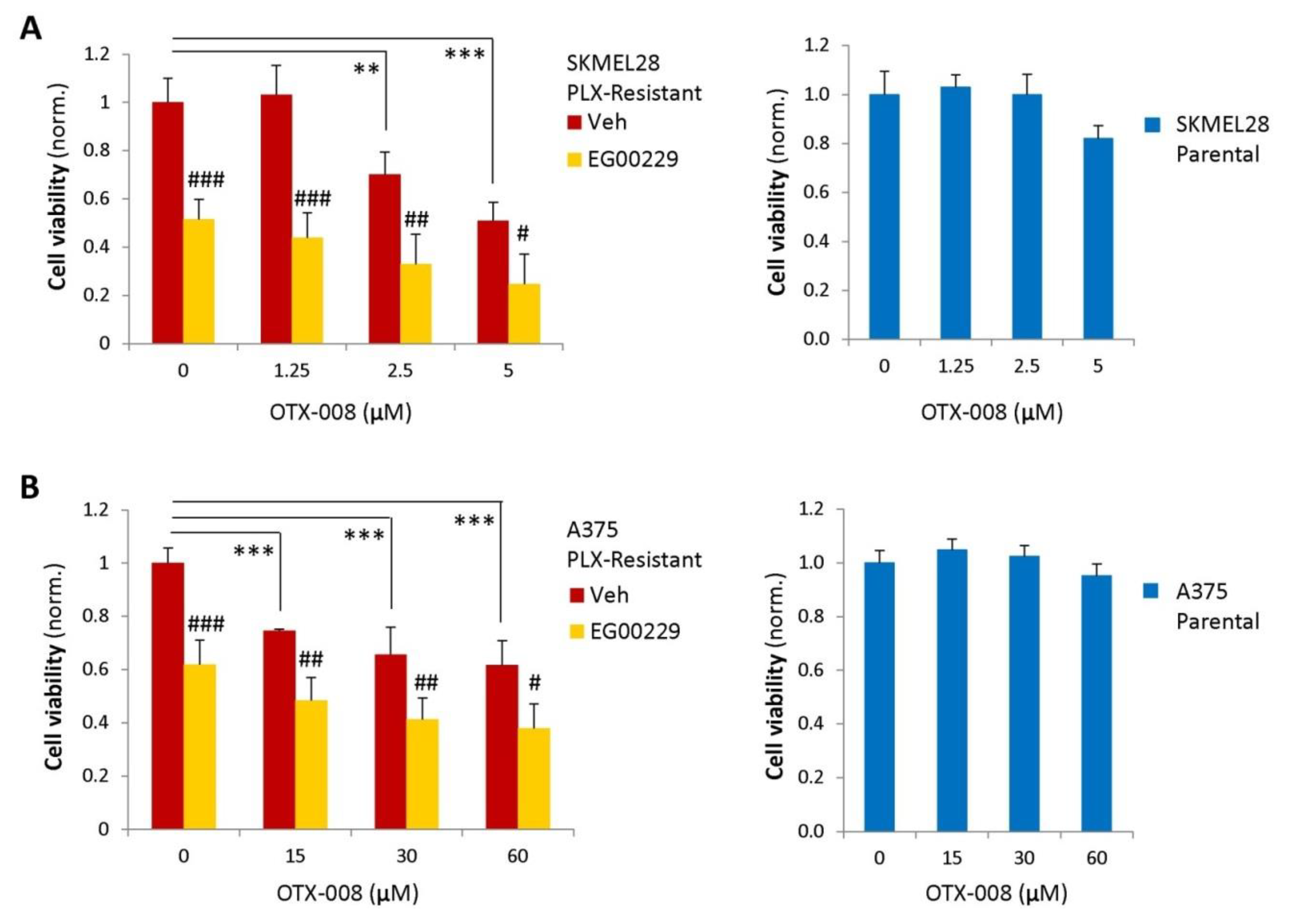
2.4. Gal-1 and NRP1 Inhibitory Drugs Cooperate to Promote Melanoma Cell Resensitization to BRAF-Targeted Therapy
3. Discussion
4. Materials and Methods
4.1. Human Cell Lines
4.2. Analysis of Galectin-1 mRNA Expression in Tumor Samples
4.3. Antibodies and Other Reagents
4.4. Establishment of Acquired Cancer Cell Resistance to Oncogene-Targeted Inhibitors
4.5. Transient Cell Transfection with cDNA and siRNA
4.6. RNA Isolation and Real-Time PCR
4.7. Cell Viability Assays
4.8. Conditioned Media Analysis by ELISA
4.9. Western Blotting Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, D.J.L.; Ribas, A. Targeted Therapy for Melanoma. Cancer Treat. Res. 2016, 167, 251–262. [Google Scholar] [CrossRef]
- Vanella, V.; Festino, L.; Trojaniello, C.; Vitale, M.G.; Sorrentino, A.; Paone, M.; Ascierto, P.A. The Role of BRAF-Targeted Therapy for Advanced Melanoma in the Immunotherapy Era. Curr. Oncol. Rep. 2019, 21, 76. [Google Scholar] [CrossRef]
- Manzano, J.L.; Layos, L.; Bugés, C.; Gil, M.D.L.L.; Vila, L.; Martínez-Balibrea, E.; Martinez-Cardus, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Herlyn, M. Resistance to BRAF inhibitors: Unraveling mechanisms and future treatment options. Cancer Res. 2011, 71, 7137–7140. [Google Scholar] [CrossRef] [PubMed]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Rizzolio, S.; Cagnoni, G.; Battistini, C.; Bonelli, S.; Isella, C.; A Van Ginderachter, J.; Bernards, R.; Di Nicolantonio, F.; Giordano, S.; Tamagnone, L. Neuropilin-1 upregulation elicits adaptive resistance to oncogene-targeted therapies. J. Clin. Investig. 2018, 128, 3976–3990. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Baek, D.-S.; Lee, S.; Park, D.; Na Kang, H.; Cho, B.C.; Kim, Y.S. Dual-targeting of EGFR and Neuropilin-1 attenuates resistance to EGFR-targeted antibody therapy in KRAS-mutant non-small cell lung cancer. Cancer Lett. 2019, 466, 23–34. [Google Scholar] [CrossRef]
- Tomida, C.; Yamagishi, N.; Nagano, H.; Uchida, T.; Ohno, A.; Hirasaka, K.; Nikawa, T.; Teshima-Kondo, S. VEGF pathway-targeting drugs induce evasive adaptation by activation of neuropilin-1/cMet in colon cancer cells. Int. J. Oncol. 2018, 52, 1350–1362. [Google Scholar] [CrossRef]
- Tse, B.W.C.; Volpert, M.; Ratther, E.; Stylianou, N.; Nouri, M.; McGowan, K.; Lehman, M.L.; McPherson, S.J.; Roshan-Moniri, M.; Butler, M.S.; et al. Neuropilin-1 is upregulated in the adaptive response of prostate tumors to androgen-targeted therapies and is prognostic of metastatic progression and patient mortality. Oncogene 2017, 36, 3417–3427. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Guttmann-Raviv, N.; Kessler, O.; Shraga-Heled, N.; Lange, T.; Herzog, Y.; Neufeld, G. The neuropilins and their role in tumorigenesis and tumor progression. Cancer Lett. 2006, 231, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major functions. Glycobiolology 2006, 16, 137R–157R. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.H.; Ying, N.W.; Wu, M.H.; Chiang, W.F.; Hsu, C.L.; Wong, T.Y.; Jin, Y.T.; Hong, T.M.; Chen, Y.L. Galectin-1, a novel ligand of neuropilin-1, activates VEGFR-2 signaling and modulates the migration of vascular endothelial cells. Oncogene 2008, 27, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef]
- Liu, F.-T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef]
- Quintá, H.R.; Pasquini, J.M.; Rabinovich, G.A.; A Pasquini, L. Glycan-dependent binding of galectin-1 to neuropilin-1 promotes axonal regeneration after spinal cord injury. Cell Death Differ. 2014, 21, 941–955. [Google Scholar] [CrossRef]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat. Rev. 2014, 40, 307–319. [Google Scholar] [CrossRef]
- Cousin, J.M.; Cloninger, M.J. The Role of Galectin-1 in Cancer Progression, and Synthetic Multivalent Systems for the Study of Galectin-1. Int. J. Mol. Sci. 2016, 17, 1566. [Google Scholar] [CrossRef]
- Dings, R.P.M.; Miller, M.C.; Nesmelova, I.V.; Astorgues-Xerri, L.; Kumar, N.; Serova, M.; Chen, X.; Raymond, E.; Hoye, T.R.; Mayo, K.H. Antitumor Agent Calixarene 0118 Targets Human Galectin-1 as an Allosteric Inhibitor of Carbohydrate Binding. J. Med. Chem. 2012, 55, 5121–5129. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, Y.; Wang, Y.; Meng, Y.; Zhu, J.; Jin, H.; Li, J.; Zhang, N.; Yu, Y.; Wu, X.; et al. A new tumour suppression mechanism by p27Kip1: EGFR down-regulation mediated by JNK/c-Jun pathway inhibition. Biochem. J. 2014, 463, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Pazarentzos, E.; Bivona, T.G. Adaptive stress signaling in targeted cancer therapy resistance. Oncogene 2015, 34, 5599–5606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Mathieu, V.; Kiss, R. Galectin-1-mediated biochemical controls of melanoma and glioma aggressive behavior. World J. Biol. Chem. 2011, 2, 193–201. [Google Scholar] [CrossRef]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Rabinovich, G.A.; Bieche, I.; Vidaud, M.; De Gramont, A.; Martinet, M.; Cvitkovic, E.; et al. OTX008, a selective small-molecule inhibitor of galectin-1, downregulates cancer cell proliferation, invasion and tumour angiogenesis. Eur. J. Cancer 2014, 50, 2463–2477. [Google Scholar] [CrossRef]
- Wu, M.H.; Ying, N.W.; Hong, T.M.; Chiang, W.F.; Lin, Y.T.; Chen, Y.L. Galectin-1 induces vascular permeability through the neuropilin-1/vascular endothelial growth factor receptor-1 complex. Angiogenesis 2014, 17, 839–849. [Google Scholar] [CrossRef]
- Gelfand, M.V.; Hagan, N.; Tata, A.; Oh, W.-J.; Lacoste, B.; Kang, K.-T.; Kopycinska, J.; Bischoff, J.; Wang, J.-H.; Gu, C. Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. eLife 2014, 3, e03720. [Google Scholar] [CrossRef]
- Rizzolio, S.; Tamagnone, L. Neuropilins as Signaling Hubs, Controlling Tyrosine Kinases and Other Cell Surface Receptors. In The Neuropilins: Role and Function in Health and Disease; Neufeld, G., Ed.; Springer Science and Business Media LLC: Cham, Switzerland, 2017; pp. 23–39. [Google Scholar]
- Li, L.; Jiang, X.; Zhang, Q.; Dong, X.; Gao, Y.; He, Y.; Qiao, H.; Xie, F.; Xie, X.-J.; Sun, X. Neuropilin-1 is associated with clinicopathology of gastric cancer and contributes to cell proliferation and migration as multifunctional co-receptors. J. Exp. Clin. Cancer Res. 2016, 35, 16. [Google Scholar] [CrossRef]
- Yoshida, A.; Shimizu, A.; Asano, H.; Kadonosono, T.; Kondoh, S.K.; Geretti, E.; Mammoto, A.; Klagsbrun, M.; Kurokawa-Seo, M. VEGF-A/NRP1 stimulates GIPC1 and Syx complex formation to promote RhoA activation and proliferation in skin cancer cells. Biol. Open 2015, 4, 1063–1076. [Google Scholar] [CrossRef]
- Blanchard, H.; Bum-Erdene, K.; Bohari, M.H.; Yu, X. Galectin-1 inhibitors and their potential therapeutic applications: A patent review. Expert Opin. Ther. Patents 2016, 26, 537–554. [Google Scholar] [CrossRef]
- Raymond, E.; Astrorgue-Xerri, L.; Serova, M.; Riveiro, M.E.; Faivre, S. Translational Rational for the Clinical Development of OTX-008: A Novel Drug That Inhibits Galectin-1 Expression. In Human Cancer Models in Galectins and Disease Implications for Targeted Therapeutics; ACS Symposium Series; ACS: Washington, DC, USA, 2012; Volume 1115, pp. 259–266. [Google Scholar]
- Chou, F.-C.; Chen, H.-Y.; Kuo, C.-C.; Sytwu, H.-K. Role of Galectins in Tumors and in Clinical Immunotherapy. Int. J. Mol. Sci. 2018, 19, 430. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, K.; Francuz, T.; Gallego-Colon, E.; Ruiz-Agamez, N.; Kubeczko, M.; Grochoła, I.; Wojnar, J. Galectin Targeted Therapy in Oncology: Current Knowledge and Perspectives. Int. J. Mol. Sci. 2018, 19, 210. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kim, J.-A.; Kim, Y.-J.; Lee, H.W.; Kim, C.-H.; Haam, S.; Kim, Y.S. A Neuropilin-1 Antagonist Exerts Antitumor Immunity by Inhibiting the Suppressive Function of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2020, 8, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, M.; Voilin, E.; Gros, G.; Corgnac, S.; De Montpréville, V.; Validire, P.; Bismuth, G.; Mami-Chouaib, F. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat. Commun. 2019, 10, 3345. [Google Scholar] [CrossRef]
- Tsai, Y.-T.; Liang, C.-H.; Yu, J.-H.; Huang, K.-C.; Tung, C.-H.; Wu, J.-E.; Wu, Y.-Y.; Chang, C.-H.; Hong, T.-M.; Chen, Y.-L. A DNA Aptamer Targeting Galectin-1 as a Novel Immunotherapeutic Strategy for Lung Cancer. Mol. Ther. Nucleic Acids 2019, 18, 991–998. [Google Scholar] [CrossRef]
- Kwong, L.N.; Boland, G.M.; Frederick, D.T.; Helms, T.L.; Akid, A.T.; Miller, J.P.; Jiang, S.; Cooper, Z.A.; Song, X.; Seth, S.; et al. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2015, 125, 1459–1470. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzolio, S.; Corso, S.; Giordano, S.; Tamagnone, L. Autocrine Signaling of NRP1 Ligand Galectin-1 Elicits Resistance to BRAF-Targeted Therapy in Melanoma Cells. Cancers 2020, 12, 2218. https://doi.org/10.3390/cancers12082218
Rizzolio S, Corso S, Giordano S, Tamagnone L. Autocrine Signaling of NRP1 Ligand Galectin-1 Elicits Resistance to BRAF-Targeted Therapy in Melanoma Cells. Cancers. 2020; 12(8):2218. https://doi.org/10.3390/cancers12082218
Chicago/Turabian StyleRizzolio, Sabrina, Simona Corso, Silvia Giordano, and Luca Tamagnone. 2020. "Autocrine Signaling of NRP1 Ligand Galectin-1 Elicits Resistance to BRAF-Targeted Therapy in Melanoma Cells" Cancers 12, no. 8: 2218. https://doi.org/10.3390/cancers12082218
APA StyleRizzolio, S., Corso, S., Giordano, S., & Tamagnone, L. (2020). Autocrine Signaling of NRP1 Ligand Galectin-1 Elicits Resistance to BRAF-Targeted Therapy in Melanoma Cells. Cancers, 12(8), 2218. https://doi.org/10.3390/cancers12082218