Chemical, Physical and Biological Triggers of Evolutionary Conserved Bcl-xL-Mediated Apoptosis

 ,
,  , , , , , ,
, , , , , ,  ,
,  , and
, and  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
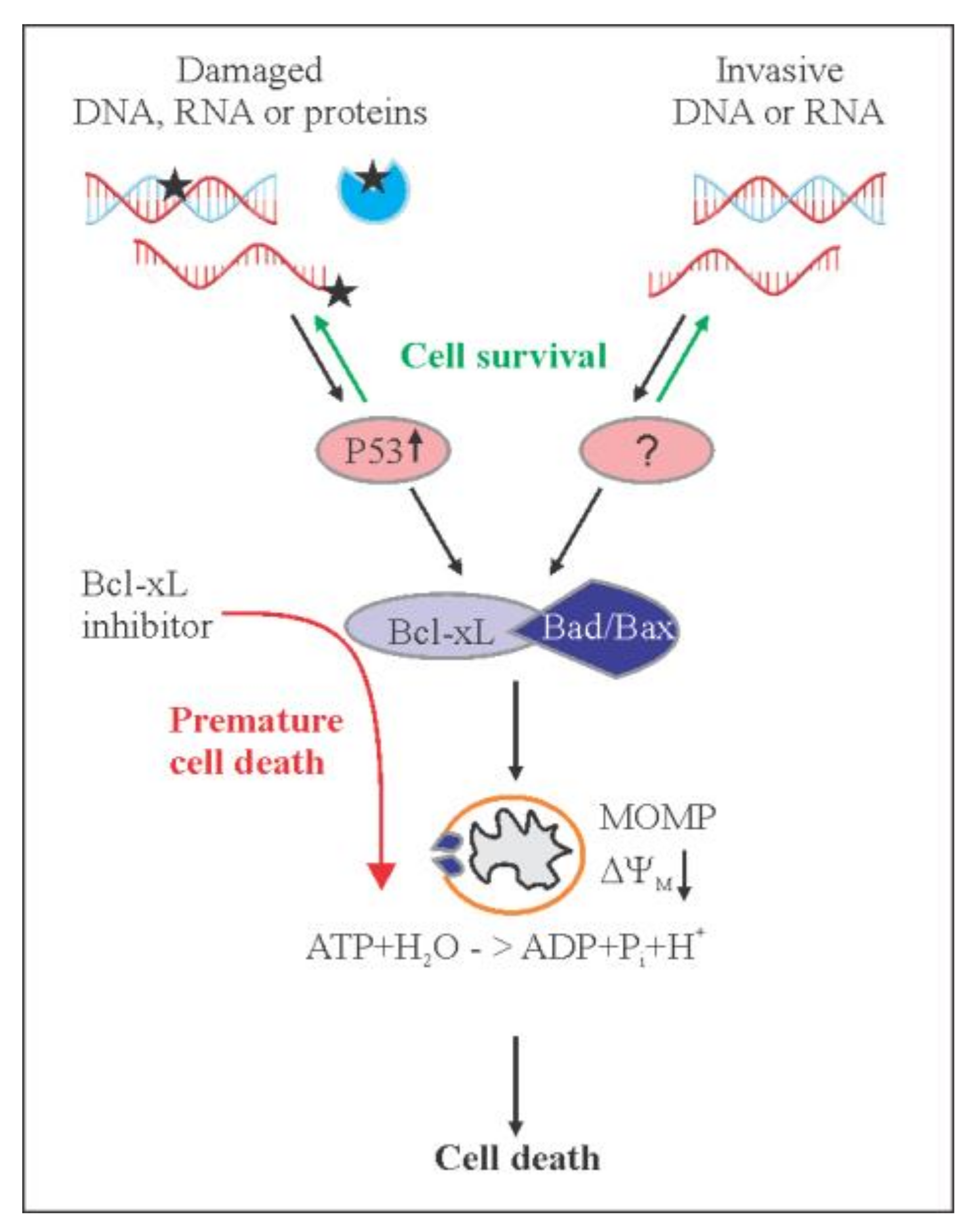
1. Introduction
2. Results
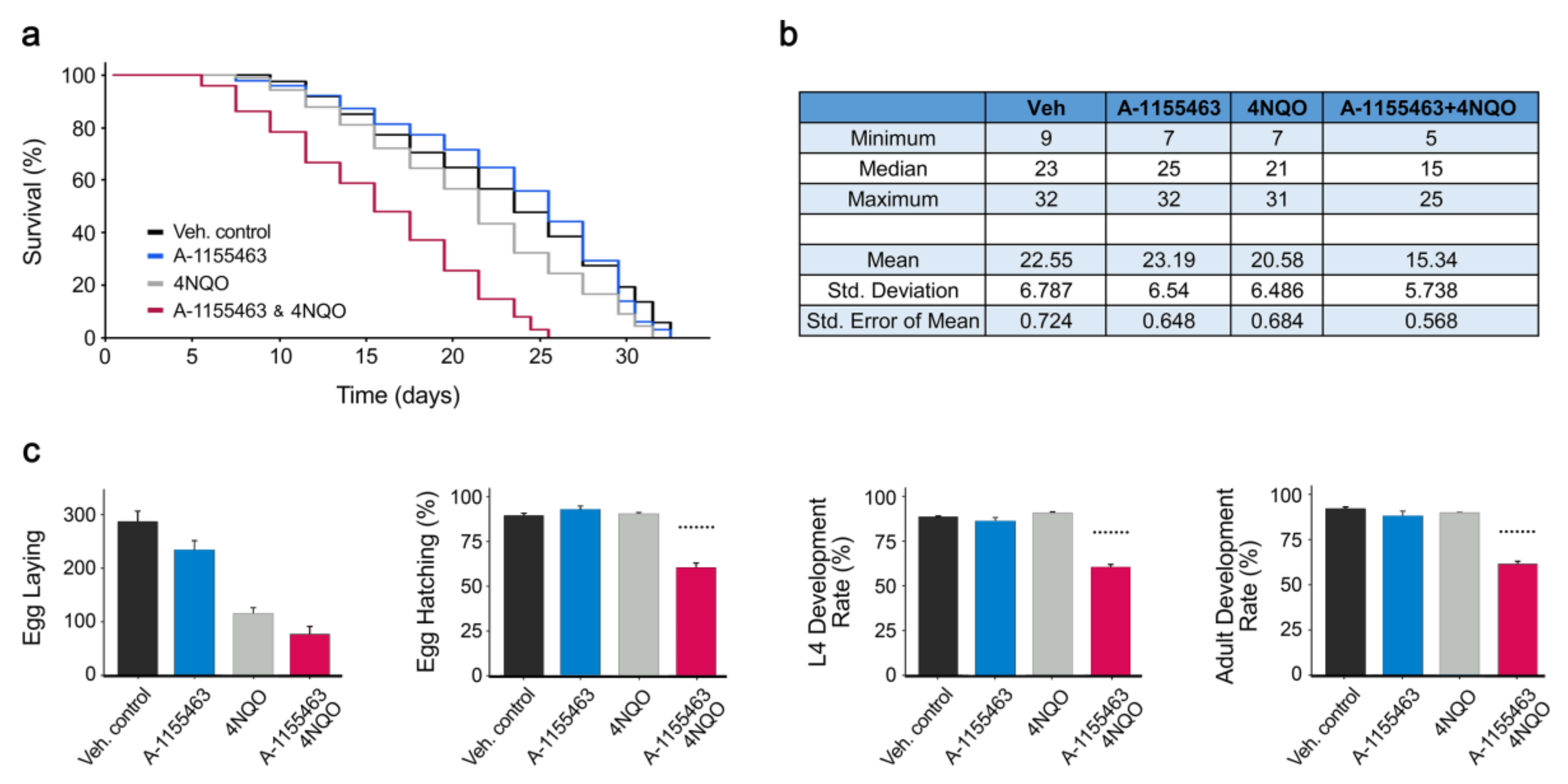
2.1. Toxicity of A-1155463-4NQO Combination in C. elegans
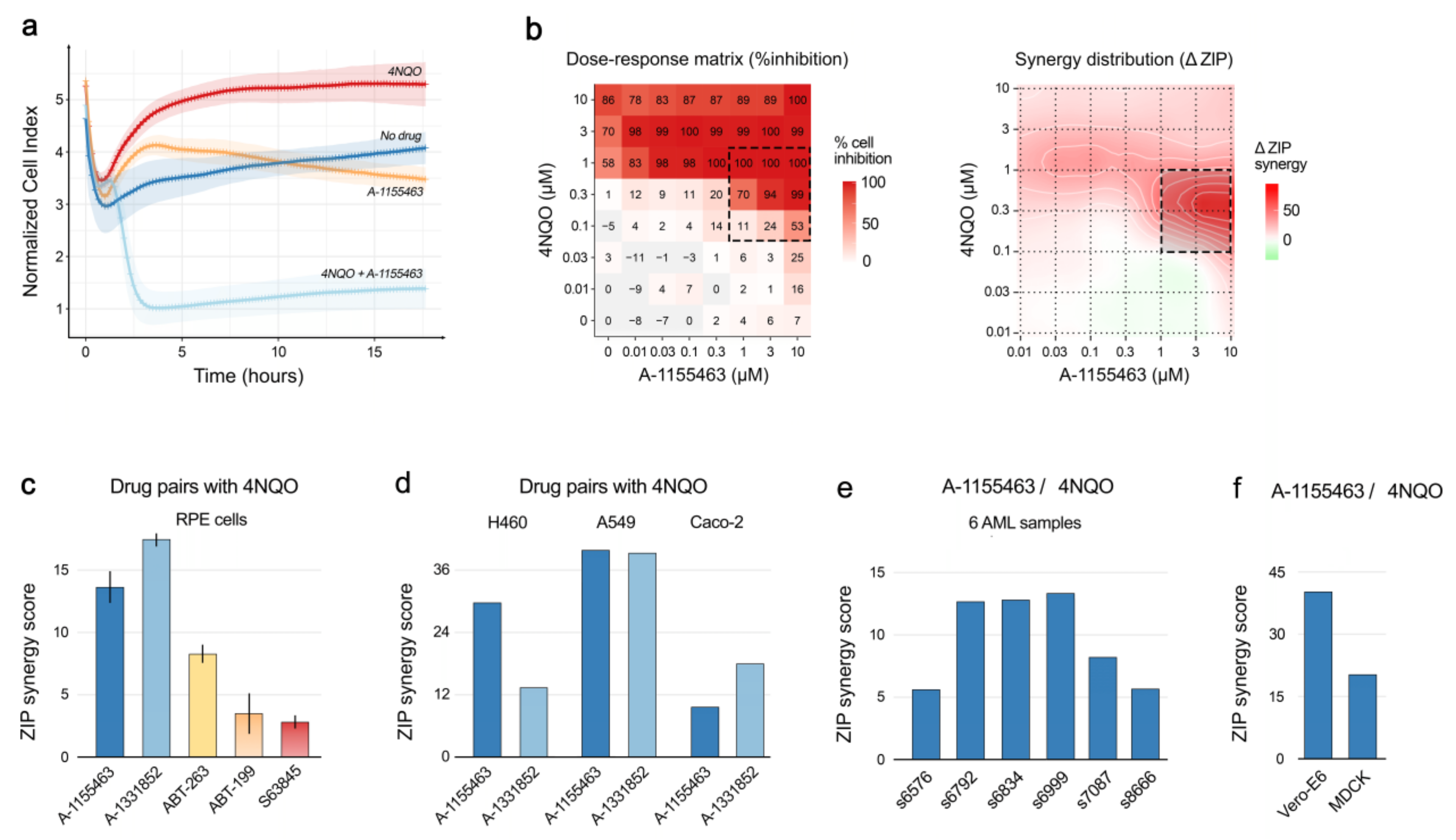
2.2. Toxicity of A-1155463-4NQO Combination for Human, Monkey and Dog Cells
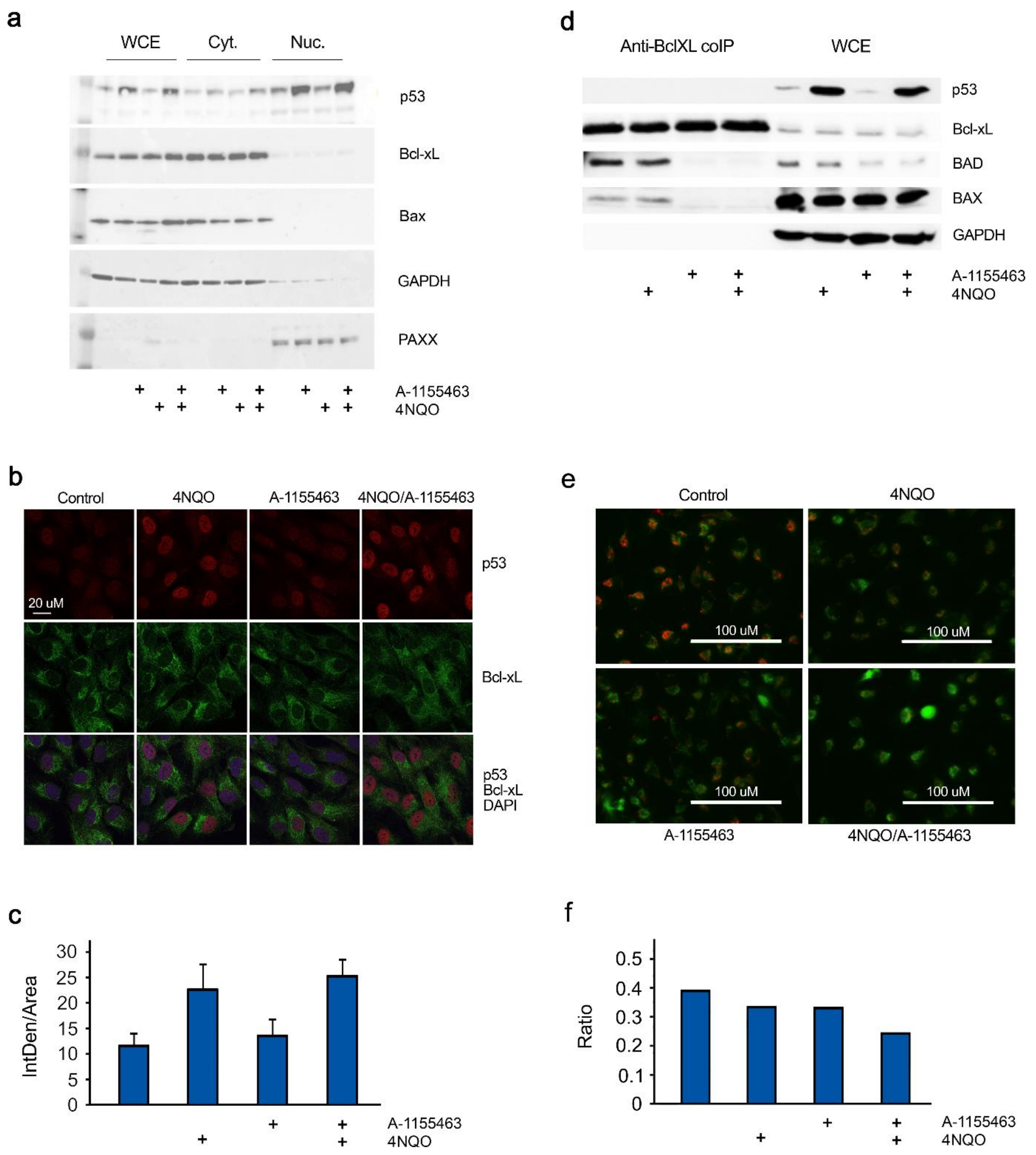
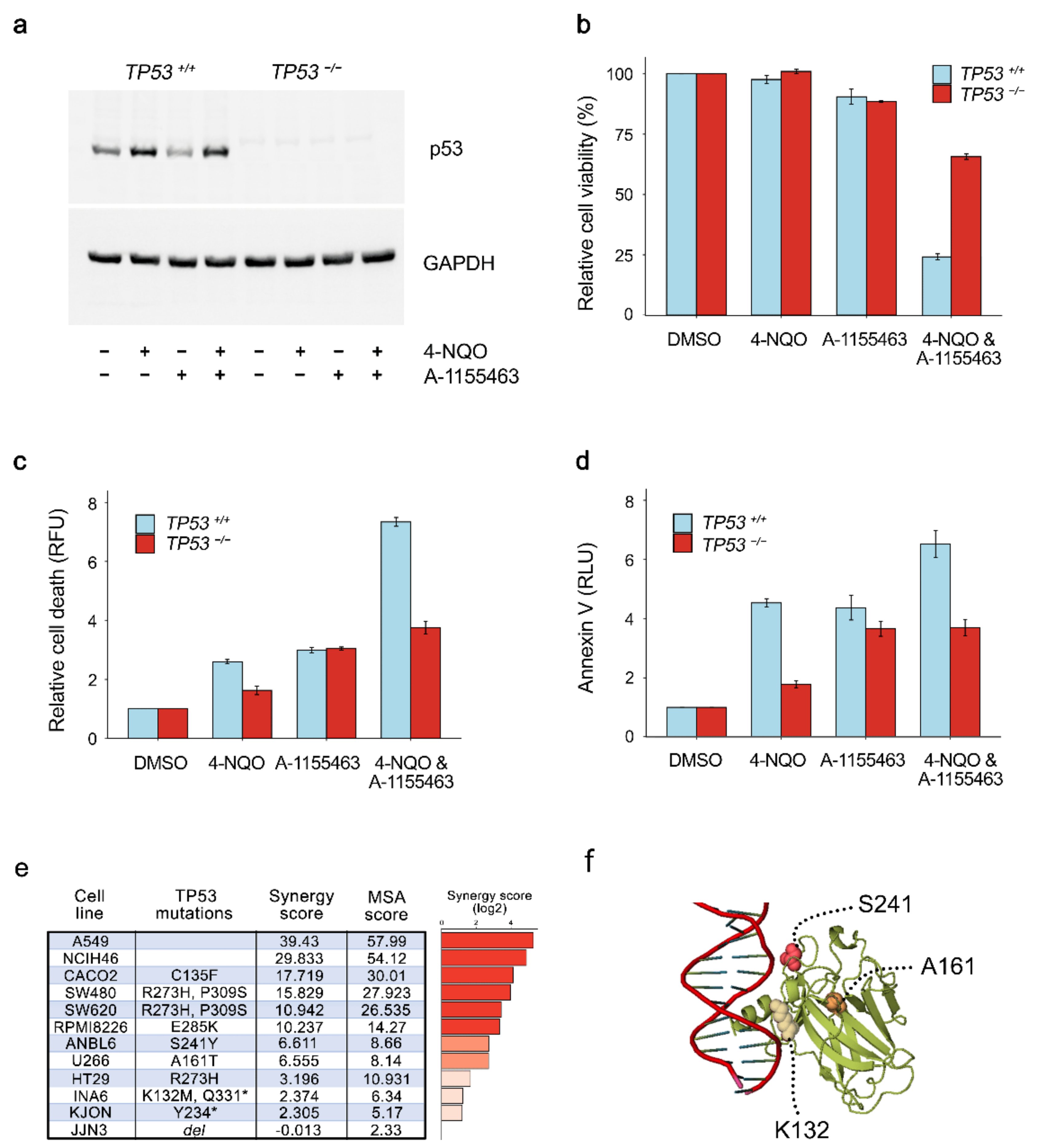
2.3. Concerted Action of 4NQO and A-1155463 Leads to Overexpression of p53, Release of Bad and Bax from Bcl-xL and Activation of MOMP
2.4. p53 is Required for A-1155463-4NQO Synergy
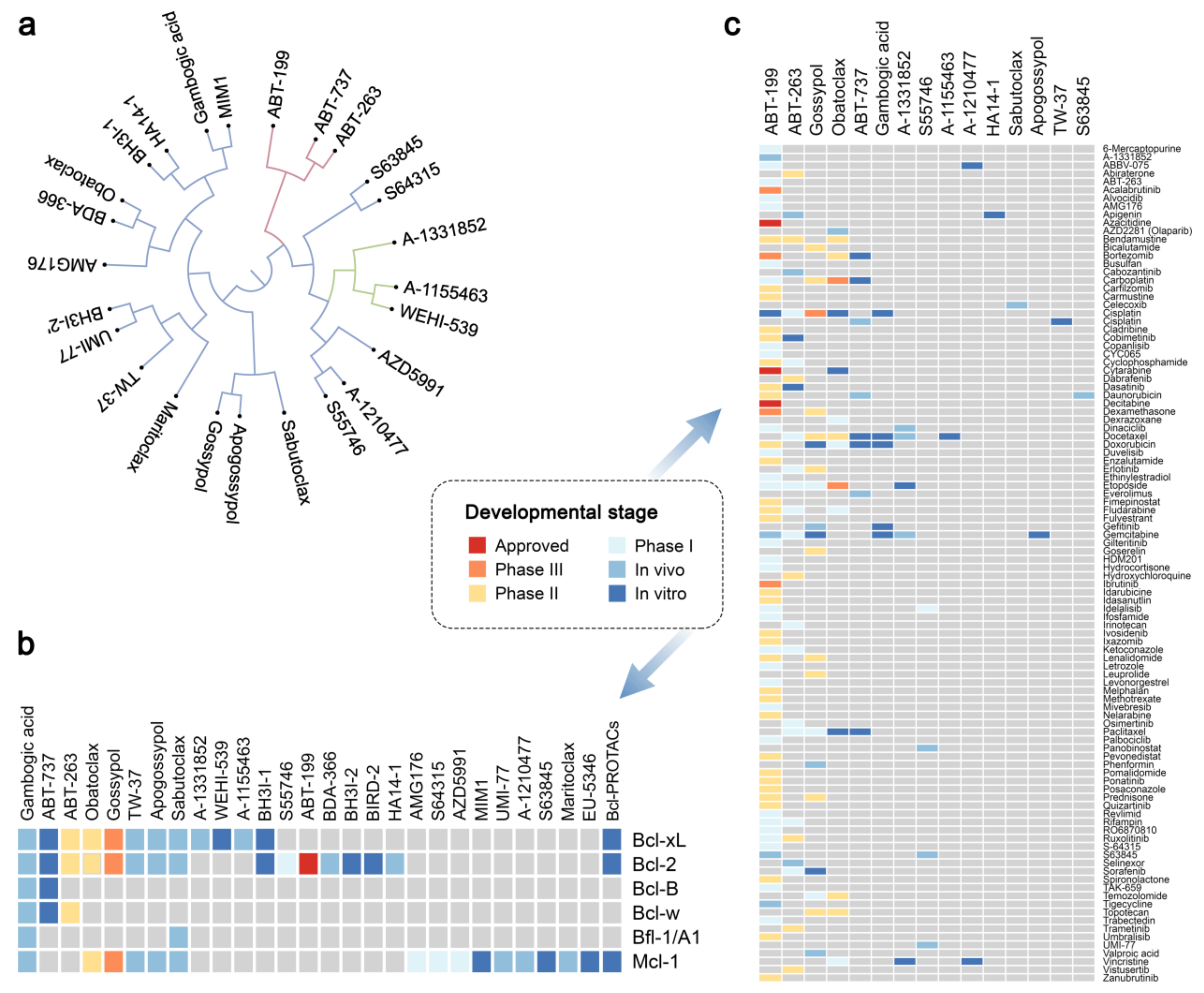
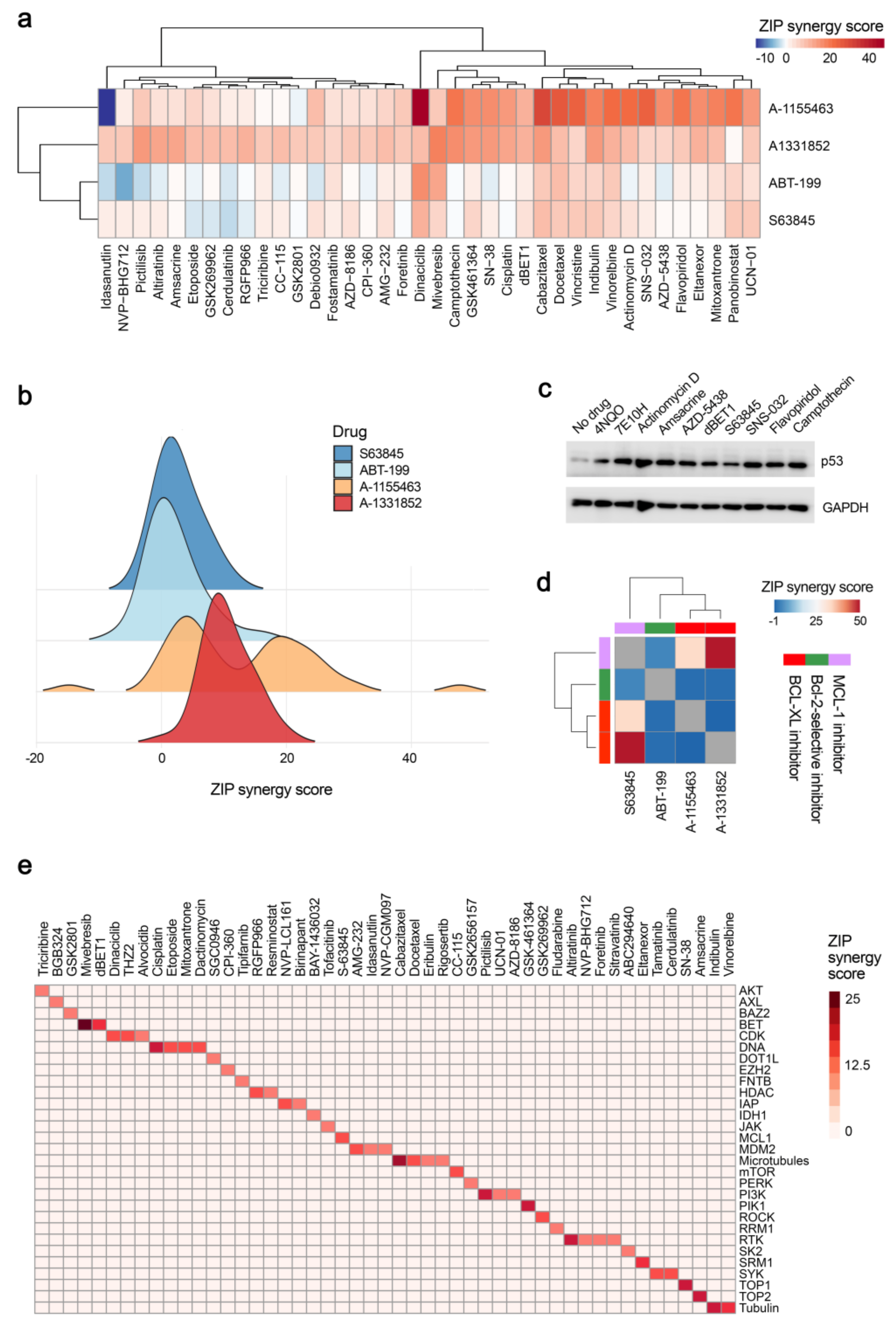
2.5. Chemical Agents Triggering p53-dependent Bcl-xL-Mediated Apoptosis
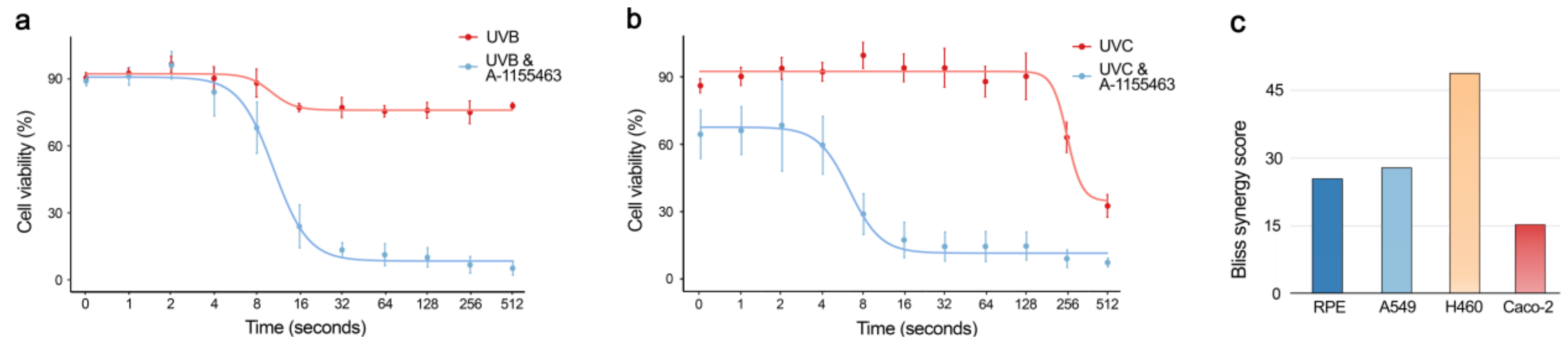
2.6. Physical Factors Triggering Bcl-xL-mediated Apoptosis
3. Discussion
4. Materials and Methods
4.1. Website
4.2. Compounds
4.3. Cells
4.4. C. elegans Maintenance, Lifespan and Toxicity Assays
4.5. Real-time Iimpedance Assay
4.6. Cell Viability Assay
4.7. Cell Toxicity Assay
4.8. Early Apoptosis Assay
4.9. Apoptosis Arrays
4.10. Metabolic Labelling of Cellular RNA and Proteins
4.11. UV Radiation Assay
4.12. Synergy Calculations
4.13. Fractionation of RPE Cells
4.14. Analysis of Mitochondrial Membrane Potential
4.15. Immuno-precipitation and Immuno-blotting
4.16. Confocal Microscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Shim, J.M.; Kim, J.; Tenson, T.; Min, J.Y.; Kainov, D.E. Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis. Viruses 2017, 9, 223. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Kuivanen, S.; Bespalov, M.M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antivir. Res. 2017, 139, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Denisova, O.V.; Kakkola, L.; Feng, L.; Stenman, J.; Nagaraj, A.; Lampe, J.; Yadav, B.; Aittokallio, T.; Kaukinen, P.; Ahola, T.; et al. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza a virus infection. J. Biol. Chem. 2012, 287, 35324–35332. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Clark-Garvey, S.; Porcu, P.; Eischen, C.M. Targeting the Bcl-2 Family in B Cell Lymphoma. Front Oncol. 2018, 8, 636. [Google Scholar] [CrossRef]
- Korycka-Wolowiec, A.; Wolowiec, D.; Kubiak-Mlonka, A.; Robak, T. Venetoclax in the treatment of chronic lymphocytic leukemia. Expert Opin. Drug Metab. Toxicol. 2019, 15, 353–366. [Google Scholar] [CrossRef]
- Timucin, A.C.; Basaga, H.; Kutuk, O. Selective targeting of antiapoptotic BCL-2 proteins in cancer. Med. Res. Rev. 2019, 39, 146–175. [Google Scholar] [CrossRef] [PubMed]
- Casara, P.; Davidson, J.; Claperon, A.; Le Toumelin-Braizat, G.; Vogler, M.; Bruno, A.; Chanrion, M.; Lysiak-Auvity, G.; Le Diguarher, T.; Starck, J.B.; et al. S55746 is a novel orally active BCL-2 selective and potent inhibitor that impairs hematological tumor growth. Oncotarget 2018, 9, 20075–20088. [Google Scholar] [CrossRef] [PubMed]
- King, A.C.; Peterson, T.J.; Horvat, T.Z.; Rodriguez, M.; Tang, L.A. Venetoclax: A First-in-Class Oral BCL-2 Inhibitor for the Management of Lymphoid Malignancies. Ann. Pharmacother. 2017, 51, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Oh, J.M.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Combination Treatment with the BRAF(V600E) Inhibitor Vemurafenib and the BH3 Mimetic Navitoclax for BRAF-Mutant Thyroid Carcinoma. Thyroid 2019, 29, 540–548. [Google Scholar] [CrossRef]
- Haikala, H.M.; Anttila, J.M.; Marques, E.; Raatikainen, T.; Ilander, M.; Hakanen, H.; Ala-Hongisto, H.; Savelius, M.; Balboa, D.; Von Eyss, B.; et al. Pharmacological reactivation of MYC-dependent apoptosis induces susceptibility to anti-PD-1 immunotherapy. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Shen, Q.; Li, J.; Mai, J.; Zhang, Z.; Fisher, A.; Wu, X.; Li, Z.; Ramirez, M.R.; Chen, S.; Shen, H. Sensitizing non-small cell lung cancer to BCL-xL-targeted apoptosis. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Chen, J.; Jin, S.; Abraham, V.; Huang, X.; Liu, B.; Mitten, M.J.; Nimmer, P.; Lin, X.; Smith, M.; Shen, Y.; et al. The Bcl-2/Bcl-X(L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 2340–2349. [Google Scholar] [CrossRef]
- Bulanova, D.; Ianevski, A.; Bugai, A.; Akimov, Y.; Kuivanen, S.; Paavilainen, H.; Kakkola, L.; Nandania, J.; Turunen, L.; Ohman, T.; et al. Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins. Viruses 2017, 9, 271. [Google Scholar] [CrossRef]
- Kakkola, L.; Denisova, O.V.; Tynell, J.; Viiliainen, J.; Ysenbaert, T.; Matos, R.C.; Nagaraj, A.; Ohman, T.; Kuivanen, S.; Paavilainen, H.; et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 2013, 4, e742. [Google Scholar] [CrossRef]
- Walker, I.G.; Sridhar, R. Alkali-labile lesions in DNA from cells treated with methylating agents, 4-nitroquinoline-N-oxide, or ultraviolet light. Basic Life Sci. 1975, 5A, 29–30. [Google Scholar]
- Lai, P.B.; Chi, T.Y.; Chen, G.G. Different levels of p53 induced either apoptosis or cell cycle arrest in a doxycycline-regulated hepatocellular carcinoma cell line in vitro. Apoptosis 2007, 12, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Koval, V.S.; Arutyunyan, A.F.; Salyanov, V.L.; Klimova, R.R.; Kushch, A.A.; Rybalkina, E.Y.; Susova, O.Y.; Zhuze, A.L. DNA sequence-specific ligands. XVII. Synthesis, spectral properties, virological and biochemical studies of fluorescent dimeric bisbenzimidazoles DBA(n). Bioorg. Med. Chem. 2018, 26, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Susova, O.Y.; Salyanov, V.I.; Kirsanov, K.I.; Zhuze, A.L. A new series of biologically active DNA minor groove binders based on bisbenzimidazole and benzimidazole-pyrrole motives. J. Biomol. Struct. Dyn. 2013, 31, 52–53. [Google Scholar] [CrossRef][Green Version]
- Kim, S.Y.; Song, X.; Zhang, L.; Bartlett, D.L.; Lee, Y.J. Role of Bcl-xL/Beclin-1 in interplay between apoptosis and autophagy in oxaliplatin and bortezomib-induced cell death. Biochem. Pharmacol. 2014, 88, 178–188. [Google Scholar] [CrossRef]
- Li, H.; Wang, H.; Deng, K.; Han, W.; Hong, B.; Lin, W. The ratio of Bcl-2/Bim as a predictor of cisplatin response provides a rational combination of ABT-263 with cisplatin or radiation in small cell lung cancer. Cancer Biomark. 2019, 24, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Schiff, D.S.; Lin, Y.; Neboori, H.J.; Goyal, S.; Feng, Z.; Haffty, B.G. Ionizing radiation sensitizes breast cancer cells to Bcl-2 inhibitor, ABT-737, through regulating Mcl-1. Radiat. Res. 2014, 182, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.; Beljanski, V.; Van Grevenynghe, J.; Richards, S.; Ben Yebdri, F.; He, Z.; Nichols, C.; Belgnaoui, S.M.; Steel, C.; Goulet, M.L.; et al. BCL-2 inhibitors sensitize therapy-resistant chronic lymphocytic leukemia cells to VSV oncolysis. Mol. Ther. 2013, 21, 1413–1423. [Google Scholar] [CrossRef]
- Ursu, O.; Holmes, J.; Bologa, C.G.; Yang, J.J.; Mathias, S.L.; Stathias, V.; Nguyen, D.T.; Schurer, S.; Oprea, T. DrugCentral 2018: An update. Nucleic Acids Res. 2019, 47, D963–D970. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Ianevski, A.; Zusinaite, E.; Kuivanen, S.; Strand, M.; Lysvand, H.; Teppor, M.; Kakkola, L.; Paavilainen, H.; Laajala, M.; Kallio-Kokko, H.; et al. Novel activities of safe-in-human broad-spectrum antiviral agents. Antivir. Res. 2018, 154, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Vatsveen, T.K.; Borset, M.; Dikic, A.; Tian, E.; Micci, F.; Lid, A.H.; Meza-Zepeda, L.A.; Coward, E.; Waage, A.; Sundan, A.; et al. VOLIN and KJON-Two novel hyperdiploid myeloma cell lines. Genes Chromosomes Cancer 2016, 55, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Guenther, A.; Bakker, F.; Schmalzing, M.; Bernand, S.; Baum, W.; Duerr, B.; Hocke, G.M.; Steininger, H.; Gebhart, E.; et al. Gp130 and ras mediated signaling in human plasma cell line INA-6: A cytokine-regulated tumor model for plasmacytoma. Hematolj. 2001, 2, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, D.F.; Ahmann, G.J.; Greipp, P.R.; Jalal, S.M.; Westendorf, J.J.; Katzmann, J.A.; Kyle, R.A.; Lust, J.A. Coexistence of aneuploid subclones within a myeloma cell line that exhibits clonal immunoglobulin gene rearrangement: Clinical implications. Cancer Res. 1993, 53, 5320–5327. [Google Scholar]
- Jackson, N.; Lowe, J.; Ball, J.; Bromidge, E.; Ling, N.R.; Larkins, S.; Griffith, M.J.; Franklin, I.M. Two new IgA1-kappa plasma cell leukaemia cell lines (JJN-1 & JJN-2) which proliferate in response to B cell stimulatory factor 2. Clin. Exp. Immunol. 1989, 75, 93–99. [Google Scholar]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Solly, K.; Wang, X.; Xu, X.; Strulovici, B.; Zheng, W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay. Drug Dev. Technol. 2004, 2, 363–372. [Google Scholar] [CrossRef]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ianevski, A.; Kulesskiy, E.; Krpina, K.; Lou, G.; Aman, Y.; Bugai, A.; Aasumets, K.; Akimov, Y.; Bulanova, D.; Gildemann, K.; et al. Chemical, Physical and Biological Triggers of Evolutionary Conserved Bcl-xL-Mediated Apoptosis. Cancers 2020, 12, 1694. https://doi.org/10.3390/cancers12061694
Ianevski A, Kulesskiy E, Krpina K, Lou G, Aman Y, Bugai A, Aasumets K, Akimov Y, Bulanova D, Gildemann K, et al. Chemical, Physical and Biological Triggers of Evolutionary Conserved Bcl-xL-Mediated Apoptosis. Cancers. 2020; 12(6):1694. https://doi.org/10.3390/cancers12061694
Chicago/Turabian StyleIanevski, Aleksandr, Evgeny Kulesskiy, Klara Krpina, Guofeng Lou, Yahyah Aman, Andrii Bugai, Koit Aasumets, Yevhen Akimov, Daria Bulanova, Kiira Gildemann, and et al. 2020. "Chemical, Physical and Biological Triggers of Evolutionary Conserved Bcl-xL-Mediated Apoptosis" Cancers 12, no. 6: 1694. https://doi.org/10.3390/cancers12061694
APA StyleIanevski, A., Kulesskiy, E., Krpina, K., Lou, G., Aman, Y., Bugai, A., Aasumets, K., Akimov, Y., Bulanova, D., Gildemann, K., Arutyunyan, A. F., Susova, O. Y., Zhuze, A. L., Ji, P., Wang, W., Holien, T., Bugge, M., Zusinaite, E., Oksenych, V., ... Kainov, D. E. (2020). Chemical, Physical and Biological Triggers of Evolutionary Conserved Bcl-xL-Mediated Apoptosis. Cancers, 12(6), 1694. https://doi.org/10.3390/cancers12061694