The Roles of Imprinted SLC22A18 and SLC22A18AS Gene Overexpression Caused by Promoter CpG Island Hypomethylation as Diagnostic and Prognostic Biomarkers for Non-Small Cell Lung Cancer Patients

 , , ,
, , , 
Abstract
1. Introduction
2. Results
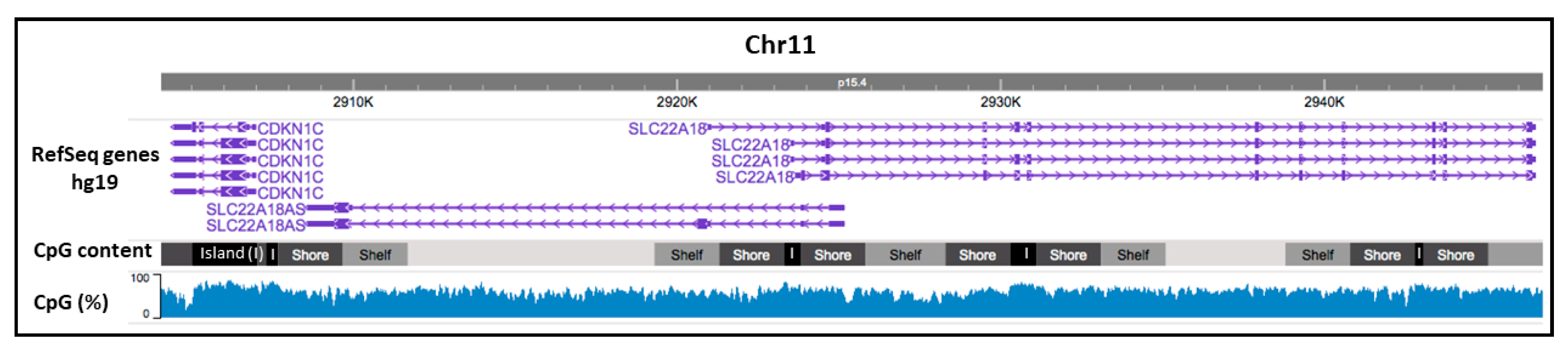
2.1. CpG Promoter Methylation Status of the Imprinted SLC22A18 and SLC22A18AS Genes in NSCLC Patients
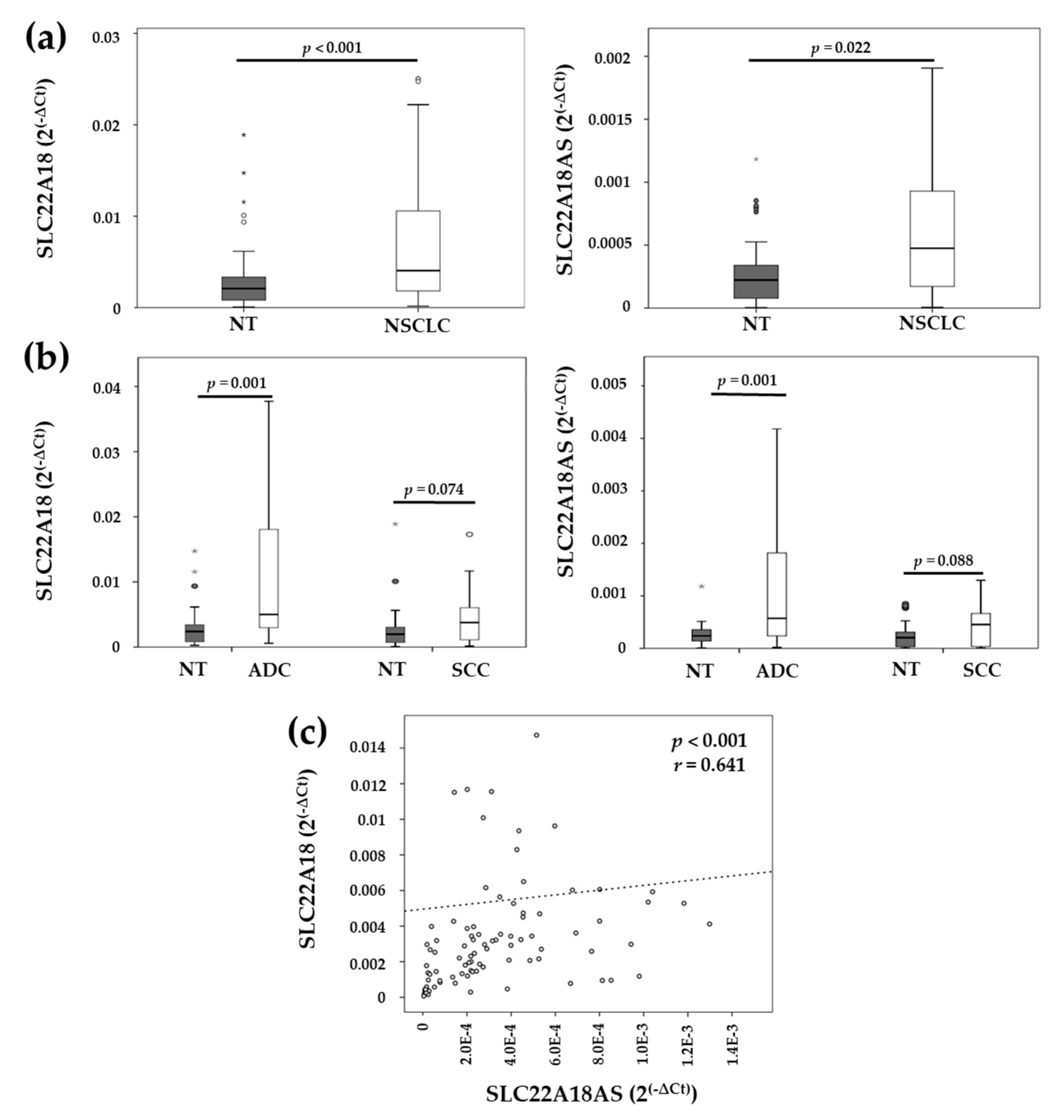
2.2. Expression Levels of the Imprinted SLC22A18 and SLC22A18AS Genes in NSCLC Tissue
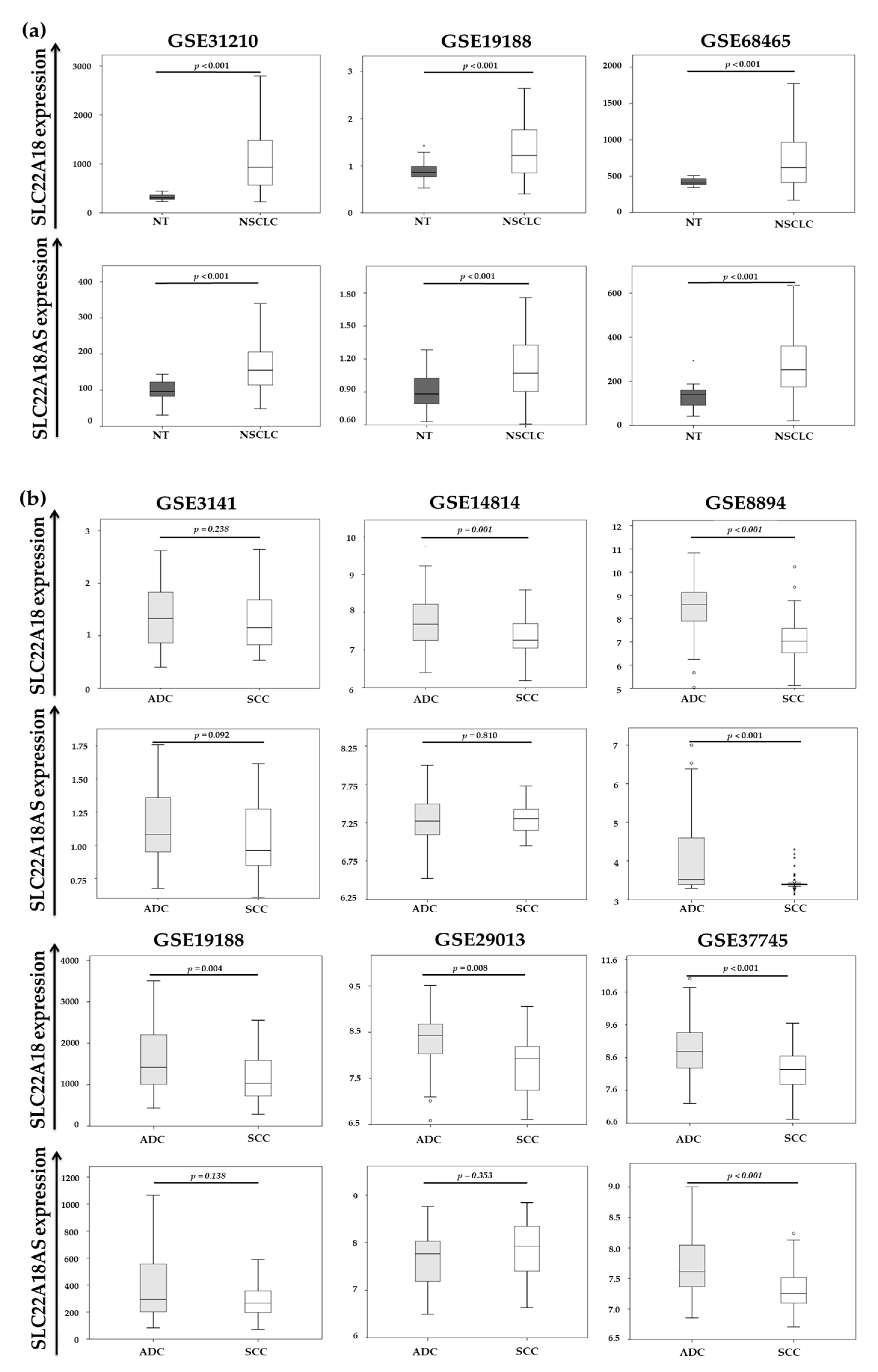
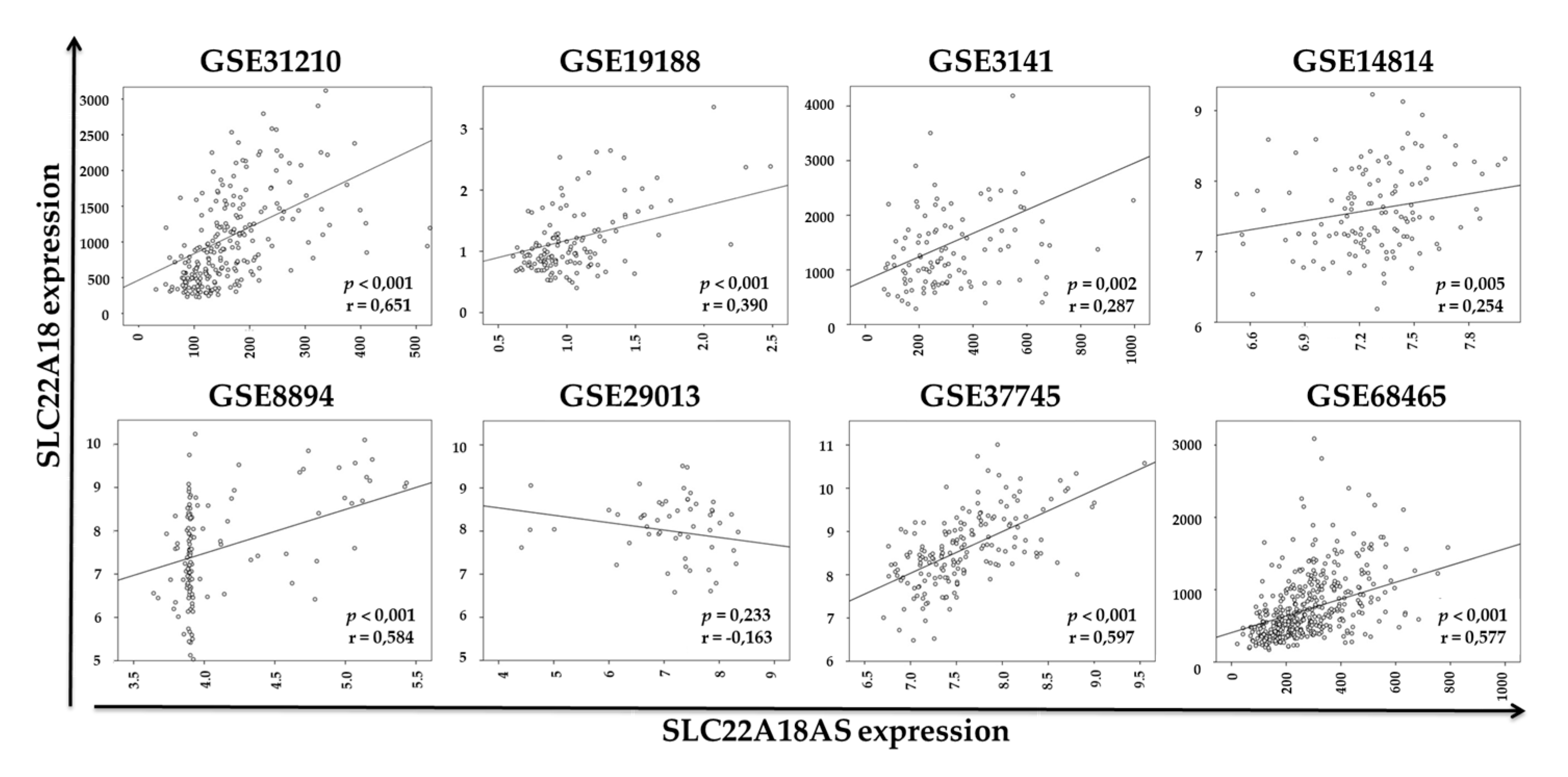
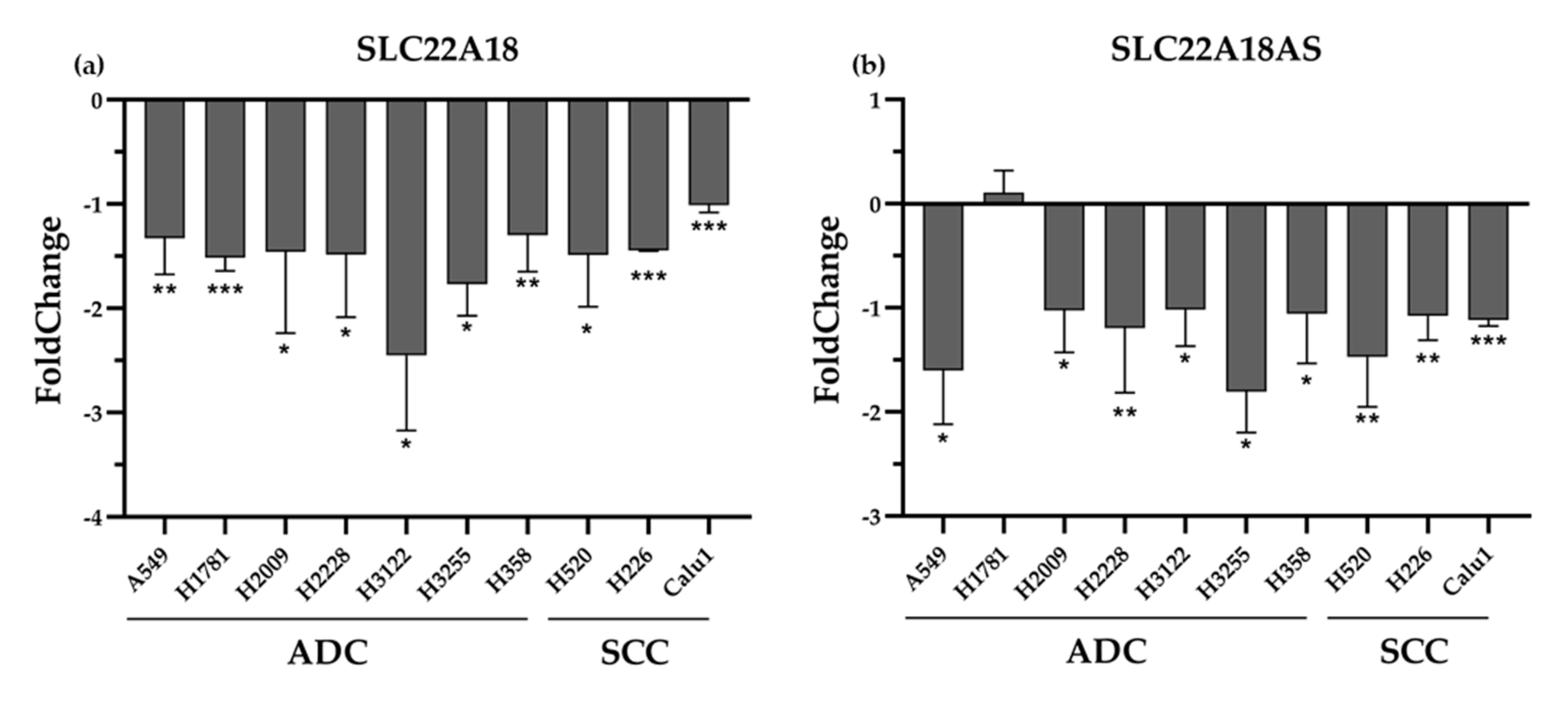
2.3. Validation of the Expression Profiles of the SLC22A18 and SLC22A18AS Genes in the NSCLC Samples in Public Databases
2.4. Rescue of DNA Methylation Status of the SLC22A18 and SLC22A18AS Genes In Vitro by Treatment with Ademetionine
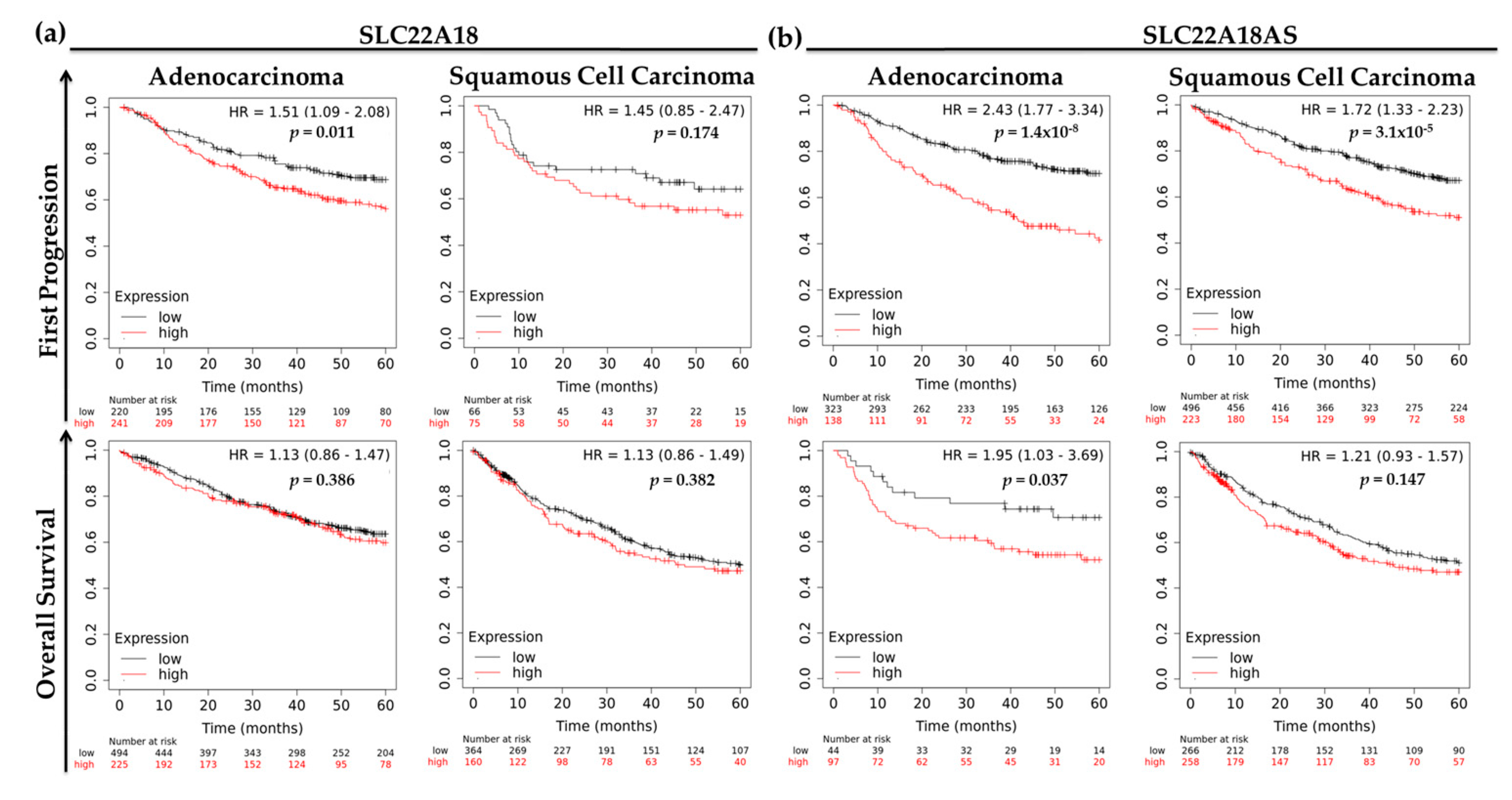
2.5. Prognostic Roles of the Imprinted SLC22A18 and SLC22A18AS Genes for NSCLC Patients
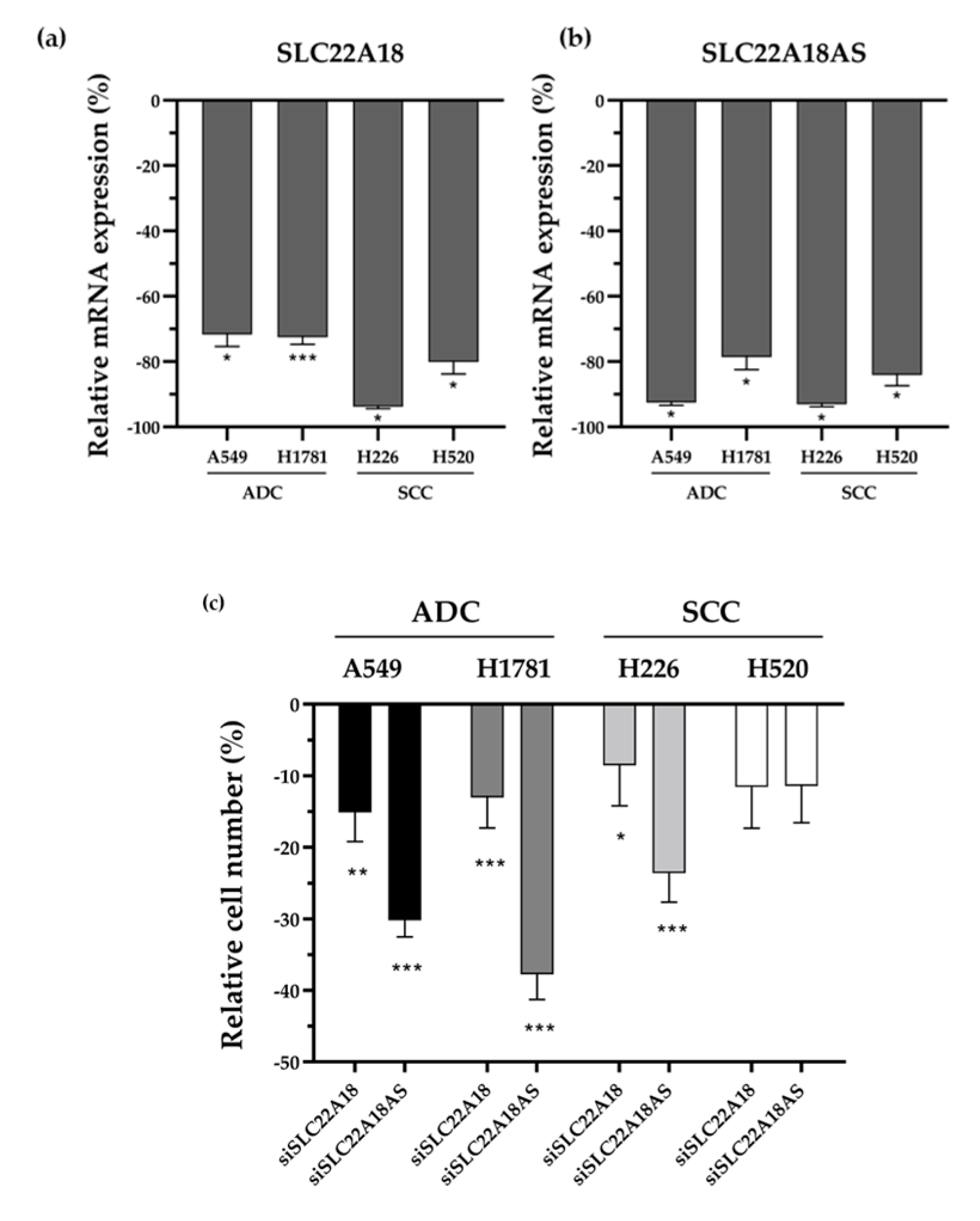
2.6. SLC22A18 and SLC22A18AS Knockdown Impairs Tumor Cell Proliferation
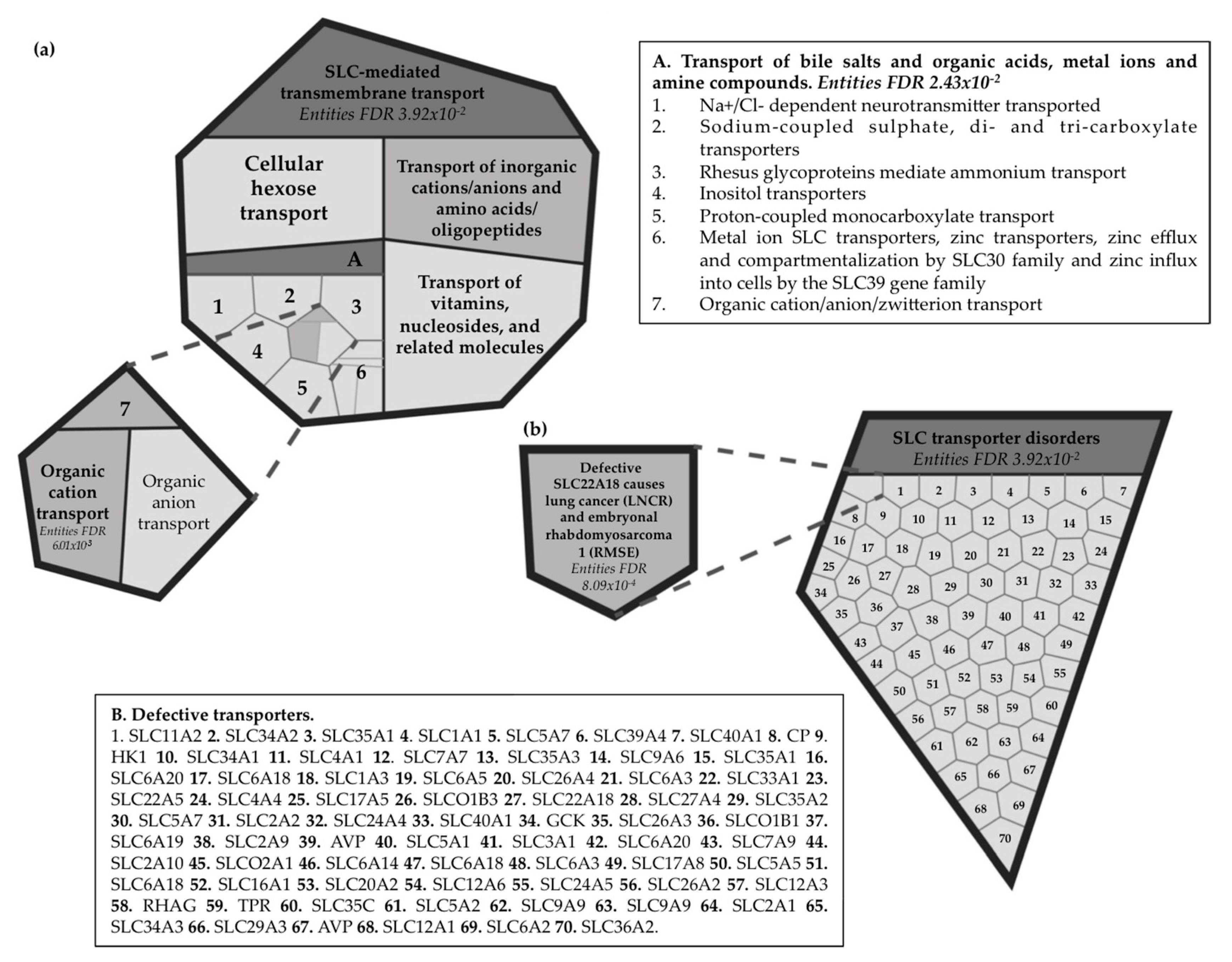
2.7. Reactome Pathway Analysis for the Imprinted SLC22A18 and SLC22A18AS Genes
3. Discussion
4. Materials and Methods
4.1. Patient and Sample Selection
4.2. Cell Culture and Treatments
4.3. SiRNA Transfections
4.4. Cell Proliferation Assay
4.5. Nucleic Acid Isolation
4.6. Bisulfite Transformation
4.7. DNA Methylation Pattern of the SLC22A18 and SLC22A18AS Genes
4.8. Expression Levels of the SLC22A18 and SLC22A18AS Genes
4.9. Validation of the Expression Analysis for SLC22A18 and SLC22A18AS in the NSCLC Patients Using Public Databases
4.10. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NSCLC | Non-small cell lung cancer | SCLC | Small cell lung cancer |
| SCC | Squamous cell carcinoma | ADC | Adenocarcinoma |
| SLC22A18 | Solute-carrier family 22 member 18 | TCGA | The Cancer Genome Atlas |
| SLC22A18AS | Solute-carrier family 22 member 18 antisense | HR | Hazard ratio |
| CI | Confidence interval | FDR | False discovery rate |
| NT | Nontumoral lung tissue | DMSO | Dimethyl Sulfoxide |
References
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015, 34, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Shackelford, R.E.; El-Osta1, H. Epigenetics in non-small cell lung cancer: From basics to therapeutics. Transl. Lung Cancer Res. 2016, 5, 155–171. [Google Scholar] [CrossRef]
- Wu, X.Y.; Chen, H.C.; Li, W.W.; Yan, J.D.; Lv, R.Y. DNMT1 promotes cell proliferation via methylating hMLH1 and hMSH2 promoters in EGFR-mutated non-small cell lung cancer. J. Biochem. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Balgkouranidou, I.; Liloglou, T.; Lianidou, E.S. Lung cancer epigenetics: Emerging biomarkers. Biomark Med. 2013, 7, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Brzeziańska, E.; Dutkowska, A.; Antczak, A. The significance of epigenetic alterations in lung carcinogenesis. Mol. Biol. Rep. 2013, 40, 309–325. [Google Scholar] [CrossRef]
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung Cancer. Med. Clin. North Am. 2019, 103, 463–473. [Google Scholar] [CrossRef]
- Pirker, R. Conquering lung cancer: Current status and prospects for the future. Pulmonology 2020, 20, 30031–30033. [Google Scholar] [CrossRef]
- O’Hagan, H.M.; Wang, W.; Sen, S.; DeStefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, Á.; Molina-Pinelo, S. Epigenetics of lung cancer: A translational perspective. Cell Oncol. 2019, 42, 739–756. [Google Scholar] [CrossRef]
- Bartolomei, M.S. Genomic imprinting: Employing and avoiding epigenetic processes. Genes Dev. 2009, 23, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Mai, M.; Yokomizo, A.; Qian, C.; Yang, P.; Tindall, D.J.; Smith, D.I.; Liu, W. Activation of p73 silent allele in lung cancer. Cancer Res. 1998, 58, 2347–2349. [Google Scholar] [PubMed]
- Molina-Pinelo, S.; Salinas, A.; Moreno-Mata, N.; Ferrer, I.; Suarez, R.; Andrés-León, E.; Rodríguez-Paredes, M.; Gutekunst, J.; Jantus-Lewintre, E.; Camps, C.; et al. Impact of DLK1-DIO3 imprinted cluster hypomethylation in smoker patients with lung cancer. Oncotarget 2018, 9, 4395–4410. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, V.; Singhmar, P.; Kumar, A. Promoter characterization and regulation of expression of an imprinted gene SLC22A18AS. Gene 2008, 424, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.; Mc Goldrick, A.; Chung, W.Y.; Mc Cormack, O.; Harrison, M.; Kerin, M.; Dervan, P.A.; Mc Cann, A. Gain of imprinting of SLC22A18 sense and antisense transcripts in human breast cancer. Genomics 2006, 88, 12–17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Yang, S.; Thangaraju, M.; Ganapathy, V. SLC transporters as a novel class of tumour suppressors: Identity, function and molecular mechanisms. Biochem. J. 2016, 473, 1113–1124. [Google Scholar] [CrossRef]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar]
- Goovaerts, T.; Steyaert, S.; Vandenbussche, C.A.; Galle, J.; Thas, O.; Van Criekinge, W.; De Meyer, T. A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat. Commun. 2018, 9, 4120. [Google Scholar] [CrossRef]
- Kim, J.; Bretz, C.L.; Lee, S. Epigenetic instability of imprinted genes in human cancers. Nucleic. Acids. Res. 2015, 43, 10689–10699. [Google Scholar] [CrossRef]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol. 2007, 211, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. IGF2 and cancer. Endocr. Relat. Cancer 2013, 20, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wu, C.H.; Zhu, X.L.; Wang, Y.J. Loss of imprinting of insulin-like growth factor 2 is associated with increased risk of primary lung cancer in the central china region. Asian Pacific J. Cancer Prev. 2014, 15, 7799–7803. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tian, F.; Tang, Z.; Song, G.; Pan, Y.; He, B.; Bao, Q.; Wang, S. Loss of imprinting of IGF2 correlates with hypomethylation of the H19 differentially methylated region in the tumor tissue of colorectal cancer patients. Mol. Med. Rep. 2012, 5, 1536–1540. [Google Scholar]
- Schagdarsurengin, U.; Lammert, A.; Schunk, N.; Sheridan, D.; Gattenloehner, S.; Steger, K.; Wagenlehner, F.; Dansranjavin, T. Impairment of IGF2 gene expression in prostate cancer is triggered by epigenetic dysregulation of IGF2-DMR0 and its interaction with KLF4. Cell Commun. Signal. 2017, 15, 40. [Google Scholar] [CrossRef]
- Setty, B.A.; Jinesh, G.G.; Arnold, M.; Pettersson, F.; Cheng, C.-H.; Cen, L.; Yoder, S.J.; Teer, J.K.; Flores, E.R.; Reed, D.R.; et al. The genomic landscape of undifferentiated embryonal sarcoma of the liver is typified by C19MC structural rearrangement and overexpression combined with TP53 mutation or loss. PLoS Genet. 2020, 16, e1008642. [Google Scholar] [CrossRef]
- Sin-Chan, P.; Mumal, I.; Suwal, T.; Ho, B.; Fan, X.; Singh, I.; Du, Y.; Lu, M.; Patel, N.; Torchia, J.; et al. A C19MC-LIN28A-MYCN Oncogenic Circuit Driven by Hijacked Super-enhancers Is a Distinct Therapeutic Vulnerability in ETMRs: A Lethal Brain Tumor. Cancer Cell 2019, 36, 51–67. [Google Scholar] [CrossRef]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef]
- Kasprzak, A.; Adamek, A. Insulin-Like Growth Factor 2 (IGF2) Signaling in Colorectal Cancer-From Basic Research to Potential Clinical Applications. Int. J. Mol. Sci. 2019, 20, 4915. [Google Scholar] [CrossRef]
- Kohda, M.; Hoshiya, H.; Katoh, M.; Tanaka, I.; Masuda, R.; Takemura, T.; Fujiwara, M.; Oshimura, M. Frequent loss of imprinting of IGF2 and MEST in lung adenocarcinoma. Mol. Carcinog. 2001, 31, 184–191. [Google Scholar] [CrossRef]
- Chu, S.H.; Feng, D.F.; Ma, Y.B.; Zhang, H.; Zhu, Z.A.; Li, Z.Q.; Jiang, P.C. Promoter methylation and downregulation of SLC22A18 are associated with the development and progression of human glioma. J. Transl. Med. 2011, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Ma, Y.B.; Feng, D.F.U.; Zhang, H.; Qiu, J.H.; Zhu, Z.A.N. Effect of 5-Aza-2′-deoxycytidine on SLC22A18 in glioma U251 cells. Mol. Med. Rep. 2012, 5, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Elsnerova, K.; Mohelnikova-Duchonova, B.; Cerovska, E.; Ehrlichova, M.; Gut, I.; Rob, L.; Skapa, P.; Hruda, M.; Bartakova, A.; Bouda, J.; et al. Gene expression of membrane transporters: Importance for prognosis and progression of ovarian carcinoma. Oncol. Rep. 2016, 35, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Mohelnikova-Duchonova, B.; Brynychova, V.; Hlavac, V.; Kocik, M.; Oliverius, M.; Hlavsa, J.; Honsova, E.; Mazanec, J.; Kala, Z.; Melichar, B.; et al. The association between the expression of solute carrier transporters and the prognosis of pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 72, 669–682. [Google Scholar] [CrossRef]
- Alberg, A.J.; Brock, M.V.; Ford, J.G.; Samet, J.M.; Spivack, S.D. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American college of chest physicians evidence-based clinical practice guidelines. Chest 2013, 143, e1S–e29S. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Meijer, T.W.H.; Schuurbiers, O.C.J.; Kaanders, J.H.A.M.; Looijen-Salamon, M.G.; de Geus-Oei, L.F.; Verhagen, A.F.T.M.; Lok, J.; van der Heijden, H.F.M.; Rademakers, S.E.; Span, P.N.; et al. Differences in metabolism between adeno- and squamous cell non-small cell lung carcinomas: Spatial distribution and prognostic value of GLUT1 and MCT4. Lung Cancer 2012, 76, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Bajaj, V.; Gopinath, K.S.; Kumar, A. Characterization of the human SLC22A18 gene promoter and its regulation by the transcription factor Sp1. Gene 2009, 429, 37–43. [Google Scholar] [CrossRef]
- Lei, M.; Cheng, Q.; Zhao, Y.; Liu, T.; Wang, X.; Deng, Y.; Yang, J.; Zhang, Z. Expression and its clinical significance of SLC22a18 in non-small cell lung cancer. Chinese J. Lung Cancer 2012, 15, 17–20. [Google Scholar]
- Zhang, B.; Liu, T.; Wu, T.; Wang, Z.; Rao, Z.; Gao, J. MicroRNA-137 functions as a tumor suppressor in human non-small cell lung cancer by targeting SLC22A18. Int. J. Biol. Macromol. 2015, 74, 111–118. [Google Scholar] [CrossRef]
- Jung, Y.; Jun, Y.; Lee, H.Y.; Kim, S.; Jung, Y.; Keum, J.; Lee, Y.S.; Cho, Y.B.; Lee, S.; Kim, J. Characterization of SLC22A18 as a tumor suppressor and novel biomarker in colorectal cancer. Oncotarget 2015, 6, 25368–25380. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Fujino, Y.; Ogata, S.; Hirayama-Kurogi, M.; Ohtsuki, S. Involvement of an Orphan Transporter, SLC22A18, in Cell Growth and Drug Resistance of Human Breast Cancer MCF7 Cells. J. Pharm. Sci. 2018, 107, 3163–3170. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Xu, C.; Zhao, Z.; Qin, X.; Xu, H.; Zhang, H. Low expression of SLC22A18 predicts poor survival outcome in patients with breast cancer after surgery. Cancer Epidemiol. 2011, 35, 279–285. [Google Scholar] [CrossRef]
- Yamada, H.Y.; Gorbsky, G.J. Tumor suppressor candidate TSSC5 is regulated by UbcH6 and a novel ubiquitin ligase RING105. Oncogene 2006, 25, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Honda, G.; Fujino, Y.; Ogata, S.; Hirayama-Kurogi, M.; Ohtsuki, S. Knockdown of Orphan Transporter SLC22A18 Impairs Lipid Metabolism and Increases Invasiveness of HepG2 Cells. Pharm. Res. 2019, 36, 1–11. [Google Scholar] [CrossRef]
- Schwienbacher, C.; Gramantieri, L.; Scelfo, R.; Veronese, A.; Calin, G.A.; Bolondi, L.; Croce, C.M.; Barbanti-Brodano, G.; Negrini, M. Gain of imprinting at chromosome 11p15: A pathogenetic mechanism identified in human hepatocarcinomas. Proc. Natl. Acad. Sci. USA 2000, 97, 5445–5449. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, F.; Lan, R. RNA-sequencing dissects the transcriptome of polyploid cancer cells that are resistant to combined treatments of cisplatin with paclitaxel and docetaxel. Mol. Biosyst. 2017, 13, 2125–2134. [Google Scholar] [CrossRef]
- Bajkowska, K.; Sumardika, I.W.; Tomonobu, N.; Chen, Y.; Yamamoto, K. ichi; Kinoshita, R.; Murata, H.; Gede Yoni Komalasari, N.L.; Jiang, F.; Yamauchi, A.; et al. Neuroplastinβ-mediated upregulation of solute carrier family 22 member 18 antisense (SLC22A18AS) plays a crucial role in the epithelial-mesenchymal transition, leading to lung cancer cells’ enhanced motility. Biochem. Biophys. Reports 2020, 22, 100768. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Niland, J.C.; Townsend, R.M.; Annechiarico, R.; Johnson, K.; Beck, J.R.; Manion, F.J.; Robbins, R.J.; Chute, C.G.; Vogel, L.H.; Saltz, J.H.; et al. The cancer biomedical informatics grid (caBIGTM): Infrastructure and applications for a worldwide research community. Stud. Health Technol. Inform. 2007, 129, 330–334. [Google Scholar]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets - Update. Nucleic Acids Res. 2013, 41, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Gordon, L.; Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Diyagama, D.; Neilson, J.; van Laar, R.; Dobrovic, A.; Holloway, A.; Smyth, G.K. Empirical array quality weights in the analysis of microarray data. BMC. Bioinformatics 2006, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, D.; Lowdon, R.F.; Costello, J.F.; Wang, T. MethylC Track: Visual integration of single-base resolution DNA methylation data on the WashU EpiGenome Browser. Bioinformatics 2014, 30, 2206–2207. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | First cohort (N = 70) | Second cohort (N = 56) | |
|---|---|---|---|
| Study group (N = 47) | Control group (N = 23) | ||
| Age (years) | 67 (60–73) | 35 (21–62) | 69 (63–75] |
| Gender | |||
| Male | 76.6 (36) | 87.0 (20) | 82.1 (46) |
| Female | 23.4 (11) | 13.0 (3) | 17.9 (10) |
| Smoking status | |||
| Smokers | 85.1 (40) | 52.2 (12) | 94.6 (53) |
| Nonsmokers | 14.9 (7) | 47.8 (11) | 5.4 (3) |
| Histology | |||
| Lung adenocarcinoma | 57.4 (27) | - | 50.0 (28) |
| Squamous cell lung carcinoma | 42.6 (20) | - | 50.0 (28) |
| Staging | |||
| I | 40.5 (19) | - | 50.0 (28) |
| II | 38.3 (18) | - | 21.4 (12) |
| III–IV | 21.2 (10) | - | 17.9 (10) |
| Subjects with COPD | 42.6 (20) | 17.4 (4) | 53.6 (30) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noguera-Uclés, J.F.; Boyero, L.; Salinas, A.; Cordero Varela, J.A.; Benedetti, J.C.; Bernabé-Caro, R.; Sánchez-Gastaldo, A.; Alonso, M.; Paz-Ares, L.; Molina-Pinelo, S. The Roles of Imprinted SLC22A18 and SLC22A18AS Gene Overexpression Caused by Promoter CpG Island Hypomethylation as Diagnostic and Prognostic Biomarkers for Non-Small Cell Lung Cancer Patients. Cancers 2020, 12, 2075. https://doi.org/10.3390/cancers12082075
Noguera-Uclés JF, Boyero L, Salinas A, Cordero Varela JA, Benedetti JC, Bernabé-Caro R, Sánchez-Gastaldo A, Alonso M, Paz-Ares L, Molina-Pinelo S. The Roles of Imprinted SLC22A18 and SLC22A18AS Gene Overexpression Caused by Promoter CpG Island Hypomethylation as Diagnostic and Prognostic Biomarkers for Non-Small Cell Lung Cancer Patients. Cancers. 2020; 12(8):2075. https://doi.org/10.3390/cancers12082075
Chicago/Turabian StyleNoguera-Uclés, José Francisco, Laura Boyero, Ana Salinas, Juan Antonio Cordero Varela, Johana Cristina Benedetti, Reyes Bernabé-Caro, Amparo Sánchez-Gastaldo, Miriam Alonso, Luis Paz-Ares, and Sonia Molina-Pinelo. 2020. "The Roles of Imprinted SLC22A18 and SLC22A18AS Gene Overexpression Caused by Promoter CpG Island Hypomethylation as Diagnostic and Prognostic Biomarkers for Non-Small Cell Lung Cancer Patients" Cancers 12, no. 8: 2075. https://doi.org/10.3390/cancers12082075
APA StyleNoguera-Uclés, J. F., Boyero, L., Salinas, A., Cordero Varela, J. A., Benedetti, J. C., Bernabé-Caro, R., Sánchez-Gastaldo, A., Alonso, M., Paz-Ares, L., & Molina-Pinelo, S. (2020). The Roles of Imprinted SLC22A18 and SLC22A18AS Gene Overexpression Caused by Promoter CpG Island Hypomethylation as Diagnostic and Prognostic Biomarkers for Non-Small Cell Lung Cancer Patients. Cancers, 12(8), 2075. https://doi.org/10.3390/cancers12082075