Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cancer-Associated Fibroblasts (CAFs) Express Epidermal Growth Factor Receptor (EGFR) but Remain Viable during EGFR Inhibition
2.2. CAFs Decrease Cancer Cell Sensitivity to Cetuximab
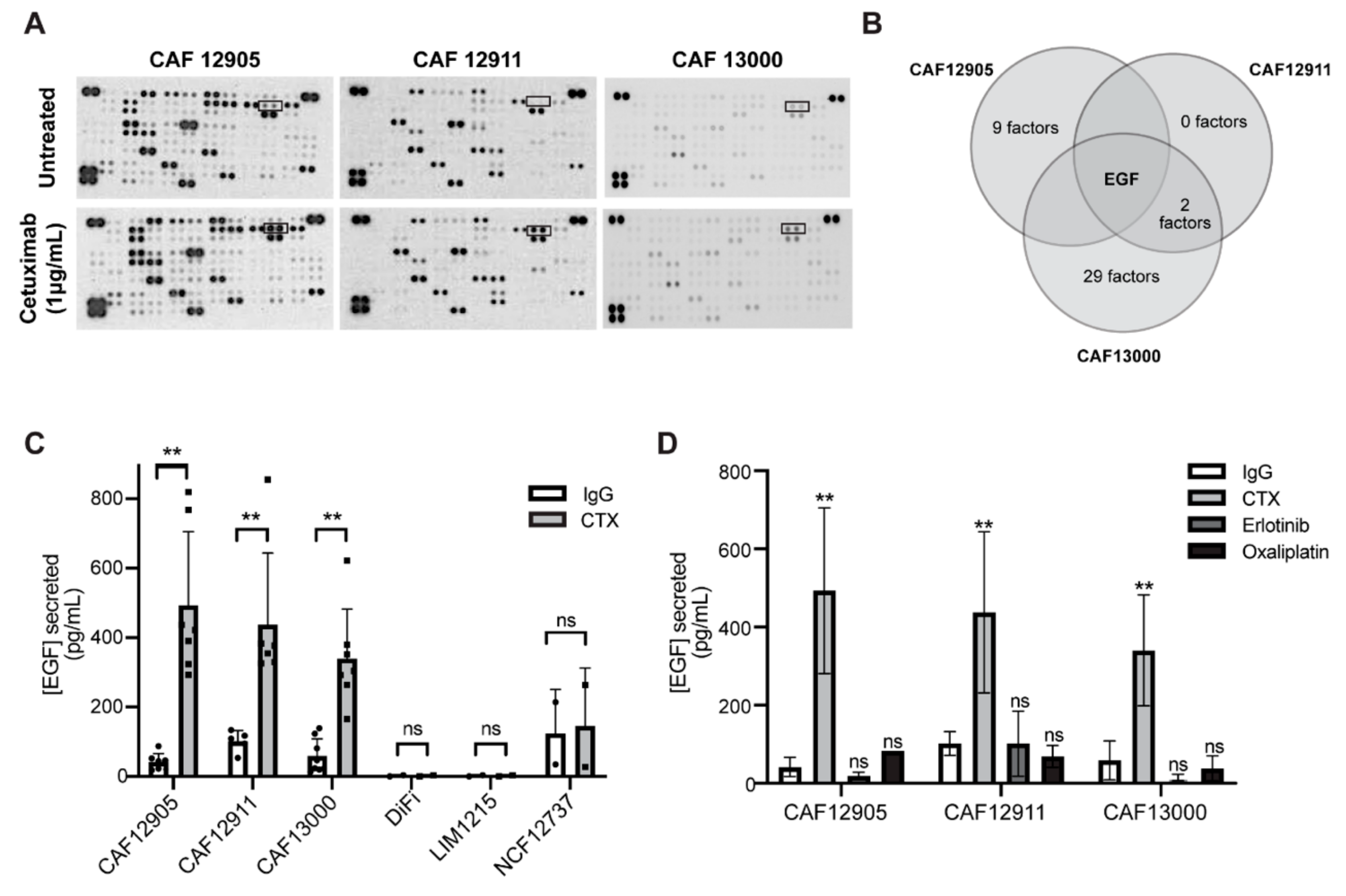
2.3. CAFs Secrete More Epidermal Growth Factor (EGF) in Response to Cetuximab Treatment
2.4. Exogenous EGF Causes Cetuximab Resistance in 2D and 3D Cell Culture Models
2.5. Secreted EGF from Cetuximab-Treated CAFs Is Sufficient to Render Cancer Cells Resistant to Cetuximab
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Primary Cell Culture: Human Tumor Organoids and Cancer-Associated Fibroblasts
4.3. Imaging Growth Rate Assays
4.4. Collection and Processing of Conditioned Media
4.5. Secretome Analysis
4.6. Western Blotting
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Trinh, A.; Trumpi, K.; De Sousa E Melo, F.; Wang, X.; de Jong, J.H.; Fessler, E.; Kuppen, P.J.; Reimers, M.S.; Swets, M.; Koopman, M.; et al. Practical and Robust Identification of Molecular Subtypes in Colorectal Cancer by Immunohistochemistry. Clin. Cancer Res. 2017, 23, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Wang, L.; Cao, L.; Wang, H.; Liu, B.; Zhang, Q.; Meng, Z.; Wu, X.; Zhou, Q.; Xu, K. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget 2017, 8, 76116–76128. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Tommelein, J.; De Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2018, 78, 659–670. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Garvey, C.M.; Spiller, E.; Lindsay, D.; Chiang, C.T.; Choi, N.C.; Agus, D.B.; Mallick, P.; Foo, J.; Mumenthaler, S.M. A high-content image-based method for quantitatively studying context-dependent cell population dynamics. Sci. Rep. 2016, 6, 29752. [Google Scholar] [CrossRef]
- Garvey, C.M.; Gerhart, T.A.; Mumenthaler, S.M. Discrimination and Characterization of Heterocellular Populations Using Quantitative Imaging Techniques. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef]
- Luraghi, P.; Bigatto, V.; Cipriano, E.; Reato, G.; Orzan, F.; Sassi, F.; De Bacco, F.; Isella, C.; Bellomo, S.E.; Medico, E.; et al. A Molecularly Annotated Model of Patient-Derived Colon Cancer Stem-Like Cells to Assess Genetic and Nongenetic Mechanisms of Resistance to Anti-EGFR Therapy. Clin. Cancer Res. 2018, 24, 807–820. [Google Scholar] [CrossRef]
- Price, T.J.; Peeters, M.; Kim, T.W.; Li, J.; Cascinu, S.; Ruff, P.; Suresh, A.S.; Thomas, A.; Tjulandin, S.; Zhang, K.; et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): A randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014, 15, 569–579. [Google Scholar] [CrossRef]
- Derer, S.; Bauer, P.; Lohse, S.; Scheel, A.H.; Berger, S.; Kellner, C.; Peipp, M.; Valerius, T. Impact of epidermal growth factor receptor (EGFR) cell surface expression levels on effector mechanisms of EGFR antibodies. J. Immunol. 2012, 189, 5230–5239. [Google Scholar] [CrossRef]
- Matsuo, T.; Nishizuka, S.S.; Ishida, K.; Iwaya, T.; Ikeda, M.; Wakabayashi, G. Analysis of the anti-tumor effect of cetuximab using protein kinetics and mouse xenograft models. BMC Res. Notes 2011, 4, 140. [Google Scholar] [CrossRef]
- Yoshida, T.; Okamoto, I.; Okabe, T.; Iwasa, T.; Satoh, T.; Nishio, K.; Fukuoka, M.; Nakagawa, K. Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. Int. J. Cancer 2008, 122, 1530–1538. [Google Scholar] [CrossRef]
- Woolston, A.; Khan, K.; Spain, G.; Barber, L.J.; Griffiths, B.; Gonzalez-Exposito, R.; Hornsteiner, L.; Punta, M.; Patil, Y.; Newey, A.; et al. Genomic and Transcriptomic Determinants of Therapy Resistance and Immune Landscape Evolution during Anti-EGFR Treatment in Colorectal Cancer. Cancer Cell 2019, 36, 35–50. [Google Scholar] [CrossRef]
- Troiani, T.; Napolitano, S.; Vitagliano, D.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; Ciardiello, D.; Ciardiello, F.; Martinelli, E. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin. Cancer Res. 2014, 20, 3775–3786. [Google Scholar] [CrossRef]
- Pranteda, A.; Piastra, V.; Stramucci, L.; Fratantonio, D.; Bossi, G. The p38 MAPK Signaling Activation in Colorectal Cancer upon Therapeutic Treatments. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef]
- Stintzing, S.; Einem, J.V.; Fueweger, C.; Haidenberger, A.; Fedorov, M.; Muavcevic, A. Long-term Survival in Patients Treated with a Robotic Radiosurgical Device for Liver Metastases. Cancer Res. Treat. 2019, 51, 187–193. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Fioravanti, A.; Orlandi, P.; Salvatore, L.; Masi, G.; Schirripa, M.; Di Desidero, T.; Antoniotti, C.; Canu, B.; et al. EGFR ligands as pharmacodynamic biomarkers in metastatic colorectal cancer patients treated with cetuximab and irinotecan. Target. Oncol. 2014, 9, 205–214. [Google Scholar] [CrossRef]
- Han, S.W.; Oh, D.Y.; Im, S.A.; Park, S.R.; Lee, K.W.; Song, H.S.; Lee, N.S.; Lee, K.H.; Choi, I.S.; Lee, M.H.; et al. Phase II study and biomarker analysis of cetuximab combined with modified FOLFOX6 in advanced gastric cancer. Br. J. Cancer 2009, 100, 298–304. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Mumenthaler, S.M.; Foo, J.; Leder, K.; Choi, N.C.; Agus, D.B.; Pao, W.; Mallick, P.; Michor, F. Evolutionary modeling of combination treatment strategies to overcome resistance to tyrosine kinase inhibitors in non-small cell lung cancer. Mol. Pharm. 2011, 8, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 6 June 2019).
- Cluster: Cluster Analysis Basics and Extensions; R package version 2.1.0. Available online: https://cran.r-project.org/web/packages/cluster/ (accessed on 6 June 2019).
- P’ng, C.; Green, J.; Chong, L.C.; Waggott, D.; Prokopec, S.D.; Shamsi, M.; Nguyen, F.; Mak, D.Y.F.; Lam, F.; Albuquerque, M.A.; et al. BPG: Seamless, automated and interactive visualization of scientific data. BMC Bioinform. 2019, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A Package for the Generation of Highly-Customizable Venn and Euler Diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garvey, C.M.; Lau, R.; Sanchez, A.; Sun, R.X.; Fong, E.J.; Doche, M.E.; Chen, O.; Jusuf, A.; Lenz, H.-J.; Larson, B.; et al. Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance. Cancers 2020, 12, 1393. https://doi.org/10.3390/cancers12061393
Garvey CM, Lau R, Sanchez A, Sun RX, Fong EJ, Doche ME, Chen O, Jusuf A, Lenz H-J, Larson B, et al. Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance. Cancers. 2020; 12(6):1393. https://doi.org/10.3390/cancers12061393
Chicago/Turabian StyleGarvey, Colleen M., Roy Lau, Alyssa Sanchez, Ren X. Sun, Emma J. Fong, Michael E. Doche, Oscar Chen, Anthony Jusuf, Heinz-Josef Lenz, Brent Larson, and et al. 2020. "Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance" Cancers 12, no. 6: 1393. https://doi.org/10.3390/cancers12061393
APA StyleGarvey, C. M., Lau, R., Sanchez, A., Sun, R. X., Fong, E. J., Doche, M. E., Chen, O., Jusuf, A., Lenz, H.-J., Larson, B., & Mumenthaler, S. M. (2020). Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance. Cancers, 12(6), 1393. https://doi.org/10.3390/cancers12061393