The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis

 and
and
Abstract
1. Introduction
2. Cancer-Associated Fibroblasts (CAFs)
3. CAF Heterogeneity
4. Stroma Characteristics in Prostate Homeostasis and Disease
4.1. The Stroma in Prostate Homeostasis
4.2. The Stroma in Benign Prostatic Hyperplasia
4.3. The Stroma in Pre-Neoplastic Lesions
4.4. The Reactive Stroma in Prostate Cancer
5. Tumor–CAF Interactions in Primary Prostate Cancer
5.1. ECM Remodeling by CAFs
5.2. CAF-Tumor Signaling: Paracrine, Systemic, Cell–Cell Direct, and Metabolic
5.3. CAF-Mediated Resistance to Androgen Deprivation and Chemotherapy
6. Stroma in PCa Bone Metastasis
Role of CAFs in PCa Bone Metastasis
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| List of Abbreviations: | Full Name: |
| α-SMA | α- smooth muscle actin |
| ADT | Androgen-deprivation therapy |
| AR | Androgen Receptor |
| ASPN | Asporin |
| BCR | Biochemical recurrence |
| BM | Bone metastasis |
| BMPs | Bone morphogenetic proteins |
| BPH | Benign prostatic hyperplasia |
| CAFs | Cancer-associated fibroblasts |
| CD | Cluster of designation antigens |
| CRPC | Castration-resistant prostate cancer |
| CXCL | Chemokine (C-X-C motif) ligand |
| CXCR | Chemokine (C-X-C motif) receptor |
| ECM | Extracellular matrix |
| EMT | Epithelial to mesenchymal transition |
| EVs | Extracellular vesicles |
| FAP | Fibroblast activation protein |
| FGFs | Fibroblast growth factors |
| FSP-1 (also known as S100A4) | Fibroblast-specific protein-1 |
| GDF15 | Growth differentiation factor 15 |
| HGF | Hepatocyte growth factor |
| HSCs | Hematopoietic stem cells |
| IGFs | Insulin-like growth factors |
| ILs | Interleukins |
| MFBs | Myofibroblasts |
| MMPs | Matrix metalloproteinases |
| OB-BMST | Osteoblastic bone metastasis-associated stroma transcriptome |
| PCa | Prostate cancer |
| PDGFR | Platelet-derived growth factor receptor |
| PIA | Proliferative inflammatory atrophy |
| PIN | Prostatic intraepithelial neoplasia |
| POSTN | Periostin |
| PSA | Prostate-specific antigen |
| ROS | Reactive-oxygen species |
| TGF-β | Transforming growth factor -β |
| TGFβRII | TGF-β receptor type II |
| TIMPs | Tissue inhibitors of matrix metalloproteinases |
| TME | Tumor microenvironment |
| TNC | Tenascin C |
| VEGF | Vascular endothelial growth factor |
References
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Oncogenic resistance to growth-limiting conditions. Nat. Rev. Cancer 2002, 2, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4567. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Humphrey, P.A. Histopathology of prostate cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Sartor, O.; Lewis, B. Beyond Just Androgen Deprivation Therapy: Novel Therapies Combined With Radiation. Semin. Radiat. Oncol. 2017, 27, 87–93. [Google Scholar] [CrossRef]
- Gamat, M.; McNeel, D.G. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr. Relat. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.N.S.; Ferraldeschi, R.; Attard, G.; De Bono, J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol. 2014, 11, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Li, H.; Zhou, J.; Chen, X.; Tang, D.G. Cellular determinants and microenvironmental regulation of prostate cancer metastasis. Semin. Cancer Biol. 2017, 44, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Eder, T.; Weber, A.; Neuwirt, H.; Grünbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-associated fibroblastsmodify the response of prostate cancer cells to androgen and anti-androgens in three-dimensional spheroid culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef]
- Kato, M.; Placencio-Hickok, V.R.; Madhav, A.; Haldar, S.; Tripathi, M.; Billet, S.; Mishra, R.; Smith, B.; Rohena-Rivera, K.; Agarwal, P.; et al. Heterogeneous cancer-associated fibroblast population potentiates neuroendocrine differentiation and castrate resistance in a CD105-dependent manner. Oncogene 2019, 38, 716–730. [Google Scholar] [CrossRef]
- Ayala, G.; Tuxhorn, J.A.; Wheeler, T.M.; Frolov, A.; Scardino, P.T.; Ohori, M.; Wheeler, M.; Spitler, J.; Rowley, D.R. Reactive Stroma as a Predictor of Biochemical-Free Recurrence in Prostate Cancer. Clin. Cancer Res. 2003, 9, 4792–4801. [Google Scholar]
- Chiarugi, P.; Paoli, P.; Cirri, P. Tumor microenvironment and metabolism in prostate cancer. Semin. Oncol. 2014, 41, 267–280. [Google Scholar] [CrossRef]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar]
- Thalmann, G.N.; Rhee, H.; Sikes, R.A.; Pathak, S.; Multani, A.; Zhau, H.E.; Marshall, F.F.; Chung, L.W.K. Human Prostate Fibroblasts Induce Growth and Confer Castration Resistance and Metastatic Potential in LNCaP Cells. Eur. Urol. 2010, 58, 162–172. [Google Scholar] [CrossRef]
- Turpin, A.; Duterque-Coquillaud, M.; Vieillard, M.H. Bone Metastasis: Current State of Play. Eur. Urol. 2010, 58, 162–172. [Google Scholar] [CrossRef]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Rucci, N.; Teti, A. Osteomimicry: How the Seed Grows in the Soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Investig. 2009, 119, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.F.; Mustoe, T.A.; Altrock, B.W.; Deuel, T.F.; Thomason, A. Role of platelet-derived growth factor in wound healing. J. Cell. Biochem. 1991, 45, 319–326. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Newman, A.C.; Nakatsu, M.N.; Chou, W.; Gershon, P.D.; Hughes, C.C.W. The requirement for fibroblasts in angiogenesis: Fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol. Biol. Cell 2011, 22, 3791–3800. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Turley, S.J. A short field guide to fibroblast function in immunity. Semin. Immunol. 2018, 35, 48–58. [Google Scholar] [CrossRef]
- Brizzi, M.F.; Tarone, G.; Defilippi, P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr. Opin. Cell Biol. 2012, 24, 645–651. [Google Scholar] [CrossRef]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Levesque, C.; Nelson, P.S. Cellular constituents of the prostate stroma: Key contributors to prostate cancer progression and therapy resistance. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Flier, J.S.; Underhill, L.H.; Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Orr, B.; Riddick, A.C.P.; Stewart, G.D.; Anderson, R.A.; Franco, O.E.; Hayward, S.W.; Thomson, A.A. Identification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts, normal fibroblasts and fetal prostate. Oncogene 2012, 31, 1130–1142. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Ishii, K.; Sasaki, T.; Iguchi, K.; Kajiwara, S.; Kato, M.; Kanda, H.; Hirokawa, Y.; Arima, K.; Mizokami, A.; Sugimura, Y. Interleukin-6 induces VEGF secretion from prostate cancer cells in a manner independent of androgen receptor activation. Prostate 2018, 78, 849–856. [Google Scholar] [CrossRef]
- Bruzzese, F.; Hägglöf, C.; Leone, A.; Sjöberg, E.; Roca, M.S.; Kiflemariam, S.; Sjöblom, T.; Hammarsten, P.; Egevad, L.; Bergh, A.; et al. Local and systemic protumorigenic effects of cancer-associatedfibroblast- derived GDF15. Cancer Res. 2014, 74, 3408–3417. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar] [PubMed]
- Ishii, K.; Mizokami, A.; Tsunoda, T.; Iguchi, K.; Kato, M.; Hori, Y.; Arima, K.; Namiki, M.; Sugimura, Y. Heterogenous induction of carcinoma-associated fibroblast-like differentiation in normal human prostatic fibroblasts by co-culturing with prostate cancer cells. J. Cell. Biochem. 2011, 112, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Hensel, J.; Secondini, C.; Wetterwald, A.; Schwaninger, R.; Fleischmann, A.; Raffelsberger, W.; Poch, O.; Delorenzi, M.; Temanni, R.; et al. The molecular signature of the stroma response in prostate cancer-induced osteoblastic bone metastasis highlights expansion of hematopoietic and prostate epithelial stem cell niches. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Rochette, A.; Boufaied, N.; Scarlata, E.; Hamel, L.; Brimo, F.; Whitaker, H.C.; Ramos-Montoya, A.; Neal, D.E.; Dragomir, A.; Aprikian, A.; et al. Asporin is a stromally expressed marker associated with prostate cancer progression. Br. J. Cancer 2017, 116, 775–784. [Google Scholar] [CrossRef]
- Ao, M.; Franco, O.E.; Park, D.; Raman, D.; Williams, K.; Hayward, S.W. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 2007, 67, 4244–4253. [Google Scholar] [CrossRef]
- Memarzadeh, S.; Xin, L.; Mulholland, D.J.; Mansukhani, A.; Wu, H.; Teitell, M.A.; Witte, O.N. Enhanced Paracrine FGF10 Expression Promotes Formation of Multifocal Prostate Adenocarcinoma and an Increase in Epithelial Androgen Receptor. Cancer Cell 2007, 12, 572–585. [Google Scholar] [CrossRef]
- Yang, F.; Strand, D.W.; Rowley, D.R. Fibroblast growth factor-2 mediates transforming growth factor-Β action in prostate cancer reactive stroma. Oncogene 2008, 27, 450–459. [Google Scholar] [CrossRef]
- Liu, A.Y.; True, L.D. Characterization of prostate cell types by CD cell surface molecules. Am. J. Pathol. 2002, 160, 37–43. [Google Scholar] [CrossRef]
- Ustach, C.V.; Taube, M.E.; Hurst, N.J.; Bhagat, S.; Bonfil, R.D.; Cher, M.L.; Schuger, L.; Kim, H.R.C. A Potential Oncogenic Activity of Platelet-Derived Growth Factor D in Prostate Cancer Progression. Cancer Res. 2004, 64, 1722–1729. [Google Scholar] [CrossRef][Green Version]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Shahriari, K.; Shen, F.; Worrede-Mahdi, A.; Liu, Q.; Gong, Y.; Garcia, F.U.; Fatatis, A. Cooperation among heterogeneous prostate cancer cells in the bone metastatic niche. Oncogene 2017, 36, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Roudier, M.P.; True, L.D. Heterogeneity in primary and metastatic prostate cancer as defined by cell surface CD profile. Am. J. Pathol. 2004, 165, 1543–1556. [Google Scholar] [CrossRef]
- Zhao, H.; Peehl, D.M. Tumor-promoting phenotype of CD90hi prostate cancer-associated fibroblasts. Prostate 2009, 69, 991–1000. [Google Scholar] [CrossRef]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S.; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-β signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef]
- Kiskowski, M.A.; Jackson, R.S.; Banerjee, J.; Li, X.; Kang, M.; Iturregui, J.M.; Franco, O.E.; Hayward, S.W.; Bhowmick, N.A. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011, 71, 3459–3470. [Google Scholar] [CrossRef]
- Vickman, R.E.; Broman, M.M.; Lanman, N.A.; Franco, O.E.; Sudyanti, P.A.G.; Ni, Y.; Ji, Y.; Helfand, B.T.; Petkewicz, J.; Paterakos, M.C.; et al. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate 2020, 80, 173–185. [Google Scholar] [CrossRef]
- Eiro, N.; Fernandez-Gomez, J.; Sacristán, R.; Fernandez-Garcia, B.; Lobo, B.; Gonzalez-Suarez, J.; Quintas, A.; Escaf, S.; Vizoso, F.J. Stromal factors involved in human prostate cancer development, progression and castration resistance. J. Cancer Res. Clin. Oncol. 2017, 143, 351–359. [Google Scholar] [CrossRef]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1238–1260. [Google Scholar] [CrossRef] [PubMed]
- Josson, S.; Matsuoka, Y.; Chung, L.W.K.; Zhau, H.E.; Wang, R. Tumor-stroma co-evolution in prostate cancer progression and metastasis. Semin. Cell Dev. Biol. 2010, 21, 26–32. [Google Scholar] [CrossRef]
- Corn, P.G. The tumor microenvironment in prostate cancer: Elucidating molecular pathways for therapy development. Cancer Manag. Res. 2012, 4, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, S.; Ishii, K.; Sasaki, T.; Kato, M.; Nishikawa, K.; Kanda, H.; Arima, K.; Watanabe, M.; Sugimura, Y. Castration-induced stromal remodeling disrupts the reconstituted prostate epithelial structure. Lab. Investig. 2019, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285. [Google Scholar]
- Leong, K.G.; Wang, B.E.; Johnson, L.; Gao, W.Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–810. [Google Scholar] [CrossRef]
- Wang, X.; Julio, M.K.; De Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef]
- Liu, A.; Wei, L.; Gardner, W.A.; Deng, C.X.; Man, Y.G. Correlated alterations in prostate basal cell layer and basement membrane. Int. J. Biol. Sci. 2009, 5, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Hayward, S.W.; Dahiya, R.; Foster, B.A. Smooth Muscle-Epithelial Interactions in Normal and Neoplastic Prostatic Development. Cells Tissues Organs 1996, 155, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Leimgruber, C.; Quintar, A.A.; Peinetti, N.; Scalerandi, M.V.; Nicola, J.P.; Miano, J.M.; Maldonado, C.A. Testosterone Rescues the De-Differentiation of Smooth Muscle Cells Through Serum Response Factor/Myocardin. J. Cell. Physiol. 2017, 232, 2806–2817. [Google Scholar] [CrossRef] [PubMed]
- Peinetti, N.; Scalerandi, M.V.; Rubio, M.M.C.; Leimgruber, C.; Nicola, J.P.; Torres, A.I.; Quintar, A.A.; Maldonado, C.A. The response of prostate smooth muscle cells to testosterone is determined by the subcellular distribution of the androgen receptor. Endocrinology 2018, 159, 945–956. [Google Scholar] [CrossRef]
- Yang, Z.; Peng, Y.C.; Gopalan, A.; Gao, D.; Chen, Y.; Joyner, A.L. Stromal hedgehog signaling maintains smooth muscle and hampers micro-invasive prostate cancer. DMM Dis. Model. Mech. 2017, 10, 39–52. [Google Scholar] [CrossRef]
- Bolt, C.C.; Negi, S.; Guimarães-Camboa, N.; Zhang, H.; Troy, J.M.; Lu, X.; Kispert, A.; Evans, S.M.; Stubbs, L. Tbx18 Regulates the Differentiation of Periductal Smooth Muscle Stroma and the Maintenance of Epithelial Integrity in the Prostate. PLoS ONE 2016, 11, e0154413. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Marchi, V.L.; Epstein, J.I.; Nelson, W.G. Proliferative inflammatory atrophy of the prostate: Implications for prostatic carcinogenesis. Am. J. Pathol. 1999, 155, 1985–1992. [Google Scholar] [CrossRef]
- Ziada, A.; Rosenblum, M.; Crawford, E.D. Benign prostatic hyperplasia: An overview. Urology 1999, 53, 1–6. [Google Scholar] [CrossRef]
- McNeal, J.E. The zonal anatomy of the prostate. Prostate 1981, 2, 35–49. [Google Scholar] [CrossRef]
- Van Der Heul-Nieuwenhuijsen, L.; Hendriksen, P.J.M.; Van Der Kwast, T.H.; Jenster, G. Gene expression profiling of the human prostate zones. BJU Int. 2006, 98, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Shaikhibrahim, Z.; Lindstrot, A.; Ellinger, J.; Rogenhofer, S.; Buettner, R.; Wernert, N. Genes differentially expressed in the peripheral zone compared to the transitional zone of the normal human prostate and their potential regulation by ETS factors. Mol. Med. Rep. 2012, 5, 32–36. [Google Scholar]
- Wang, X.; Wang, Y.; Gratzke, C.; Sterr, C.; Yu, Q.; Li, B.; Strittmatter, F.; Herlemann, A.; Tamalunas, A.; Rutz, B.; et al. Ghrelin Aggravates Prostate Enlargement in Rats with Testosterone-Induced Benign Prostatic Hyperplasia, Stromal Cell Proliferation, and Smooth Muscle Contraction in Human Prostate Tissues. Oxid. Med. Cell. Longev. 2019, 2019, 4748312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gratzke, C.; Tamalunas, A.; Rutz, B.; Ciotkowska, A.; Strittmatter, F.; Herlemann, A.; Janich, S.; Waidelich, R.; Liu, C.; et al. Smooth muscle contraction and growth of stromal cells in the human prostate are both inhibited by the Src family kinase inhibitors, AZM475271 and PP2. Br. J. Pharmcol. 2016, 173, 3342–3358. [Google Scholar] [CrossRef] [PubMed]
- Schwinn, D.A. The role of α1-adrenergic receptor subtypes in lower urinary tract symptoms. BJU Int. 2001, 88, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Larson, J.A.; Andriole, G.L. Management of Benign Prostatic Hyperplasia. Annu. Rev. Med. 2016, 67, 137–151. [Google Scholar] [CrossRef]
- Li, F.; Pascal, L.E.; Stolz, D.B.; Wang, K.; Zhou, Y.; Chen, W.; Xu, Y.; Chen, Y.; Dhir, R.; Parwani, A.V.; et al. E-cadherin is downregulated in benign prostatic hyperplasia and required for tight junction formation and permeability barrier in the prostatic epithelial cell monolayer. Prostate 2019, 79, 1226–1237. [Google Scholar] [CrossRef]
- Schauer, I.G.; Rowley, D.R. The functional role of reactive stroma in benign prostatic hyperplasia. Differentiation 2011, 82, 200–210. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef]
- Begley, L.A.; Kasina, S.; MacDonald, J.; Macoska, J.A. The inflammatory microenvironment of the aging prostate facilitates cellular proliferation and hypertrophy. Cytokine 2008, 43, 194–199. [Google Scholar] [CrossRef]
- Schauer, I.G.; Ressler, S.J.; Tuxhorn, J.A.; Dang, T.D.; Rowley, D.R. Elevated Epithelial Expression of Interleukin-8 Correlates with Myofibroblast Reactive Stroma in Benign Prostatic Hyperplasia. Urology 2008, 72, 205–213. [Google Scholar] [CrossRef]
- Ho, C.K.M.; Habib, F.K. Estrogen and androgen signaling in the pathogenesis of BPH. Nat. Rev. Urol. 2011, 8, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.P.; Huang, C.K.; Fang, L.Y.; Izumi, K.; Lo, C.W.; Wood, R.; Kindblom, J.; Yeh, S.; Chang, C. Targeting stromal androgen receptor suppresses prolactin-driven benign prostatic hyperplasia (BPH). Mol. Endocrinol. 2013, 27, 1617–1631. [Google Scholar] [CrossRef][Green Version]
- Izumi, K.; Mizokami, A.; Lin, W.J.; Lai, K.P.; Chang, C. Androgen receptor roles in the development of benign prostate hyperplasia. Am. J. Pathol. 2013, 182, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Gangkak, G.; Bhattar, R.; Mittal, A.; Yadav, S.S.; Tomar, V.; Yadav, A.; Mehta, J. Immunohistochemical analysis of estrogen receptors in prostate and clinical correlation in men with benign prostatic hyperplasia. Investig. Clin. Urol. 2017, 58, 117–126. [Google Scholar] [CrossRef]
- Ruska, K.M.; Sauvageot, J.; Epstein, J.I. Histology and cellular kinetics of prostatic atrophy. Am. J. Surg. Pathol. 1998, 22, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M. High-grade prostatic intraepithelial neoplasia, PIN-like carcinoma, ductal carcinoma, and intraductal carcinoma of the prostate. Mod. Pathol. 2018, 31, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Häggman, M.J.; Macoska, J.A.; Wojno, K.J.; Oesterling, J.E. The relationship between prostatic intraepithelial neoplasia and prostate cancer: Critical issues. J. Urol. 1997, 158, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187. [Google Scholar] [CrossRef]
- Kryza, T.; Silva, L.M.; Bock, N.; Fuhrman-Luck, R.A.; Stephens, C.R.; Gao, J.; Samaratunga, H.; Australian Prostate Cancer BioResource; Lawrence, M.G.; Hooper, J.D.; et al. Kallikrein-related peptidase 4 induces cancer-associated fibroblast features in prostate-derived stromal cells. Mol. Oncol. 2017, 11, 1307–1329. [Google Scholar] [CrossRef]
- Wegner, K.A.; Mueller, B.R.; Unterberger, C.J.; Avila, E.J.; Ruetten, H.; Turco, A.E.; Oakes, S.R.; Girardi, N.M.; Halberg, R.B.; Swanson, S.M.; et al. Prostate epithelial-specific expression of activated PI3K drives stromal collagen production and accumulation. J. Pathol. 2020, 250, 231–242. [Google Scholar] [CrossRef]
- Wang, M.; Nagle, R.B.; Knudsen, B.S.; Rogers, G.C.; Cress, A.E. A basal cell defect promotes budding of prostatic intraepithelial neoplasia. J. Cell Sci. 2017, 130, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.W.; Wang, Y.; Cao, M.; Hom, Y.K.; Zhang, B.; Grossfeld, G.D.; Sudilovsky, D.; Cunha, G.R. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001, 61, 8135–8142. [Google Scholar] [PubMed]
- Gleave, M.; Hsieh, J.T.; Gao, C.A.; von Eschenbach, A.C.; Chung, L.W. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Res. 1991, 51, 3753–3761. [Google Scholar]
- Bianchi-Frias, D.; Basom, R.; Delrow, J.J.; Coleman, I.M.; Dakhova, O.; Qu, X.; Fang, M.; Franco, O.E.; Ericson, N.G.; Bielas, J.H.; et al. Cells comprising the prostate cancer microenvironment lack recurrent clonal somatic genomic aberrations. Mol. Cancer Res. 2016, 14, 374–384. [Google Scholar] [CrossRef]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef] [PubMed]
- Tomas, D.; Ulamec, M.; Hudolin, T.; Bulimbašić, S.; Belicza, M.; Krušlin, B. Myofibroblastic stromal reaction and expression of tenascin-C and laminin in prostate adenocarcinoma. Prostate Cancer Prostatic Dis. 2006, 9, 414–419. [Google Scholar] [CrossRef]
- Tuxhorn, J.A.; McAlhany, S.J.; Dang, T.D.; Ayala, G.E.; Rowley, D.R. Stromal Cells Promote Angiogenesis and Growth of Human Prostate Tumors in a Differential Reactive Stroma (DRS) Xenograft Model. Cancer Res. 2002, 62, 3298–3307. [Google Scholar]
- Mahal, B.A.; Alshalalfa, M.; Zhao, S.G.; Beltran, H.; Chen, W.S.; Chipidza, F.; Davicioni, E.; Karnes, R.J.; Ku, S.Y.; Lotan, T.L.; et al. Genomic and clinical characterization of stromal infiltration markers in prostate cancer. Cancer 2020, 126, 1407–1412. [Google Scholar] [CrossRef]
- Mo, F.; Lin, D.; Takhar, M.; Ramnarine, V.R.; Dong, X.; Bell, R.H.; Volik, S.V.; Wang, K.; Xue, H.; Wang, Y.; et al. Stromal Gene Expression is Predictive for Metastatic Primary Prostate Cancer. Eur. Urol. 2018, 73, 524–532. [Google Scholar] [CrossRef]
- Andersen, M.K.; Rise, K.; Giskeødegård, G.F.; Richardsen, E.; Bertilsson, H.; Størkersen, Ø.; Bathen, T.F.; Rye, M.; Tessem, M.B. Integrative metabolic and transcriptomic profiling of prostate cancer tissue containing reactive stroma. Sci. Rep. 2018, 8, 14269. [Google Scholar] [CrossRef]
- Ersvær, E.; Hveem, T.S.; Vlatkovic, L.; Brennhovd, B.; Kleppe, A.; Tobin, K.A.R.; Pradhan, M.; Cyll, K.; Wæhre, H.; Kerr, D.J.; et al. Prognostic value of DNA ploidy and automated assessment of stroma fraction in prostate cancer. Int. J. Cancer 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mccandless, J.R.; Cress, A.E.; Rabinovitz, I.; Payne, C.M.; Bowden, G.T.; Knox, J.D.; Nagle, R.B. A human xenograft model for testing early events of epithelial neoplastic invasion. Int. J. Oncol. 1997, 10, 279–285. [Google Scholar] [CrossRef]
- Richards, Z.; McCray, T.; Marsili, J.; Zenner, M.L.; Manlucu, J.T.; Garcia, J.; Kajdacsy-Balla, A.; Murray, M.; Voisine, C.; Murphy, A.B.; et al. Prostate Stroma Increases the Viability and Maintains the Branching Phenotype of Human Prostate Organoids. iScience 2019, 12, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ivich, F.; Tahsin, S.; Tran, M.; Frank, S.B.; Miranti, C.K.; Zohar, Y. Human stroma and epithelium co-culture in a microfluidic model of a human prostate gland. Biomicrofluidics 2019, 13, 064116. [Google Scholar] [CrossRef] [PubMed]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikström, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011, 179, 1961–1968. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef]
- Nguyen, E.V.; Pereira, B.A.; Lawrence, M.G.; Ma, X.; Rebello, R.J.; Chan, H.; Niranjan, B.; Wu, Y.; Ellem, S.; Guan, X.; et al. Proteomic profiling of human prostate cancer-associated fibroblasts (CAF) reveals LOXL2-dependent regulation of the tumor microenvironment. Mol. Cell. Proteom. 2019, 18, 1410–1427. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Gong, Y.; Chippada-Venkata, U.D.; Oh, W.K. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers 2014, 6, 1298–1327. [Google Scholar] [CrossRef]
- Wood, M.; Fudge, K.; Mohler, J.L.; Frost, A.R.; Garcia, F.; Wang, M.; Stearns, M.E. In situ hybridization studies of metalloproteinases 2 and 9 and TIMP-1 and TIMP-2 expression in human prostate cancer. Clin. Exp. Metastasis 1997, 15, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Al-Shareef, Z.; Gorroño-Etxebarria, I.; Atkins, S.; Turrell, F.; Chhetri, J.; Bengoa-Vergniory, N.; Zenzmaier, C.; Berger, P.; Waxman, J.; et al. Dickkopf-3 regulates prostate epithelial cell acinar morphogenesis and prostate cancer cell invasion by limiting TGF-β-dependent activation of matrix metalloproteases. Carcinogenesis 2016, 37, 18–29. [Google Scholar] [CrossRef]
- Al Shareef, Z.; Kardooni, H.; Murillo-Garzón, V.; Domenici, G.; Stylianakis, E.; Steel, J.H.; Rabano, M.; Gorroño-Etxebarria, I.; Zabalza, I.; Vivanco, M.M.; et al. Protective effect of stromal Dickkopf-3 in prostate cancer: Opposing roles for TGFBI and ECM-1. Oncogene 2018, 37, 5305–5324. [Google Scholar] [CrossRef]
- Sampson, N.; Brunner, E.; Weber, A.; Puhr, M.; Schäfer, G.; Szyndralewiez, C.; Klocker, H. Inhibition of Nox4-dependent ROS signaling attenuates prostate fibroblast activation and abrogates stromal-mediated protumorigenic interactions. Int. J. Cancer 2018, 143, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, L.; Lin, H.K.; Kan, P.Y.; Xie, S.; Tsai, M.Y.; Wang, P.H.; Chen, Y.T.; Chang, C. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem. Biophys. Res. Commun. 2003, 305, 462–469. [Google Scholar] [CrossRef]
- Culig, Z.; Puhr, M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol. Cell. Endocrinol. 2018, 462, 25–30. [Google Scholar] [CrossRef]
- Joesting, M.S.; Perrin, S.; Elenbaas, B.; Fawell, S.E.; Rubin, J.S.; Franco, O.E.; Hayward, S.W.; Cunha, G.R.; Marker, P.C. Identification of SFRP1 as a candidate mediator of stromal-to-epithelial signaling in prostate cancer. Cancer Res. 2005, 65, 10423–10430. [Google Scholar] [CrossRef]
- Henke, A.; Franco, O.; Stewart, G.; Riddick, A.; Katz, E.; Hayward, S.; Thomson, A. Reduced Contractility and Motility of Prostatic Cancer-Associated Fibroblasts after Inhibition of Heat Shock Protein 90. Cancers 2016, 8, 77. [Google Scholar] [CrossRef]
- Wilkinson, S.E.; Furic, L.; Buchanan, G.; Larsson, O.; Pedersen, J.; Frydenberg, M.; Risbridger, G.P.; Taylor, R.A. Hedgehog signaling is active in human prostate cancer stroma and regulates proliferation and differentiation of adjacent epithelium. Prostate 2013, 73, 1810–1823. [Google Scholar] [CrossRef]
- Shen, T.; Li, Y.; Zhu, S.; Yu, J.; Zhang, B.; Chen, X.; Zhang, Z.; Ma, Y.; Niu, Y.; Shang, Z. YAP1 plays a key role of the conversion of normal fibroblasts into cancer-associated fibroblasts that contribute to prostate cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 36. [Google Scholar] [CrossRef]
- Vlaeminck-Guillem, V. Extracellular vesicles in prostate cancer carcinogenesis, diagnosis, and management. Front. Oncol. 2018, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Josson, S.; Gururajan, M.; Sung, S.Y.; Hu, P.; Shao, C.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; Li, Q.; et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2015, 34, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Johnen, H.; Lin, S.; Kuffner, T.; Brown, D.A.; Tsai, V.W.W.; Bauskin, A.R.; Wu, L.; Pankhurst, G.; Jiang, L.; Junankar, S.; et al. Tumor-induced anorexia and weight loss are mediated by the TGF-β superfamily cytokine MIC-1. Nat. Med. 2007, 13, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat. Cell Biol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhang, B.; Zhang, L.; Dang, T.; Rowley, D.; Ittmann, M.; Xin, L. Jagged1 upregulation in prostate epithelial cells promotes formation of reactive stroma in the Pten null mouse model for prostate cancer. Oncogene 2017, 36, 618–627. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Cioni, B.; Zwart, W.; Bergman, A.M. Androgen receptor moonlighting in the prostate cancer microenvironment. Endocr. Relat. Cancer 2018, 25, R331–R349. [Google Scholar] [CrossRef]
- Cioni, B.; Nevedomskaya, E.; Melis, M.H.M.; van Burgsteden, J.; Stelloo, S.; Hodel, E.; Spinozzi, D.; de Jong, J.; van der Poel, H.; de Boer, J.P.; et al. Loss of androgen receptor signaling in prostate cancer-associated fibroblasts (CAFs) promotes CCL2- and CXCL8-mediated cancer cell migration. Mol. Oncol. 2018, 12, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Jha, R.; Melamed, J.; Shapiro, E.; Hayward, S.W.; Lee, P. Stromal androgen receptor in prostate development and cancer. Am. J. Pathol. 2014, 184, 2598–2607. [Google Scholar] [CrossRef] [PubMed]
- Wikström, P.; Marusic, J.; Stattin, P.; Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 2009, 69, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, C.; Choong, C.S.; Buchanan, G.; Vivekanandan, S.; Neufing, P.; Stahl, J.; Marshall, V.R.; Horsfall, D.J.; Tilley, W.D. Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate 2005, 63, 19–28. [Google Scholar] [CrossRef]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Augsten, M.; Rundqvist, H.; Bianchi, J.; Sarne, V.; Egevad, L.; Bykov, V.J.; Östman, A.; Wiman, K.G. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 2017, 8, e2848. [Google Scholar] [CrossRef]
- Ammirante, M.; Shalapour, S.; Kang, Y.; Jamieson, C.A.M.; Karin, M. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl. Acad. Sci. USA 2014, 111, 14776–14781. [Google Scholar] [CrossRef]
- Manna, F.; La Karkampouna, S.; Zoni, E.; De Menna, M.; Hensel, J.; Thalmann, G.N.; Kruithof-de Julio, M. Metastases in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2019, 9, a033688. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Engl, T.; Relja, B.; Marian, D.; Blumenberg, C.; Müller, I.; Beecken, W.D.; Jones, J.; Ringel, E.M.; Bereiter-Hahny, J.; Jonas, D.; et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through α5 and β3 integrins. Neoplasia 2006, 8, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Dubyk, C.W.; Riesenberger, T.A.; Shein, D.; Keller, E.T.; Van Golen, K.L. Type I collagen receptor (α2β1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia 2008, 10, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Kostenuik, P.J.; Sanchez-Sweatman, O.; William Orr, F.; Singh, G. Bone cell matrix promotes the adhesion of human prostatic carcinoma cells via the α2β1 integrin. Clin. Exp. Metastasis 1996, 14, 19–26. [Google Scholar] [CrossRef]
- Koeneman, K.S.; Yeung, F.; Chung, L.W.K. Osteomimetic properties of prostate cancer cells: A hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate 1999, 39, 246–261. [Google Scholar] [CrossRef]
- Hensel, J.; Thalmann, G.N. Biology of Bone Metastases in Prostate Cancer. Urology 2016, 92, 6–13. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Kingsley, L.A.; Fournier, P.G.J.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Tharp, D.; Nandana, S. How prostate cancer cells use strategy instead of brute force to achieve metastasis. Cancers 2019, 11, 1928. [Google Scholar] [CrossRef]
- Holen, I.; Croucher, P.I.; Hamdy, F.C.; Eaton, C.L. Osteoprotegerin (OPG) Is a Survival Factor for Human Prostate Cancer Cells. Cancer Res. 2002, 62, 1619–1623. [Google Scholar]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Itoh, T.; Ito, Y.; Ohtsuki, Y.; Ando, M.; Tsukamasa, Y.; Yamada, N.; Naoe, T.; Akao, Y. Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. J. Mol. Histol. 2012, 43, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T.; Lundholm, M.; Widmark, A.; Persson, E. Tumor cell-derived exosomes from the prostate cancer cell line TRAMP-C1 impair osteoclast formation and differentiation. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Renzulli, J.F.; Del Tatto, M.; Dooner, G.; Aliotta, J.; Goldstein, L.; Dooner, M.; Colvin, G.; Chatterjee, D.; Quesenberry, P. Microvesicle induction of prostate specific gene expression in normal human bone marrow cells. J. Urol. 2010, 184, 2165–2171. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.M.; Woodson, K.; Wang, Y.; Rodriguez-Canales, J.; Erickson, H.S.; Tangrea, M.A.; Novakovic, K.; Gonzalez, S.; Velasco, A.; Kawasaki, E.S.; et al. Global Expression Analysis of Prostate Cancer-associated Stroma and Epithelia. Diagn. Mol. Pathol. 2007, 16, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Ashida, S.; Nakagawa, H.; Katagiri, T.; Furihata, M.; Iiizumi, M.; Anazawa, Y.; Tsunoda, T.; Takata, R.; Kasahara, K.; Miki, T.; et al. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: Genome-wide gene-expression profiles of prostate cancers and PINs. Cancer Res. 2004, 64, 5963–5972. [Google Scholar] [CrossRef] [PubMed]
- Baryawno, N.; Kfoury, Y.; Severe, N.; Mei, S.; Hirz, T.; Gustafsson, K.U.; Brouse, T.; Scadden, E.; Choi, B.; Barkas, N.; et al. Impact of metastatic prostate cancer on human bone marrow. bioRxiv Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef]
- Karkampouna, S.; Filippo, M.R.; De Ng, C.Y.; Klima, I.; Zoni, E.; Spahn, M.; Stein, F.; Haberkant, P.; Thalmann, G.N.; Julio, M.K. Stroma transcriptomic and proteomic profile of prostate cancer metastasis xenograft models reveals conservation of bone microenvironment signatures. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hensel, J.; Wetterwald, A.; Temanni, R.; Keller, I.; Riether, C.; Pluijm, G.; van der Cecchini, M.G.; Thalmann, G.N. Osteolytic cancer cells induce vascular/axon guidance processes in the bone/bone marrow stroma. Oncotarget 2018, 9, 28877–28896. [Google Scholar] [CrossRef]
- Martin, R.S.; Pathak, R.; Jain, A.; Jung, S.Y.; Hilsenbeck, S.G.; Piña-Barba, M.C.; Sikora, A.G.; Pienta, K.J.; Rowley, D.R. Tenascin-C and integrin α9 mediate interactions of prostate cancer with the bone microenvironment. Cancer Res. 2017, 77, 5977–5988. [Google Scholar] [CrossRef]
- Kiebish, M.A.; Cullen, J.; Mishra, P.; Ali, A.; Milliman, E.; Rodrigues, L.O.; Chen, E.Y.; Tolstikov, V.; Zhang, L.; Panagopoulos, K.; et al. Multi-omic serum biomarkers for prognosis of disease progression in prostate cancer. J. Transl. Med. 2020, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sterling, J.A.; Fan, K.H.; Vessella, R.L.; Shyr, Y.; Hayward, S.W.; Matrisian, L.M.; Bhowmick, N.A. Loss of TGF-β responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/ osteolytic bone lesions. Mol. Cancer Res. 2012, 10, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.F.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massagué, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 154, 1060–1073. [Google Scholar] [CrossRef]
- Blom, S.; Erickson, A.; Östman, A.; Rannikko, A.; Mirtti, T.; Kallioniemi, O.; Pellinen, T. Fibroblast as a critical stromal cell type determining prognosis in prostate cancer. Prostate 2019, 79, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Tyekucheva, S.; Bowden, M.; Bango, C.; Giunchi, F.; Huang, Y.; Zhou, C.; Bondi, A.; Lis, R.; Van Hemelrijck, M.; Andrén, O.; et al. Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nat. Commun. 2017, 8, 420. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
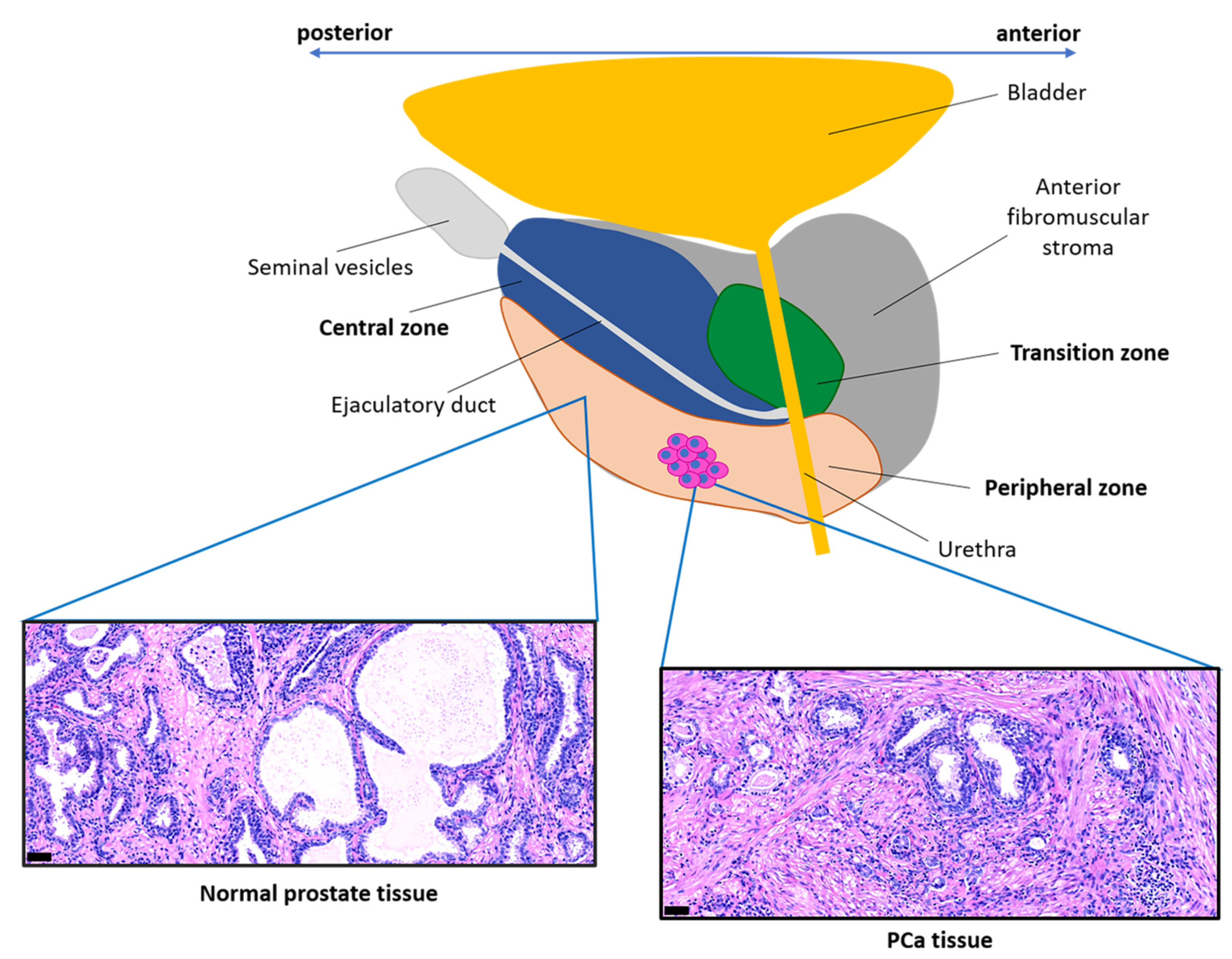{kind=link}
{kind=link}
| Gene Symbol | Protein Name | Expression in CAFs vs Normal Fibroblasts | Methods for Gene Expression Evaluation | Reference |
|---|---|---|---|---|
| ACTA2 | α-smooth actin (α-SMA) | Upregulated | RT-qPCR, IHC on tissue microarrays | [42,43] |
| ASPN | Asporin | Upregulated | Tag-based RNA profiling, microarray profiling, IHC, RT-qPCR | [38,44,45] |
| CAV1 | Caveolin-1 | Downregulated | Tag profiling, IHC, RT-qPCR | [38] |
| COL1A1 | Collagen Type-I | Upregulated | RT-qPCR, IHC | [42,43] |
| CXCL12 | Stromal cell-derived factor 1 (SDF1)/ (C-X-C motif chemokine ligand 12 (CXCL12) | Upregulated | Tag-profiling, RT-qPCR, ELISA | [38,46] |
| FAP | Fibroblast activation protein | Upregulated | IHC | [42] |
| FGF2, FGF7, FGF10 | Fibroblast growth factor-2/-7/-10 | Upregulated | RT-qPCR, Western Blot, IHC | [43,47,48] |
| FN1 | Fibronectin | Upregulated | Tag profiling, IHC, RT-qPCR | [38] |
| ITGA1 | Integrin-α1 (CD49a) | Upregulated | IHC | [49] |
| OGN | Osteoglycin | Upregulated | Tag-profiling, IHC, RT-qPCR | [38] |
| PDGFRB | Platelet-derived growth factor receptor β | Upregulated | Microarray profiling | [44,50] |
| POSTN | Periostin | Upregulated | Microarray profiling, IHC | [44] |
| S100A4 | Fibroblast-specific protein 1 (FSP1)/S100 Calcium Binding Protein A4 (S100A4) | Upregulated | Immunofluorescence, RT-qPCR, Western Blot | [51,52,53] |
| S100A6 | S100 Calcium Binding Protein A6 | Downregulated | Tag profiling, IHC, RT-qPCR | [38] |
| SPARC | Secreted Protein Acidic and Cysteine Rich | Up/downregulated | Microarray profiling, Tag-profiling | [38,44] |
| STC1 | Stanniocalcin 1 | Downregulated | Tag profiling, IHC, RT-qPCR | [38] |
| THY1 | Cluster of differentiation 90 (CD90) antigen | Upregulated | IHC | [54] |
| TNC | Tenascin C | Upregulated | RT-qPCR, IHC | [42,43] |
| VIM | Vimentin | Upregulated | IHC | [42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. https://doi.org/10.3390/cancers12071887
Bonollo F, Thalmann GN, Kruithof-de Julio M, Karkampouna S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers. 2020; 12(7):1887. https://doi.org/10.3390/cancers12071887
Chicago/Turabian StyleBonollo, Francesco, George N. Thalmann, Marianna Kruithof-de Julio, and Sofia Karkampouna. 2020. "The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis" Cancers 12, no. 7: 1887. https://doi.org/10.3390/cancers12071887
APA StyleBonollo, F., Thalmann, G. N., Kruithof-de Julio, M., & Karkampouna, S. (2020). The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers, 12(7), 1887. https://doi.org/10.3390/cancers12071887