CXCR4 Inhibition Enhances Efficacy of FLT3 Inhibitors in FLT3-Mutated AML Augmented by Suppressed TGF-β Signaling


 and
and 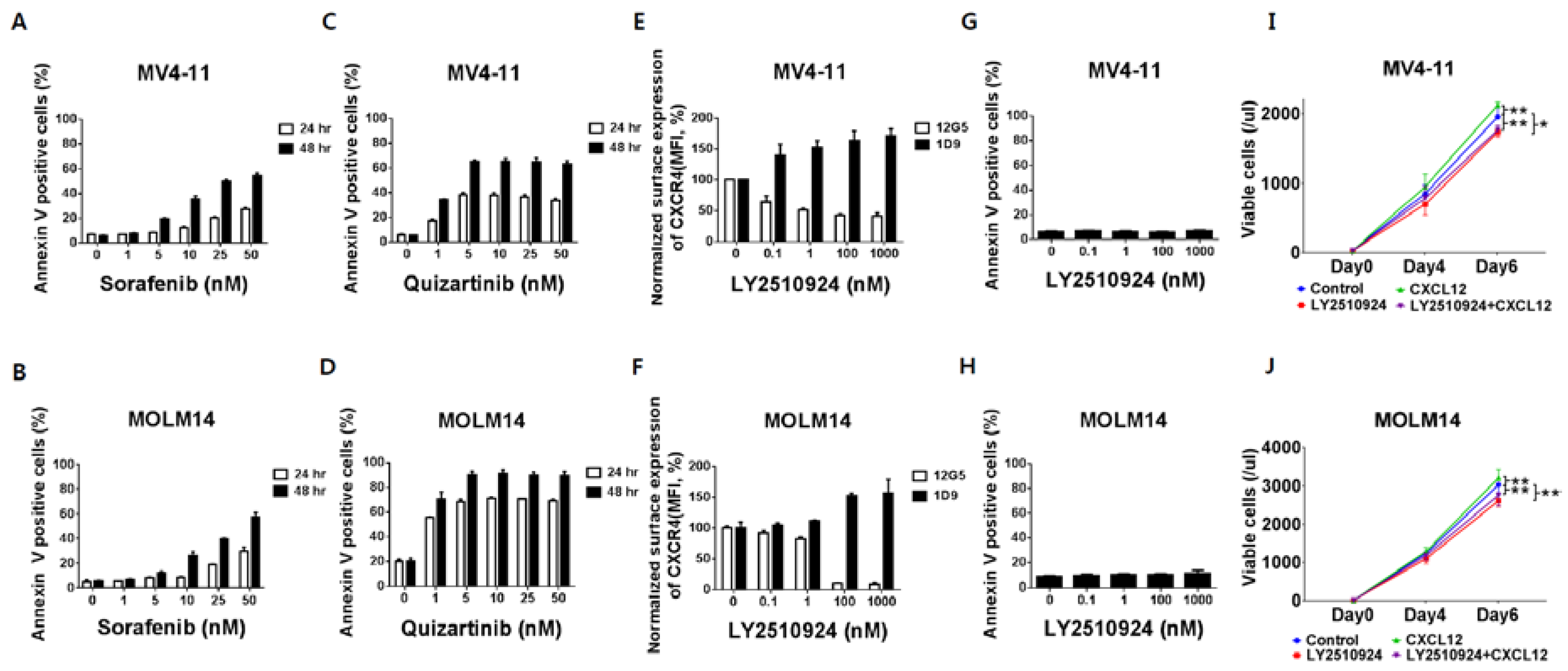{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. FLT3 Inhibitors Induce Apoptosis in FLT3-ITD-AML While LY2510924 Blocks Surface CXCR4 and Inhibits Proliferation Rather Than Causing Apoptosis
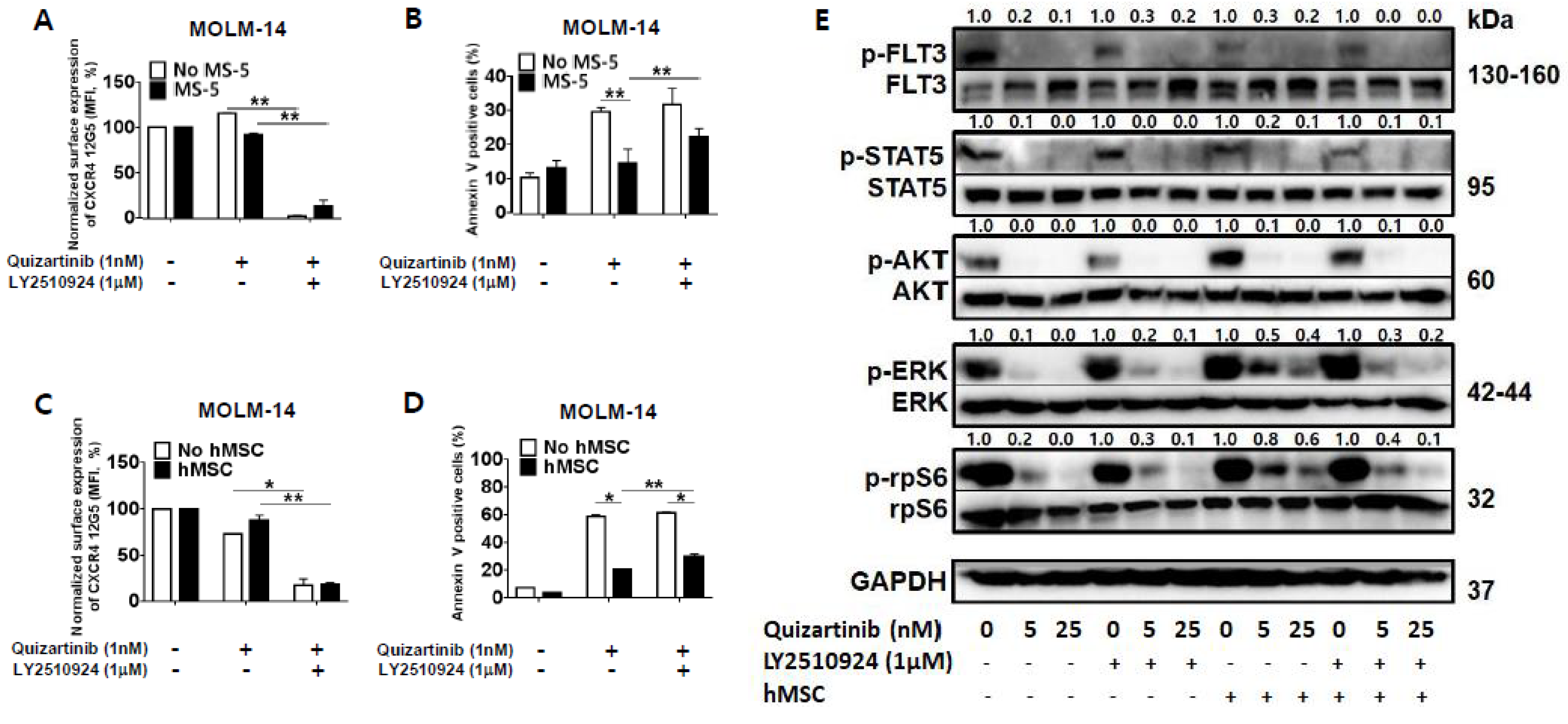
2.2. CXCR4 Inhibition by LY2510924 Significantly Reverses Stroma-Mediated Resistance to Quizartinib In Vitro, Mainly Through the MAPK Pathway
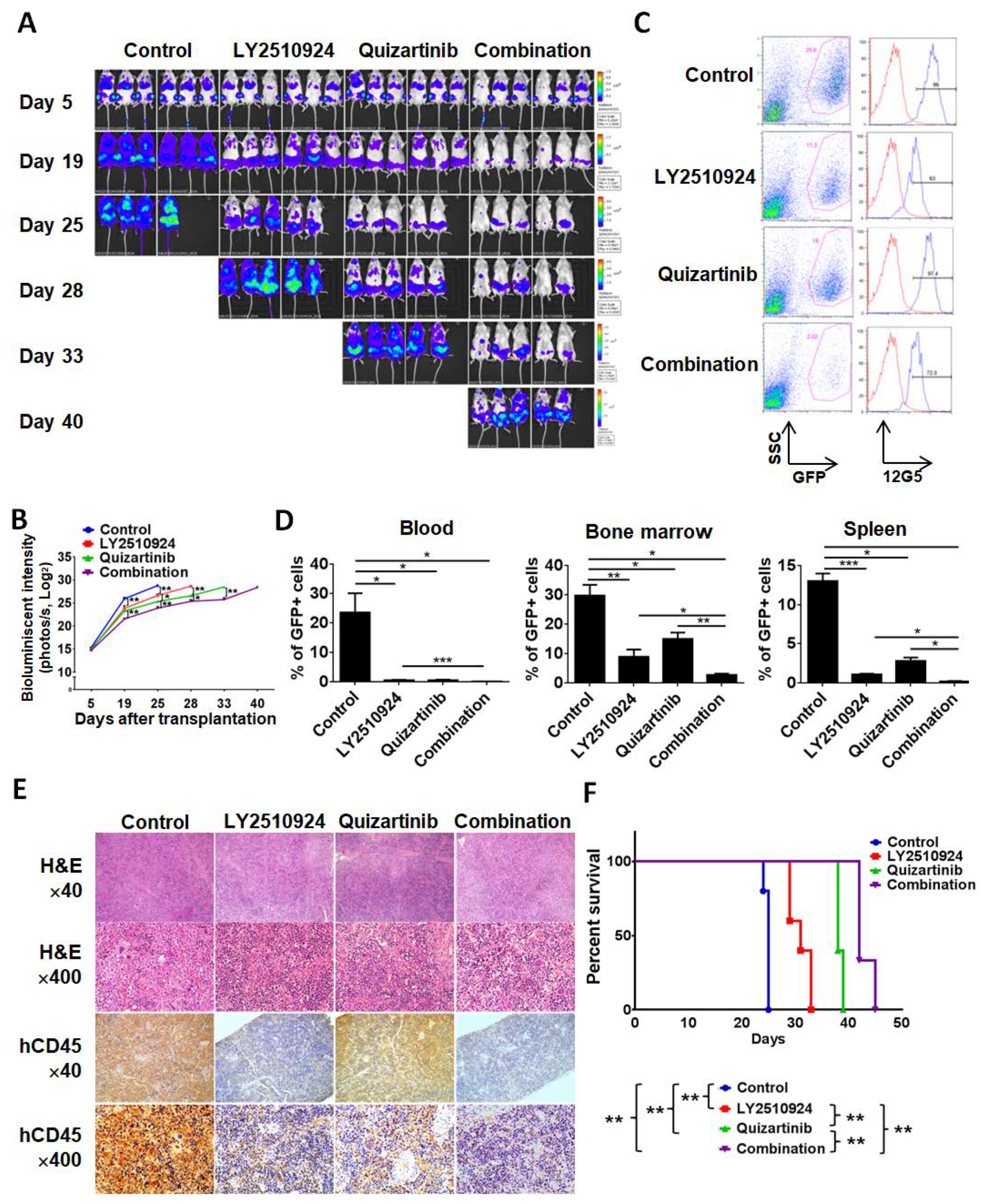
2.3. LY2510924 Enhances Anti-Leukemia Effects in Combination with Quizartinib In Vivo
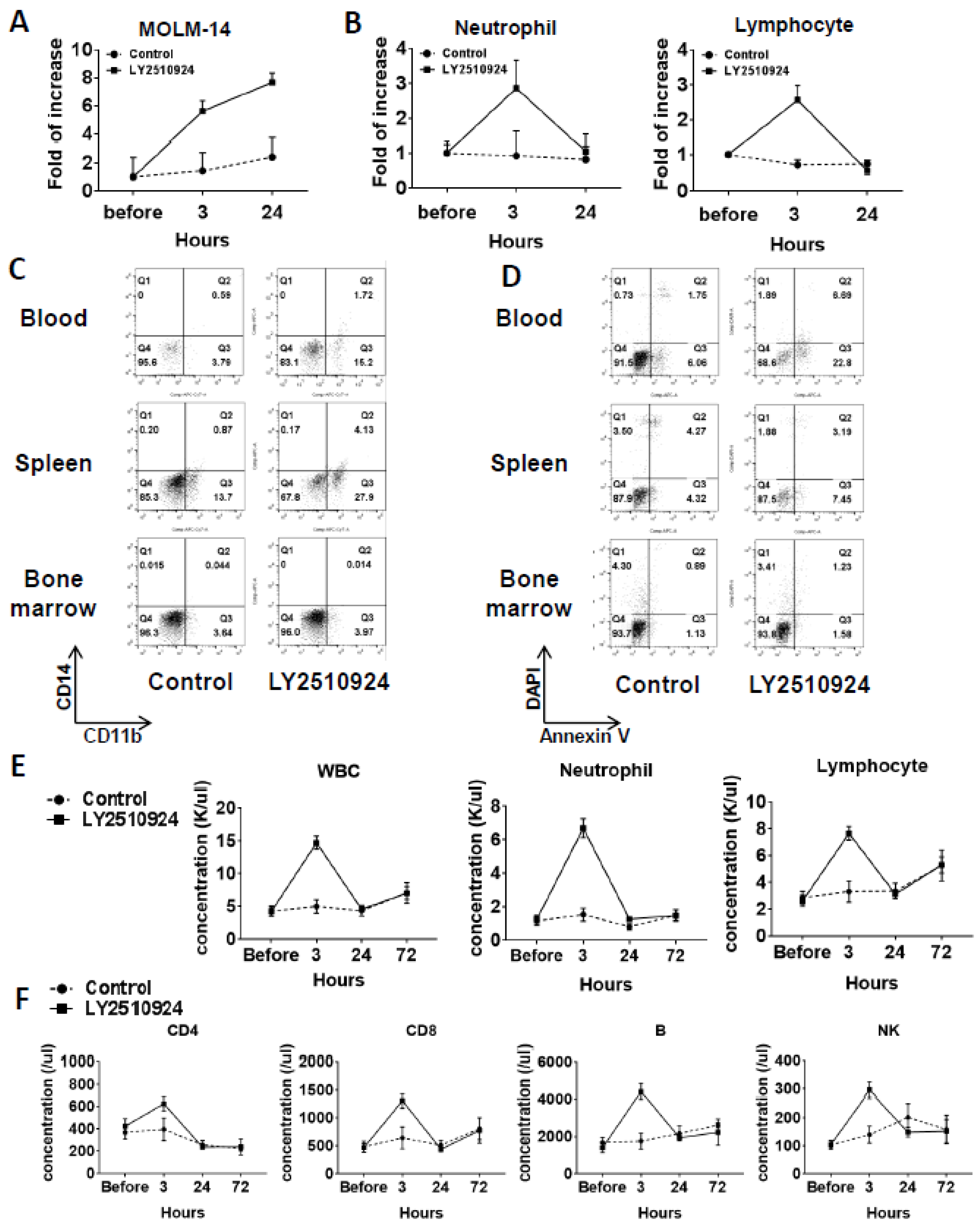
2.4. LY2510924 Induces Durable Mobilization of Leukemia Cells and Differentiation Without Causing Apoptosis In Vivo, with Only Transient Effects on Non-Leukemic Blood Cells
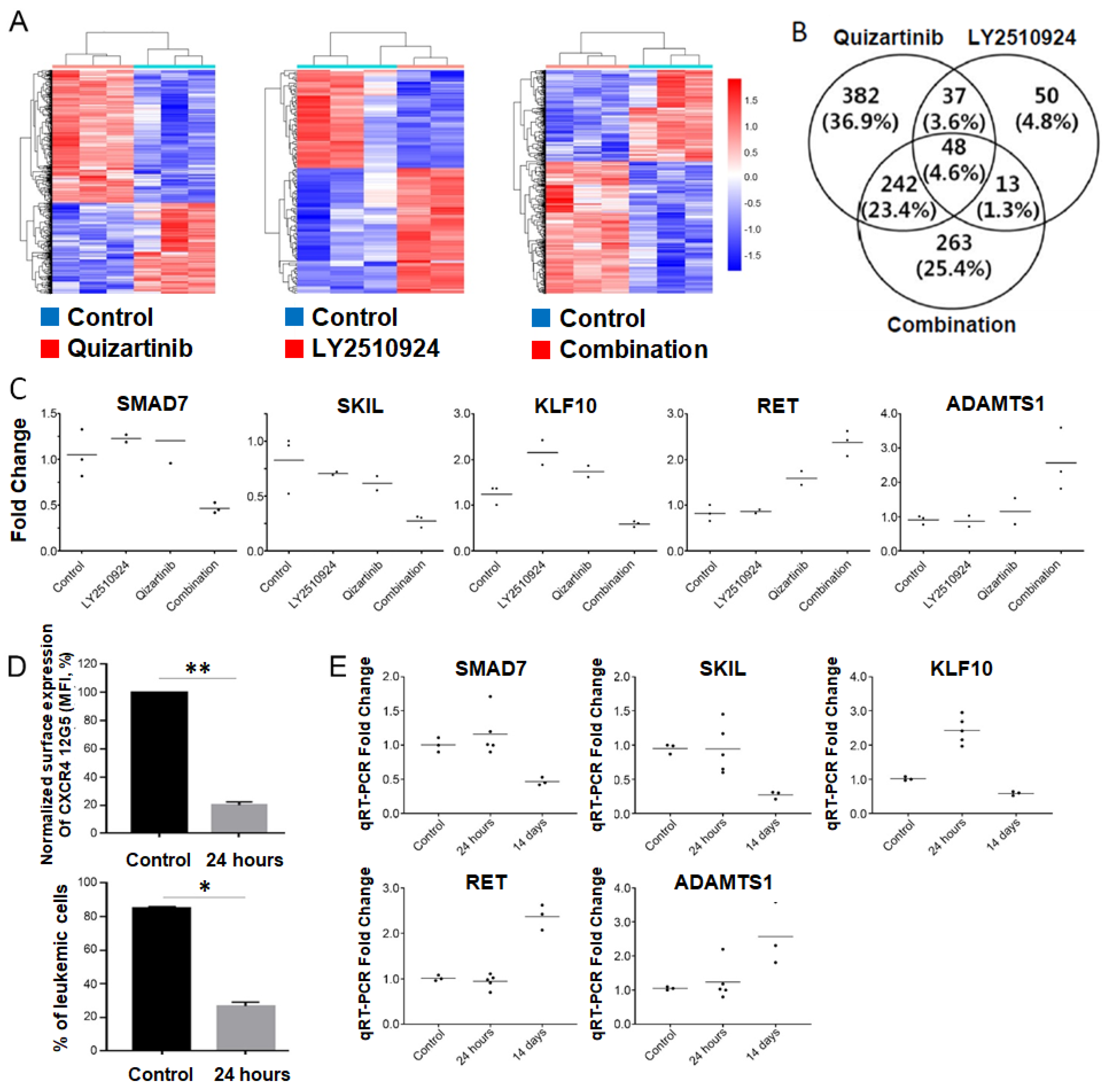
2.5. Transcriptome Analysis of the Differentially Expressed Genes upon Combination of FLT3 and CXCR4 Inhibitors in AML
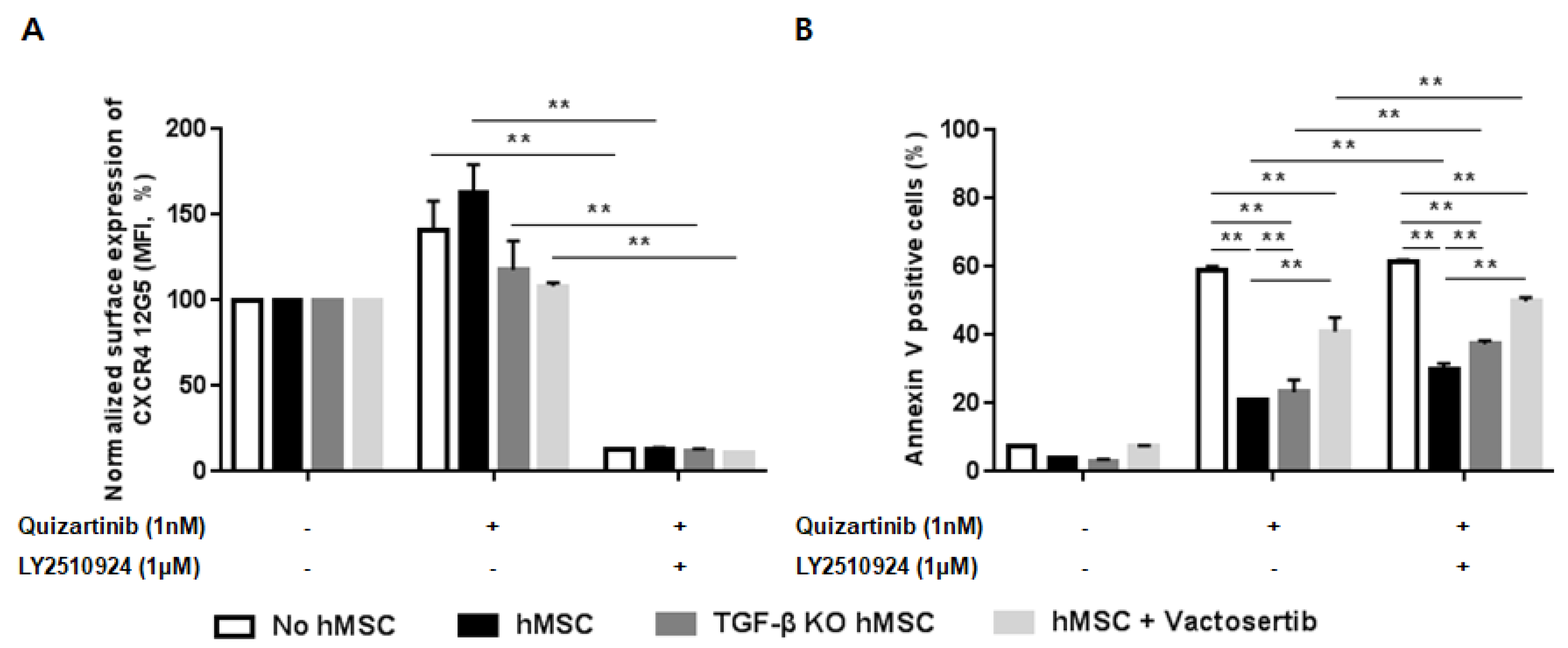
2.6. Silencing of TGF-β Signaling Reduces the Stroma-Mediated Protective Effects and Further Enhances Apoptosis by the Combined Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Materials
4.2. Flow Cytometry and Mouse Blood Cell Count
4.3. Western Blot Analysis
4.4. Co-Culture with Stromal Cells
4.5. AML Mouse Models
4.6. RNA Sequencing and Validation of Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kindler, T.; Lipka, D.B.; Fischer, T. FLT3 as a therapeutic target in AML: Still challenging after all these years. Blood 2010, 116, 5089–5102. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Brandts, C.H.; Sargin, B.; Rode, M.; Biermann, C.; Lindtner, B.; Schwable, J.; Buerger, H.; Muller-Tidow, C.; Choudhary, C.; McMahon, M.; et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005, 65, 9643–9650. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, M.; Fenski, R.; Halfter, H.; Matsumura, I.; Schmidt, R.; Muller, C.; Gruning, W.; Kratz-Albers, K.; Serve, S.; Steur, C.; et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000, 96, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Schwable, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Friedman, A.D.; Levis, M.; Li, L.; Weir, E.G.; Small, D. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expression. Blood 2004, 103, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009, 113, 6206–6214. [Google Scholar] [CrossRef]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef]
- Cho, B.S.; Zeng, Z.; Mu, H.; Wang, Z.; Konoplev, S.; McQueen, T.; Protopopova, M.; Cortes, J.; Marszalek, J.R.; Peng, S.B.; et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015, 126, 222–232. [Google Scholar] [CrossRef]
- Peled, A.; Klein, S.; Beider, K.; Burger, J.A.; Abraham, M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine 2018, 109, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Parmar, A.; Marz, S.; Rushton, S.; Holzwarth, C.; Lind, K.; Kayser, S.; Dohner, K.; Peschel, C.; Oostendorp, R.A.; Gotze, K.S. Stromal niche cells protect early leukemic FLT3-ITD+ progenitor cells against first-generation FLT3 tyrosine kinase inhibitors. Cancer Res. 2011, 71, 4696–4706. [Google Scholar] [CrossRef]
- Burger, J.A.; Peled, A. CXCR4 antagonists: Targeting the microenvironment in leukemia and other cancers. Leukemia 2009, 23, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, E.J.; Pavic, B.; Lowenberg, B.; Ploemacher, R.E. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 2004, 104, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Konoplev, S.; Rassidakis, G.Z.; Estey, E.; Kantarjian, H.; Liakou, C.I.; Huang, X.; Xiao, L.; Andreeff, M.; Konopleva, M.; Medeiros, L.J. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer 2007, 109, 1152–1156. [Google Scholar] [CrossRef]
- Fukuda, S.; Broxmeyer, H.E.; Pelus, L.M. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood 2005, 105, 3117–3126. [Google Scholar] [CrossRef]
- Klein, R.S.; Rubin, J.B. Immune and nervous system CXCL12 and CXCR4: Parallel roles in patterning and plasticity. Trends Immunol. 2004, 25, 306–314. [Google Scholar] [CrossRef]
- Boddu, P.; Borthakur, G.; Koneru, M.; Huang, X.; Naqvi, K.; Wierda, W.; Bose, P.; Jabbour, E.; Estrov, Z.; Burger, J.; et al. Initial Report of a Phase I Study of LY2510924, Idarubicin, and Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 369. [Google Scholar] [CrossRef]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef]
- Yang, X.; Sexauer, A.; Levis, M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br. J. Haematol. 2014, 164, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, D.; Nogami, A.; Okada, K.; Akiyama, H.; Umezawa, Y.; Miura, O. FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers 2019, 11, 1827. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chng, W.J. Resistance to FLT3 inhibitors in acute myeloid leukemia: Molecular mechanisms and resensitizing strategies. World J. Clin. Oncol. 2018, 9, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.; Klein, S.; Bulvik, B.; Wald, H.; Weiss, I.D.; Olam, D.; Weiss, L.; Beider, K.; Eizenberg, O.; Wald, O.; et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia 2017, 31, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Saavedra, E.; Tang, R.; Gu, Y.; Lappin, P.; Trajkovic, D.; Liu, S.H.; Smeal, T.; Fantin, V.; De Botton, S.; et al. Targeting primary acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143). Sci. Rep. 2017, 7, 7305. [Google Scholar] [CrossRef]
- Andreeff, M.; Borthakur, G.; Zeng, Z.; Kelly, M.A.; Wang, R.-Y.; McQueen, T.J.; Qiu, Y.; Mak, D.; Burger, J.A.; Daver, N.; et al. Mobilization and elimination of FLT3-ITD+ acute myelogenous leukemia (AML) stem/progenitor cells by plerixafor, G-CSF, and sorafenib: Phase I trial results in relapsed/refractory AML patients. J. Clin. Oncol. 2014, 32, 7033. [Google Scholar] [CrossRef]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Nebreda, A.R.; Gavin, A.C. Perspectives: Signal transduction. Cell survival demands some Rsk. Science 1999, 286, 1309–1310. [Google Scholar] [CrossRef]
- Tilton, B.; Ho, L.; Oberlin, E.; Loetscher, P.; Baleux, F.; Clark-Lewis, I.; Thelen, M. Signal transduction by CXC chemokine receptor 4. Stromal cell-derived factor 1 stimulates prolonged protein kinase B and extracellular signal-regulated kinase 2 activation in T lymphocytes. J. Exp. Med. 2000, 192, 313–324. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal selection with Ras pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Borthakur, G.; Gao, C.; Chen, Y.; Mu, H.; Ruvolo, V.R.; Nomoto, K.; Zhao, N.; Konopleva, M.; Andreeff, M. The Dual MEK/FLT3 Inhibitor E6201 Exerts Cytotoxic Activity against Acute Myeloid Leukemia Cells Harboring Resistance-Conferring FLT3 Mutations. Cancer Res. 2016, 76, 1528–1537. [Google Scholar] [CrossRef]
- Tavor, S.; Eisenbach, M.; Jacob-Hirsch, J.; Golan, T.; Petit, I.; Benzion, K.; Kay, S.; Baron, S.; Amariglio, N.; Deutsch, V.; et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia 2008, 22, 2151–5158. [Google Scholar] [CrossRef]
- Yan, X.; Chen, Y.G. Smad7: Not only a regulator, but also a cross-talk mediator of TGF-beta signalling. Biochem. J. 2011, 434, 1–10. [Google Scholar] [CrossRef]
- Deheuninck, J.; Luo, K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009, 19, 47–57. [Google Scholar] [CrossRef]
- Memon, A.; Lee, W.K. KLF10 as a Tumor Suppressor Gene and Its TGF-beta Signaling. Cancers (Basel) 2018, 10, 161. [Google Scholar] [CrossRef]
- Miyazono, K.; Katsuno, Y.; Koinuma, D.; Ehata, S.; Morikawa, M. Intracellular and extracellular TGF-beta signaling in cancer: Some recent topics. Front. Med. 2018, 12, 387–411. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Integrative genomic analyses of CXCR4: Transcriptional regulation of CXCR4 based on TGFbeta, Nodal, Activin signaling and POU5F1, FOXA2, FOXC2, FOXH1, SOX17, and GFI1 transcription factors. Int. J. Oncol. 2010, 36, 415–420. [Google Scholar] [CrossRef]
- Buckley, C.D.; Amft, N.; Bradfield, P.F.; Pilling, D.; Ross, E.; Arenzana-Seisdedos, F.; Amara, A.; Curnow, S.J.; Lord, J.M.; Scheel-Toellner, D.; et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J. Immunol. 2000, 165, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Shi, Y.X.; Zeng, Z.; Jin, L.; Shikami, M.; Hatanaka, Y.; Miida, T.; Hsu, F.J.; Andreeff, M.; Konopleva, M. TGF-beta-Neutralizing Antibody 1D11 Enhances Cytarabine-Induced Apoptosis in AML Cells in the Bone Marrow Microenvironment. PLoS ONE 2013, 8, e62785. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2018, 15, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Barabe, F.; Gil, L.; Celton, M.; Bergeron, A.; Lamontagne, V.; Roques, E.; Lagace, K.; Forest, A.; Johnson, R.; Pecheux, L.; et al. Modeling human MLL-AF9 translocated acute myeloid leukemia from single donors reveals RET as a potential therapeutic target. Leukemia 2017, 31, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.; Ellegast, J.M.; Buhler, C.; Marcantonio, D.D.; Martinez, E.; Gollner, S.; Wickenhauser, C.; et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef]
- Tan Ide, A.; Ricciardelli, C.; Russell, D.L. The metalloproteinase ADAMTS1: A comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer 2013, 133, 2263–2276. [Google Scholar]
- Chen, Y.; Jacamo, R.; Konopleva, M.; Garzon, R.; Croce, C.; Andreeff, M. CXCR4 downregulation of let-7a drives chemoresistance in acute myeloid leukemia. J. Clin. Investig. 2013, 123, 2395–2407. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, M.S.; Jung, C.K.; Park, H.C.; Kim, S.Y.; Liu, J.; Bae, J.S.; Lee, S.H.; Kim, T.M.; Lee, S.H.; et al. Mutational burdens and evolutionary ages of thyroid follicular adenoma are comparable to those of follicular carcinoma. Oncotarget 2016, 7, 69638–69648. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-R.; Jung, S.-H.; Han, A.-R.; Park, G.; Kim, H.-J.; Yuan, B.; Battula, V.L.; Andreeff, M.; Konopleva, M.; Chung, Y.-J.; et al. CXCR4 Inhibition Enhances Efficacy of FLT3 Inhibitors in FLT3-Mutated AML Augmented by Suppressed TGF-β Signaling. Cancers 2020, 12, 1737. https://doi.org/10.3390/cancers12071737
Kim B-R, Jung S-H, Han A-R, Park G, Kim H-J, Yuan B, Battula VL, Andreeff M, Konopleva M, Chung Y-J, et al. CXCR4 Inhibition Enhances Efficacy of FLT3 Inhibitors in FLT3-Mutated AML Augmented by Suppressed TGF-β Signaling. Cancers. 2020; 12(7):1737. https://doi.org/10.3390/cancers12071737
Chicago/Turabian StyleKim, Bo-Reum, Seung-Hyun Jung, A-Reum Han, Gyeongsin Park, Hee-Je Kim, Bin Yuan, Venkata Lokesh Battula, Michael Andreeff, Marina Konopleva, Yeun-Jun Chung, and et al. 2020. "CXCR4 Inhibition Enhances Efficacy of FLT3 Inhibitors in FLT3-Mutated AML Augmented by Suppressed TGF-β Signaling" Cancers 12, no. 7: 1737. https://doi.org/10.3390/cancers12071737
APA StyleKim, B.-R., Jung, S.-H., Han, A.-R., Park, G., Kim, H.-J., Yuan, B., Battula, V. L., Andreeff, M., Konopleva, M., Chung, Y.-J., & Cho, B.-S. (2020). CXCR4 Inhibition Enhances Efficacy of FLT3 Inhibitors in FLT3-Mutated AML Augmented by Suppressed TGF-β Signaling. Cancers, 12(7), 1737. https://doi.org/10.3390/cancers12071737