The Potential of MLN3651 in Combination with Selumetinib as a Treatment for Merlin-Deficient Meningioma

 , , , and
, , , and
Abstract
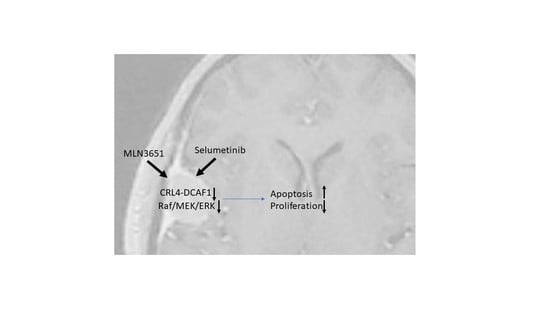
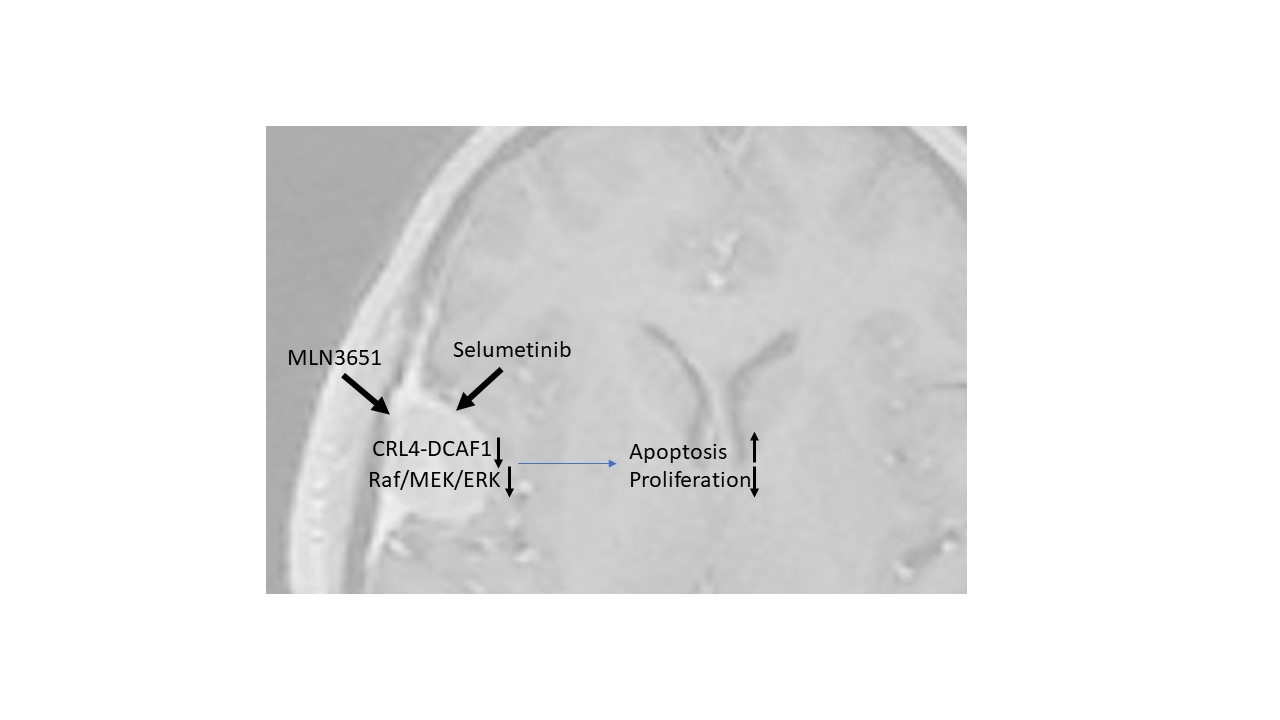{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
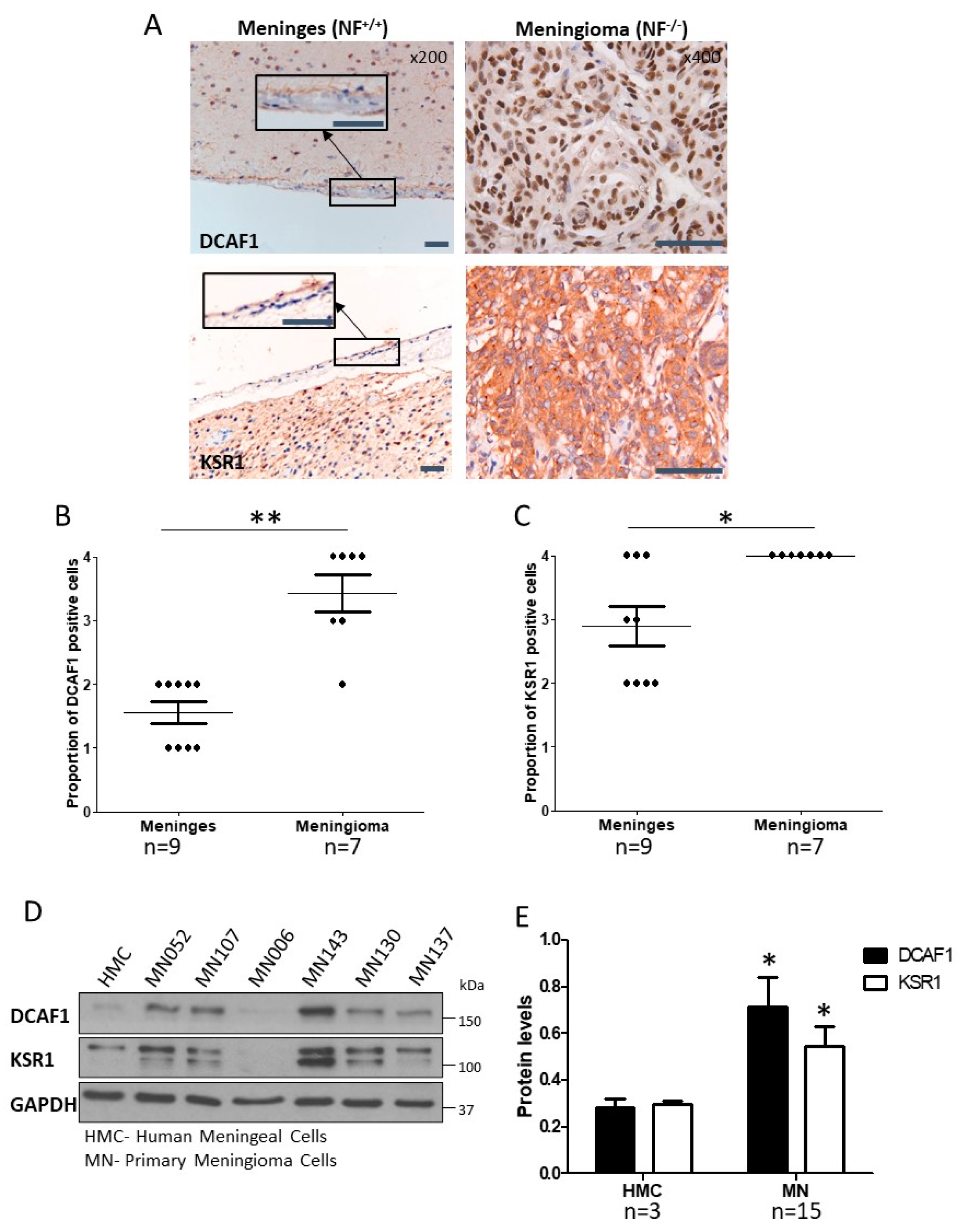
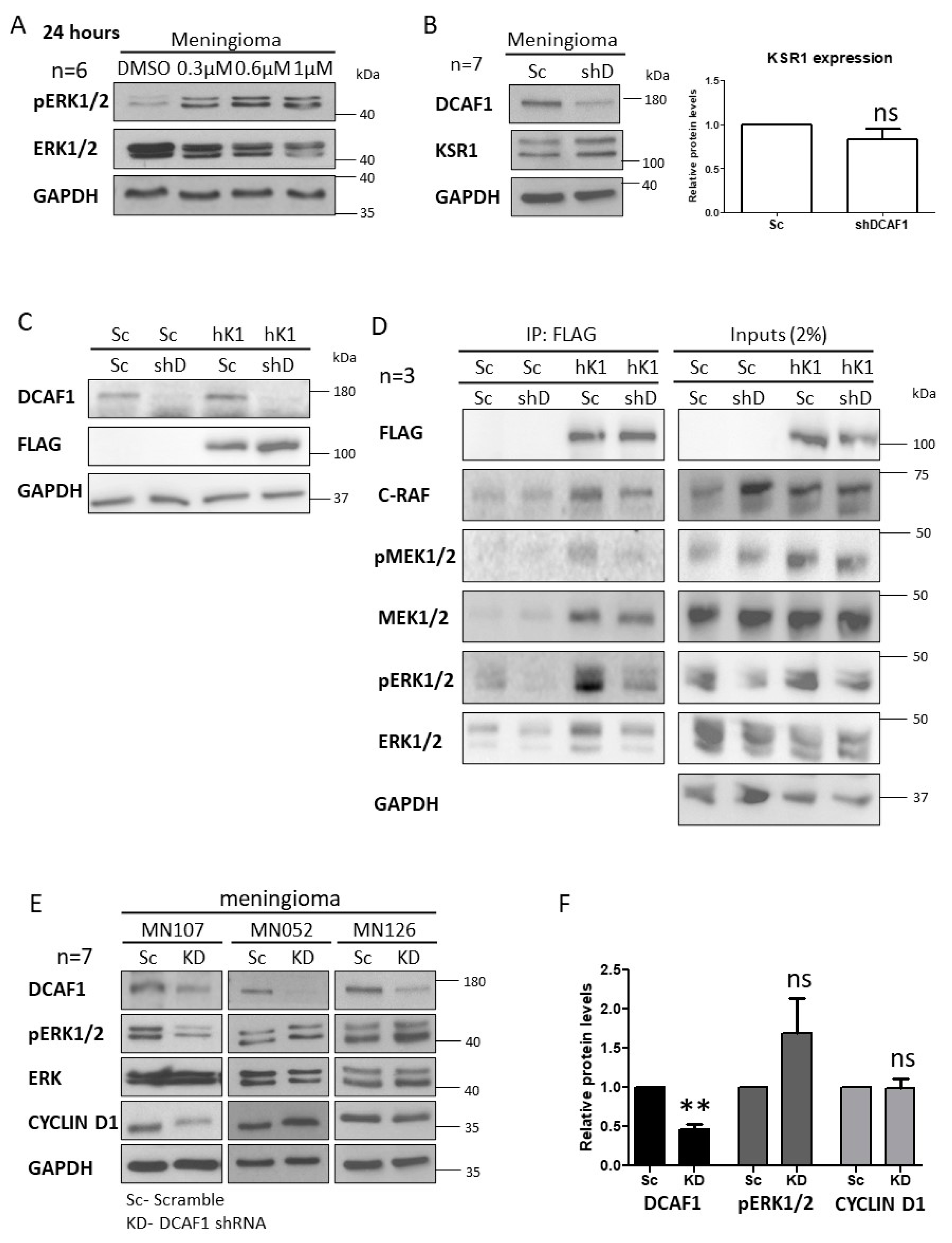
2.1. CRL4-DCAF1 and KSR1 Are Overexpressed in Meningioma Compared to Normal Cells
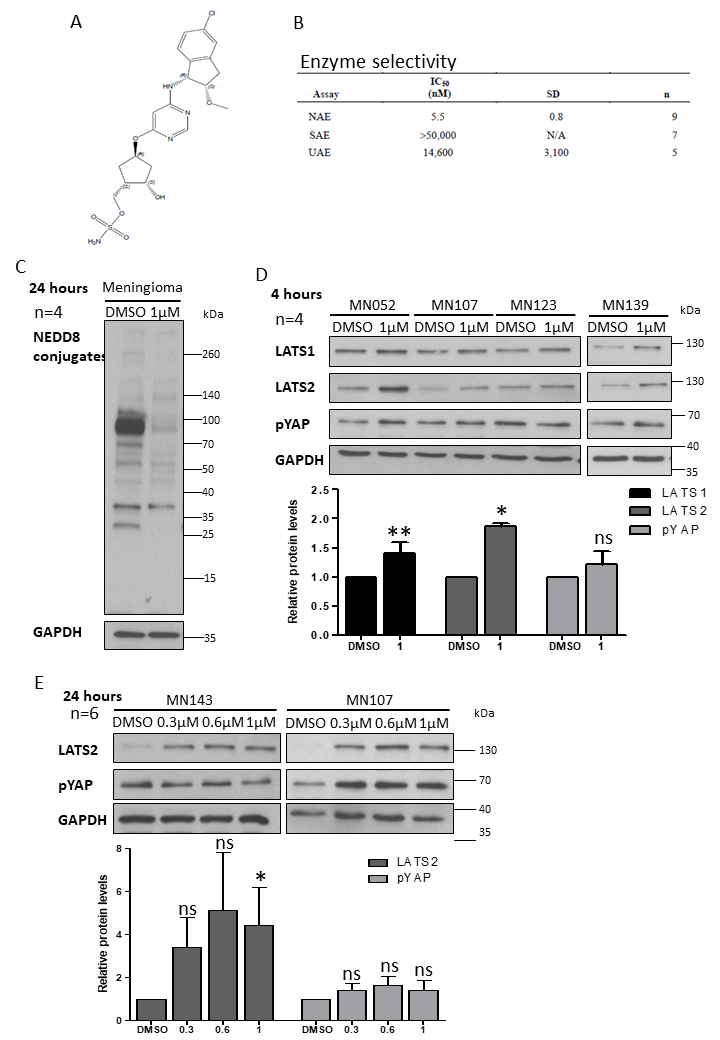
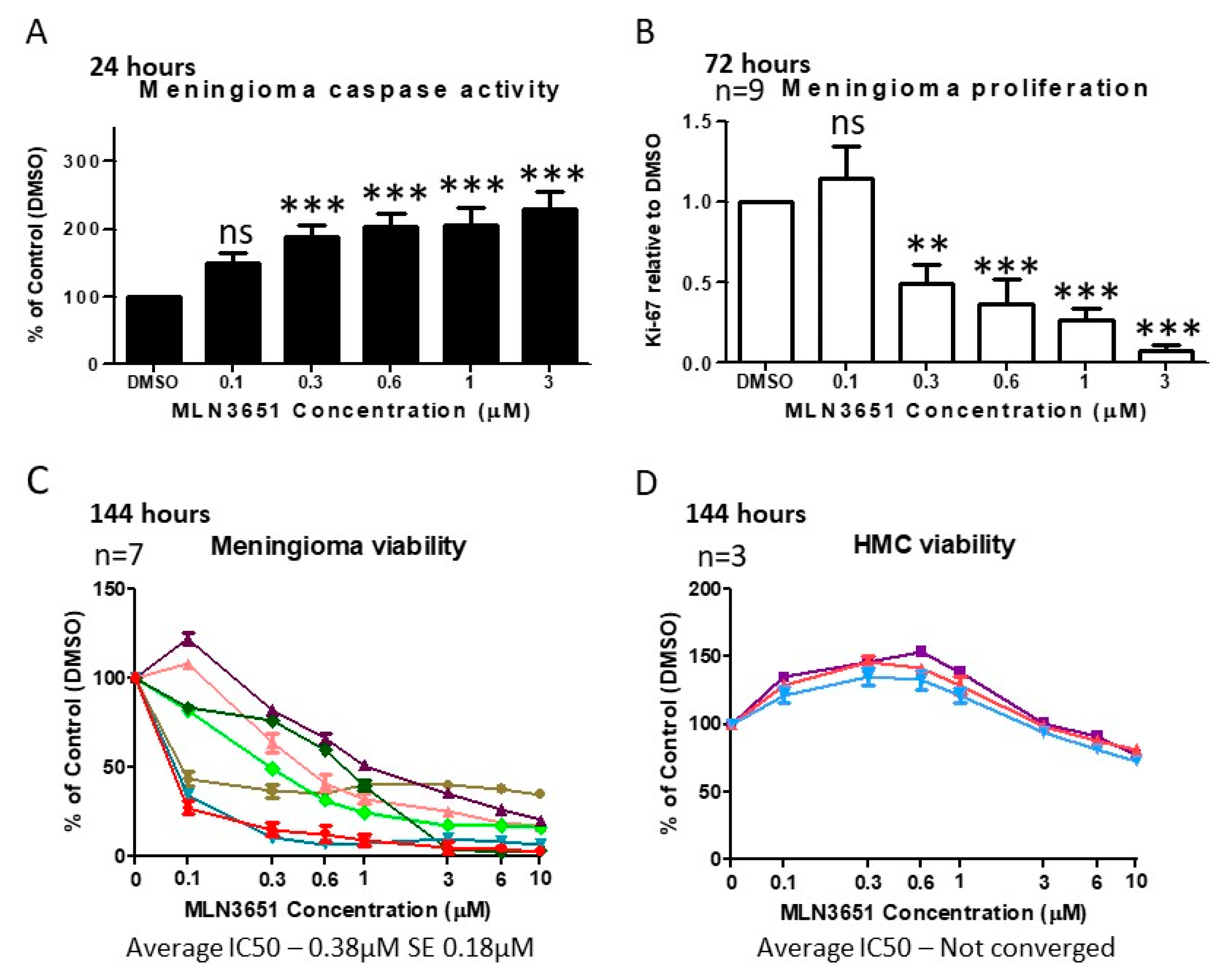
2.2. Inhibition of CRL4-DCAF1 Neddylation by MLN3651 Induces Apoptosis and Inhibits Proliferation to Reduce Cell Viability in Meningioma
2.3. MLN3651 and DCAF1 Knockdown Effect on the Raf/MEK/ERK Pathway in Meningioma
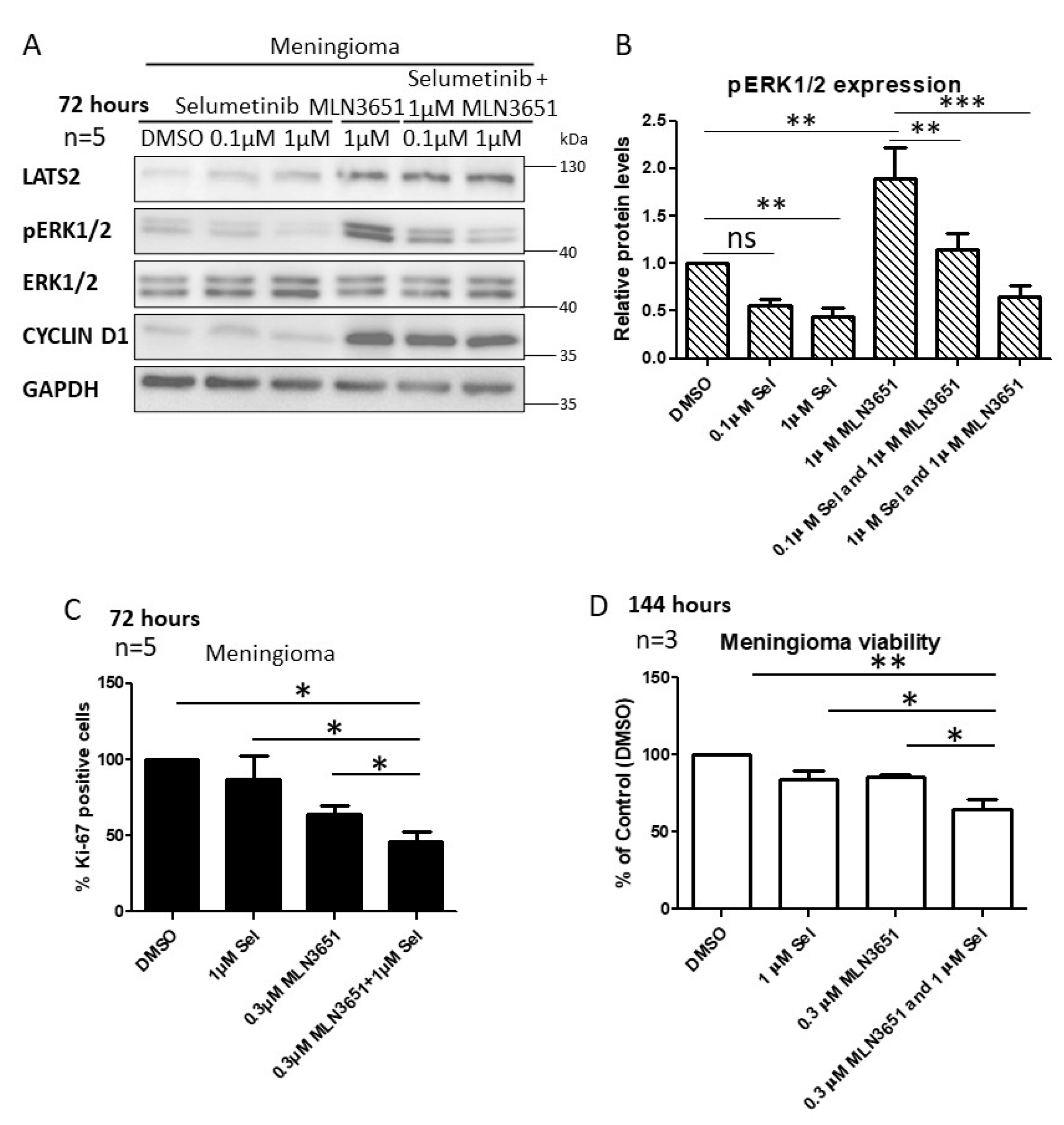
2.4. Simultaneous Inhibition of CRL4-DCAF1 and MEK1/2 Leads to an Enhanced Therapeutic Response in Meningioma
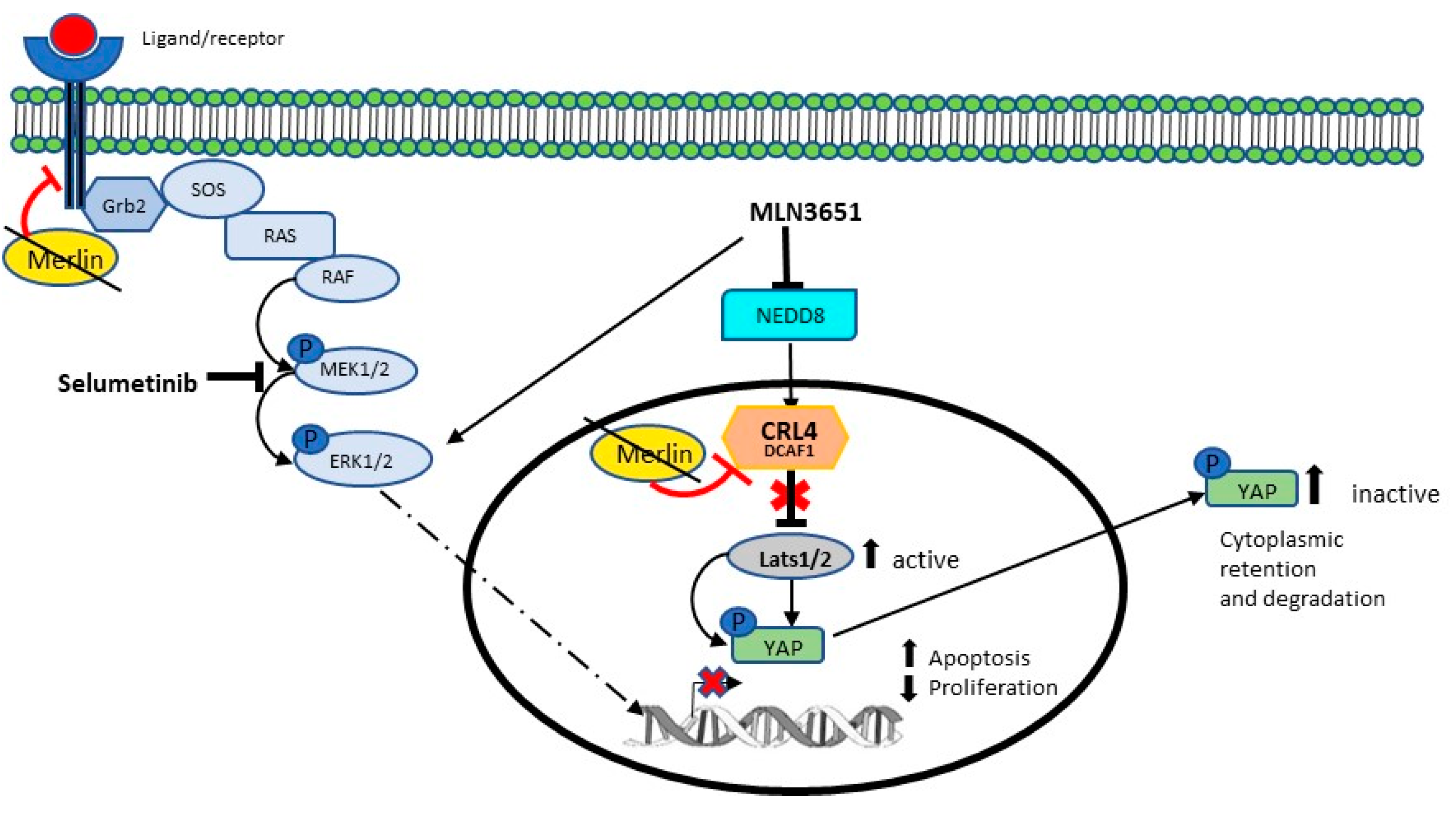
3. Discussion
4. Materials and Methods
4.1. Human Tissue
4.2. Co-Immunoprecipitation
4.3. Drug Treatments
4.4. Immunocytochemistry
4.5. Immunohistochemistry
4.6. Lentivirus Production and Infection
4.7. Western Blotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Wrensch, M.; Claus, E.B. Epidemiology and etiology of meningioma. J. Neuro-Oncol. 2010, 99, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Niwa-Kawakita, M.; Chareyre, F.; Taranchon, E.; Han, Z.-Y.; Martinelli, C.; Lusis, E.A.; Hegedus, B.; Gutmann, D.H.; et al. Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene 2011, 30, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Garcia, M.A.; Magill, S.T.; Chen, W.; Vasudevan, H.N.; Perry, A.; Theodosopoulos, P.V.; McDermott, M.W.; Braunstein, S.E.; Raleigh, D.R. Presenting Symptoms and Prognostic Factors for Symptomatic Outcomes Following Resection of Meningioma. World Neurosurg. 2018, 111, e149–e159. [Google Scholar] [CrossRef]
- Gousias, K.; Schramm, J.; Simon, M. The Simpson grading revisited: Aggressive surgery and its place in modern meningioma management. J. Neurosurg. 2016, 125, 551–560. [Google Scholar] [CrossRef]
- Bianchi, A.B.; Hara, T.; Ramesh, V.; Gao, J.; Klein-Szanto, A.J.P.; Morin, F.; Menon, A.G.; Trofatter, J.A.; Gusella, J.F.; Seizinger, B.R.; et al. Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nat. Genet. 1994, 6, 185–192. [Google Scholar] [CrossRef]
- Evans, D.G. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J. Rare Dis. 2009, 4, 16. [Google Scholar] [CrossRef]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2015, 35, 537–548. [Google Scholar] [CrossRef]
- Morrison, H.; Sperka, T.; Manent, J.; Giovannini, M.; Ponta, H.; Herrlich, P. Merlin/Neurofibromatosis Type 2 Suppresses Growth by Inhibiting the Activation of Ras and Rac. Cancer Res. 2007, 67, 520–527. [Google Scholar] [CrossRef]
- Ammoun, S.; Flaiz, C.; Ristić, N.; Schuldt, J.; O Hanemann, C. Dissecting and Targeting the Growth Factor-Dependent and Growth Factor-Independent Extracellular Signal-Regulated Kinase Pathway in Human Schwannoma. Cancer Res. 2008, 68, 5236–5245. [Google Scholar] [CrossRef]
- Ammoun, S.; Schmid, M.C.; Ristic, N.; Zhou, L.; Hilton, D.; Ercolano, E.; Carroll, C.; Hanemann, C.O. The role of insulin-like growth factors signaling in merlin-deficient human schwannomas. Glia 2012, 60, 1721–1733. [Google Scholar] [CrossRef]
- Li, W.; You, L.; Cooper, J.; Schiavon, G.; Pepe-Caprio, A.; Zhou, L.; Ishii, R.; Giovannini, M.; O Hanemann, C.; Long, S.B.; et al. Merlin/NF2 Suppresses Tumorigenesis by Inhibiting the E3 Ubiquitin Ligase CRL4DCAF1 in the Nucleus. Cell 2010, 140, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cooper, J.; Zhou, L.; Yang, C.; Erdjument-Bromage, H.; Zagzag, D.; Snuderl, M.; Ladanyi, M.; Hanemann, C.O.; Zhou, P.; et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014, 26, 48–60. [Google Scholar] [CrossRef]
- Muranen, T.; Grӧnholm, M.; Renkema, H.; Carpén, O. Cell cycle-dependent nucleocytoplasmic shuttling of the neurofibromatosis 2 tumour suppressor merlin. Oncogene 2004, 24, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.; Zheng, N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; McCall, C.M.; Hu, J.; Zeng, Y.; Xiong, Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4–ROC1 ubiquitin ligases. Genes Dev. 2006, 20, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Xu, Q.; Zhou, L.; Pavlovic, M.; Ojeda, V.; Moulick, K.; De Stanchina, E.; Poirier, J.T.; Zauderer, M.G.; Rudin, C.M.; et al. Combined Inhibition of NEDD8-Activating Enzyme and mTOR Suppresses NF2 Loss–Driven Tumorigenesis. Mol. Cancer Ther. 2017, 16, 1693–1704. [Google Scholar] [CrossRef]
- Zhou, L.; Lyons-Rimmer, J.; Ammoun, S.; Müller, J.; Lasonder, E.; Sharma, V.; Ercolano, E.; Hilton, D.; Taiwo, I.; Barczyk, M.; et al. The scaffold protein KSR1, a novel therapeutic target for the treatment of Merlin-deficient tumors. Oncogene 2015, 35, 3443–3453. [Google Scholar] [CrossRef]
- Dougherty, M.K.; Ritt, D.A.; Zhou, M.; Specht, S.I.; Monson, D.M.; Veenstra, T.D.; Morrison, D.K. KSR2 Is a Calcineurin Substrate that Promotes ERK Cascade Activation in Response to Calcium Signals. Mol. Cell 2009, 34, 652–662. [Google Scholar] [CrossRef]
- Yeh, T.C.; Marsh, V.; Bernat, B.A.; Ballard, J.; Colwell, H.; Evans, R.J.; Parry, J.; Smith, D.; Brandhuber, B.J.; Gross, S.; et al. Biological Characterization of ARRY-142886 (AZD6244), a Potent, Highly Selective Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor. Clin. Cancer Res. 2007, 13, 1576–1583. [Google Scholar] [CrossRef]
- Chen, J.J.; Tsu, C.A.; Gavin, J.M.; Milhollen, M.A.; Bruzzese, F.J.; Mallender, W.D.; Sintchak, M.D.; Bump, N.J.; Yang, X.; Ma, J.; et al. Mechanistic Studies of Substrate-assisted Inhibition of Ubiquitin-activating Enzyme by Adenosine Sulfamate Analogues. J. Biol. Chem. 2011, 286, 40867–40877. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Angus, S.P.; Oblinger, J.L.; Stuhlmiller, T.J.; A DeSouza, P.; Beauchamp, R.L.; Witt, L.; Chen, X.; Jordan, J.T.; Gilbert, T.S.K.; Stemmer-Rachamimov, A.; et al. EPH receptor signaling as a novel therapeutic target in NF2-deficient meningioma. Neuro-Oncology 2018, 20, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Wan, Y.-W.; Al-Ouran, R.; Revelli, J.-P.; Cardenas, M.F.; Oneissi, M.; Xi, L.; Jalali, A.; Magnotti, J.F.; Muzny, D.M.; et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc. Natl. Acad. Sci. USA 2019, 116, 21715–21726. [Google Scholar] [CrossRef] [PubMed]
- Harmancı, A.S.; Youngblood, M.W.; Clark, V.E.; Coskun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat. Commun. 2017, 8, 14433. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Lau, Y.-K.I.; Murray, L.B.; Houshmandi, S.S.; Xu, Y.; Gutmann, D.H.; Yu, Q. Merlin is a potent inhibitor of glioma growth. Cancer Res. 2008, 68, 5733–5742. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, Z.; Long, J.; Li, H.; Zhang, B.; Chen, G.; Li, X.; Lv, T.; Zhang, W.; Ou, X.; et al. Molecular alterations of the NF2 gene in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Oncol. Rep. 2017, 38, 3650–3658. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, J.; Lin, X.; Wu, D.; Li, G.; Zhong, C.; Fang, L.; Jiang, P.; Yin, L.; Zhang, L.; et al. Inhibition of neddylation modification by MLN4924 sensitizes hepatocellular carcinoma cells to sorafenib. Oncol. Rep. 2019, 41, 3257–3269. [Google Scholar] [CrossRef]
- Casaluce, F.; Sgambato, A.; Maione, P.; Sacco, P.C.; Santabarbara, G.; Gridelli, C. Selumetinib for the treatment of non-small cell lung cancer. Expert Opin. Investig. Drugs 2017, 26, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, D.; Giovannini, M.; Saint-Amaux, A.L. Tumor-suppression functions of merlin are independent of its role as an organizer of the actin cytoskeleton in Schwann cells. J. Cell Sci. 2009, 122, 4141–4149. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyons Rimmer, J.; Ercolano, E.; Baiz, D.; Makhija, M.; Berger, A.; Sells, T.; Stroud, S.; Hilton, D.; Adams, C.L.; Hanemann, C.O. The Potential of MLN3651 in Combination with Selumetinib as a Treatment for Merlin-Deficient Meningioma. Cancers 2020, 12, 1744. https://doi.org/10.3390/cancers12071744
Lyons Rimmer J, Ercolano E, Baiz D, Makhija M, Berger A, Sells T, Stroud S, Hilton D, Adams CL, Hanemann CO. The Potential of MLN3651 in Combination with Selumetinib as a Treatment for Merlin-Deficient Meningioma. Cancers. 2020; 12(7):1744. https://doi.org/10.3390/cancers12071744
Chicago/Turabian StyleLyons Rimmer, Jade, Emanuela Ercolano, Daniele Baiz, Mahindra Makhija, Allison Berger, Todd Sells, Steve Stroud, David Hilton, Claire L. Adams, and C Oliver Hanemann. 2020. "The Potential of MLN3651 in Combination with Selumetinib as a Treatment for Merlin-Deficient Meningioma" Cancers 12, no. 7: 1744. https://doi.org/10.3390/cancers12071744
APA StyleLyons Rimmer, J., Ercolano, E., Baiz, D., Makhija, M., Berger, A., Sells, T., Stroud, S., Hilton, D., Adams, C. L., & Hanemann, C. O. (2020). The Potential of MLN3651 in Combination with Selumetinib as a Treatment for Merlin-Deficient Meningioma. Cancers, 12(7), 1744. https://doi.org/10.3390/cancers12071744