Exploiting Cancer’s Tactics to Make Cancer a Manageable Chronic Disease


{kind=link}
{kind=link}
Abstract
1. Past and Present Cancer Therapeutics
1.1. Cancer Treatments Discovered over the Last 70 Years
1.2. Chemo- and Targeted-Therapies Co-Exist in Today’s Cancer Clinic
1.3. Immunotherapy Has Begun to Appear in Upfront Regimens
1.4. Current Limitations of Anti-Angiogenesis Therapy
1.5. Therapeutic Strategies Based on Targeting Cancer Stem Cells
2. Advances in Neurosurgical Oncology Reflect Advances in Surgical Oncology
3. Exploiting Cancer’s Evolutionary Tactics for Future Cancer Therapeutics
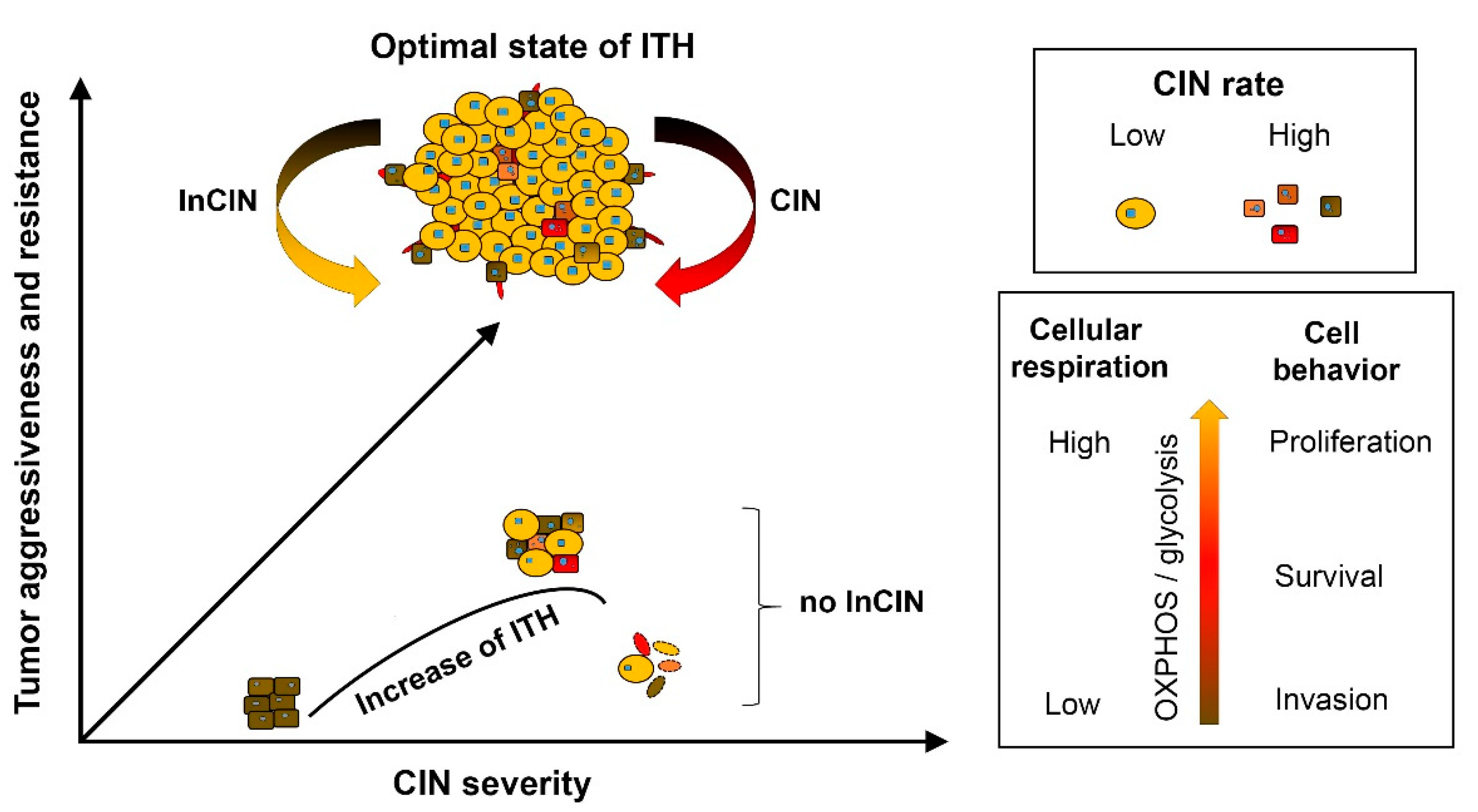
3.1. Control of Chromosome Instability Rate and Modulating Intra-Tumoral Heterogeneity
3.2. Cancer-Specific Metabolism and Energetics
3.3. Cancer Therapeutic Development by Exploiting Chromosome Instability
3.4. Future Cancer Therapeutics Targeting Natural Cancer Evolution Tactics
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Einhorn, J. Nitrogen mustard: The origin of chemotherapy for cancer. Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 1375–1378. [Google Scholar] [CrossRef]
- Christakis, P. The birth of chemotherapy at Yale. Bicentennial lecture series: Surgery Grand Round. Yale J. Biol. Med. 2011, 84, 169–172. [Google Scholar] [PubMed]
- Smith, S.L. Toxic Exposures: Mustard Gas and the Health Consequences of World War II in the United States; Critical Issues in Health and Medicine Series; Rutgers University Press: New Brunswick, NJ, USA, 2017. [Google Scholar]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Karnofsky, D.A.; Burchenal, J.H.; Ormsler, R.A.; Corman, I.; Rhoads, C.P. Experimental observations on the use of nitrogen mustard in the treatment of neoplastic diseases. In Approaches to Tumor Chemotherapy; Moulton, F.R., Ed.; American Association for the Advancement of Science: Washington, DC, USA, 1947; pp. 298–305. [Google Scholar]
- Goodman, L.S.; Wintrobe, M.M.; Dameshek, W.; Goodman, M.J.; Gilman, A.; McLennan, T. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946, 132, 126–132. [Google Scholar] [CrossRef]
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 1963, 105, 574–578. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [CrossRef]
- Diethelm-Varela, B.; Ai, Y.; Liang, D.; Xue, F. Nitrogen Mustards as Anticancer Chemotherapies: Historic Perspective, Current Developments and Future Trends. Curr. Top. Med. Chem. 2019, 19, 691–712. [Google Scholar] [CrossRef]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef]
- Miura, K.; Kinouchi, M.; Ishida, K.; Fujibuchi, W.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; et al. 5-fu metabolism in cancer and orally-administrable 5-fu drugs. Cancers 2010, 2, 1717. [Google Scholar] [CrossRef]
- Frei, E., III; Karon, M.; Levin, R.H.; Freireich, E.J.; Taylor, R.J.; Hananian, J.; Selawry, O.; Holland, J.F.; Hoogstraten, B.; Wolman, I.J.; et al. The effectiveness of combinations of antileukemic agents in inducing and maintaining remission in children with acute leukemia. Blood 1965, 26, 642–656. [Google Scholar] [CrossRef]
- Gehan, E.A.; Schneiderman, M.A. Historical and methodological developments in clinical trials at the National Cancer Institute. Stat. Med. 1990, 9, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.H.; Donohue, J. Cis-diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann. Intern. Med. 1977, 87, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.S. The radical radiotherapy of regionally localized Hodgkin’s disease. Radiology 1962, 78, 553–561. [Google Scholar] [CrossRef]
- Condit, P.T.; Levitt, S.H.; Snow, J.B., Jr.; Knox, G.S.; Williams, G.R.; Bogardus, C.R., Jr.; Eaton, B.G. Combination chemotherapy and radiation therapy of squamous cell carcinoma of the head and neck. Trans. Am. Acad. Ophthalmol. Otolaryngol. 1966, 70, 627–639. [Google Scholar]
- Shapiro, W.R.; Young, D.F. Treatment of malignant glioma. A controlled study of chemotherapy and irradiation. Arch. Neurol. 1976, 33, 450–494. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Rosenberg, S.A. Two hundred years of cancer research. N. Engl. J. Med. 2012, 366, 2207–2214. [Google Scholar] [CrossRef]
- Berenblum, I. A speculative review; the probable nature of promoting action and its significance in the understanding of the mechanism of carcinogenesis. Cancer Res. 1954, 14, 471–477. [Google Scholar]
- Ganesan, A. Epigenetics: The first 25 centuries. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug. Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2018, 15, 150. [Google Scholar] [CrossRef] [PubMed]
- Gasser, M.; Waaga-Gasser, A.M. Therapeutic Antibodies in Cancer Therapy. Adv. Exp. Med. Biol. 2016, 917, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Ayen, A.; Jimenez Martinez, Y.; Marchal, J.A.; Boulaiz, H. Recent Progress in Gene Therapy for Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 1930. [Google Scholar] [CrossRef]
- Farmer, Z.L.; Kim, E.S.; Carrizosa, D.R. Gene Therapy in Head and Neck Cancer. Oral Maxillofac. Surg. Clin. N. Am. 2019, 31, 117–124. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef]
- Unger, J.M.; Cook, E.; Tai, E.; Bleyer, A. The Role of Clinical Trial Participation in Cancer Research: Barriers, Evidence, and Strategies. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 185–198. [Google Scholar] [CrossRef]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Cancer: The Evolutionary Legacy; Oxford University Press: Oxford, NY, USA, 2001; Volume 12. [Google Scholar]
- Gutzmer, R.; Harrington, K.J.; Hoeller, C.; Lebbe, C.; Malvehy, J.; Ohrling, K.; Downey, G.; Dummer, R. Practical clinical guide on the use of talimogene laherparepvec monotherapy in patients with unresectable melanoma in Europe. Eur. J. Dermatol. 2018, 28, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Nahas, G.R.; Walker, N.D.; Bryan, M.; Rameshwar, P. A Perspective of Immunotherapy for Breast Cancer: Lessons Learned and Forward Directions for All Cancers. Breast Cancer 2015, 9, 35–43. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef]
- Brahm, C.G.; van Linde, M.E.; Enting, R.H.; Schuur, M.; Otten, R.H.J.; Heymans, M.W.; Verheul, H.M.W.; Walenkamp, A.M.E. The Current Status of Immune Checkpoint Inhibitors in Neuro-Oncology: A Systematic Review. Cancers 2020, 12, 586. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250–261. [Google Scholar] [CrossRef]
- Bedognetti, D.; Ceccarelli, M.; Galluzzi, L.; Lu, R.; Palucka, K.; Samayoa, J.; Spranger, S.; Warren, S.; Wong, K.K.; Ziv, E.; et al. Toward a comprehensive view of cancer immune responsiveness: A synopsis from the SITC workshop. J. Immunother. Cancer 2019, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Ferrara, N.; Chen, H.; Davis-Smyth, T.; Gerber, H.P.; Nguyen, T.N.; Peers, D.; Chisholm, V.; Hillan, K.J.; Schwall, R.H. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat. Med. 1998, 4, 336–340. [Google Scholar] [CrossRef]
- Shojaei, F. Anti-angiogenesis therapy in cancer: Current challenges and future perspectives. Cancer Lett. 2012, 320, 130–137. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Abrey, L.E.; Beal, K.; Gutin, P.H.; Rosenblum, M.K.; Reuter, V.E.; DeAngelis, L.M.; Lassman, A.B. Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology 2009, 73, 1200–1206. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef]
- Yang, W.H.; Xu, J.; Mu, J.B.; Xie, J. Revision of the concept of anti-angiogenesis and its applications in tumor treatment. Chronic Dis. Transl. Med. 2017, 3, 33–40. [Google Scholar] [CrossRef]
- Wagenblast, E.; Soto, M.; Gutierrez-Angel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.; Simms, N.; Polanski, R.; et al. Vasculogenic mimicry in small cell lung cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Liao, Y.D.; Mai, D.M.; Xie, P.; Qiang, Y.Y.; Zheng, L.S.; Wang, M.Y.; Mei, Y.; Meng, D.F.; Xu, L.; et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: A meta-analysis. Angiogenesis 2016, 19, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Ke, Y.Q.; Lu, G.H.; Song, Z.H.; Yu, L.; Xiao, S.; Sun, X.L.; Jiang, X.D.; Yang, Z.L.; Hu, C.C. Vasculogenic mimicry is a prognostic factor for postoperative survival in patients with glioblastoma. J. Neurooncol. 2013, 112, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.S.; Zhang, Q.P.; Chen, J.L.; Wang, J.; Chen, F.R.; NG, H.K.; Chen, Z.P. Vasculogenic mimicry persists during glioblastoma xenograft growth. Glioma 2018, 1, 16–21. [Google Scholar]
- Mei, X.; Chen, Y.S.; Chen, F.R.; Xi, S.Y.; Chen, Z.P. Glioblastoma stem cell differentiation into endothelial cells evidenced through live-cell imaging. Neuro. Oncol. 2017, 19, 1109–1118. [Google Scholar] [CrossRef]
- Sood, A.K.; Fletcher, M.S.; Zahn, C.M.; Gruman, L.M.; Coffin, J.E.; Seftor, E.A.; Hendrix, M.J. The clinical significance of tumor cell-lined vasculature in ovarian carcinoma: Implications for anti-vasculogenic therapy. Cancer Biol. Ther. 2002, 1, 661–664. [Google Scholar] [CrossRef]
- Arbab, A.S.; Jain, M.; Achyut, B.R. Vascular Mimicry: The Next Big Glioblastoma Target. Biochem. Physiol. 2015, 4. [Google Scholar] [CrossRef]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, D.; Yao, Z.; Lin, X.; Liu, J.; Gu, Q.; Dong, X.; Liu, F.; Wang, Y.; Yao, N.; et al. Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol. Ther. 2017, 18, 205–213. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro-Oncology 2010, 12, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef]
- Ke, C.; Luo, J.R.; Cen, Z.W.; Li, Y.; Cai, H.P.; Wang, J.; Chen, F.R.; Siegel, E.R.; Le, K.N.; Winokan, J.R.; et al. Dual antivascular function of human fibulin-3 variant, a potential new drug discovery strategy for glioblastoma. Cancer Sci. 2020, 111, 940–950. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Y.; Liu, C.; Wang, Q.; Han, X.; Han, Y.; Xie, X.S.; Chen, X.H.; Li, X.; Siegel, E.R.; et al. Human fibulin-3 protein variant expresses anti-cancer effects in the malignant glioma extracellular compartment in intracranial xenograft models. Oncotarget 2017, 8, 106311–106323. [Google Scholar] [CrossRef]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Yasargil, M.G. Intracranial microsurgery. Clin. Neurosurg. 1970, 17, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Yasargil, M.G. Clinical experiences with microtechnic. Schweiz Arch. Neurol. Neurochir. Psychiatr. 1972, 111, 493–504. [Google Scholar] [PubMed]
- Epstein, F. The Cavitron Ultrasonic Aspirator in Tumor Surgery. Neurosurgery 1984, 31, 497–505. [Google Scholar] [CrossRef]
- Sandeman, D.R.; Patel, N.; Chandler, C.; Nelson, R.J.; Coakham, H.B.; Griffith, H.B. Advances in image-directed neurosurgery: Preliminary experience with the ISG Viewing Wand compared with the Leksell G frame. Br. J. Neurosurg. 1994, 8, 529–544. [Google Scholar] [CrossRef]
- Smith, K.R.; Frank, K.J.; Bucholz, R.D. The NeuroStation--a highly accurate, minimally invasive solution to frameless stereotactic neurosurgery. Comput. Med. Imaging. Graph. 1994, 18, 247–256. [Google Scholar] [CrossRef]
- Knauth, M.; Wirtz, C.R.; Tronnier, V.M.; Aras, N.; Kunze, S.; Sartor, K. Intraoperative MR imaging increases the extent of tumor resection in patients with high-grade gliomas. AJNR Am. J. Neuroradiol. 1999, 20, 1642–1646. [Google Scholar]
- Schneider, J.P.; Schulz, T.; Schmidt, F.; Dietrich, J.; Lieberenz, S.; Trantakis, C.; Seifert, V.; Kellermann, S.; Schober, R.; Schaffranietz, L.; et al. Gross-total surgery of supratentorial low-grade gliomas under intraoperative MR guidance. AJNR Am. J. Neuroradiol. 2001, 22, 89–98. [Google Scholar]
- Stummer, W.; Stocker, S.; Wagner, S.; Stepp, H.; Fritsch, C.; Goetz, C.; Goetz, A.E.; Kiefmann, R.; Reulen, H.J. Intraoperative detection of malignant gliomas by 5-aminolevulinic acid-induced porphyrin fluorescence. Neurosurgery 1998, 42, 518–525. [Google Scholar] [CrossRef]
- Stummer, W.; Stepp, H.; Wiestler, O.D.; Pichlmeier, U. Randomized, Prospective Double-Blinded Study Comparing 3 Different Doses of 5-Aminolevulinic Acid for Fluorescence-Guided Resections of Malignant Gliomas. Neurosurgery 2017, 81, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Chaichana, K.L.; Jusue-Torres, I.; Navarro-Ramirez, R.; Raza, S.M.; Pascual-Gallego, M.; Ibrahim, A.; Hernandez-Hermann, M.; Gomez, L.; Ye, X.; Weingart, J.D.; et al. Establishing percent resection and residual volume thresholds affecting survival and recurrence for patients with newly diagnosed intracranial glioblastoma. Neuro. Oncol. 2014, 16, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef]
- Orringer, D.; Lau, D.; Khatri, S.; Zamora-Berridi, G.J.; Zhang, K.; Wu, C.; Chaudhary, N.; Sagher, O. Extent of resection in patients with glioblastoma: Limiting factors, perception of resectability, and effect on survival. J. Neurosurg. 2012, 117, 851–859. [Google Scholar] [CrossRef]
- Smith, J.S.; Chang, E.F.; Lamborn, K.R.; Chang, S.M.; Prados, M.D.; Cha, S.; Tihan, T.; Vandenberg, S.; McDermott, M.W.; Berger, M.S. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J. Clin. Oncol. 2008, 26, 1338–1345. [Google Scholar] [CrossRef]
- Duffau, H.; Moritz-Gasser, S.; Gatignol, P. Functional outcome after language mapping for insular World Health Organization Grade II gliomas in the dominant hemisphere: Experience with 24 patients. Neurosurg. Focus 2009, 27, E7. [Google Scholar] [CrossRef]
- Cagan, R.; Meyer, P. Rethinking cancer: Current challenges and opportunities in cancer research. Dis. Model Mech. 2017, 10, 349–352. [Google Scholar] [CrossRef]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef]
- Chakraborty, S.; Dhakshinamurthy, G.S.; Misra, S.K. Tailoring of physicochemical properties of nanocarriers for effective anti-cancer applications. J. Biomed. Mater. Res. A 2017, 105, 2906–2928. [Google Scholar] [CrossRef]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Câtoi, C. Tumor Cell Morphology. In Comparative Oncology; The Publishing House of the Romanian Academy: Bucharest, Romania, 2007; Chapter 3. [Google Scholar]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front. Oncol. 2018, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell. Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Zur Frage der Entstehung Maligner Tumoren; Fischer: Jena, Germany, 1914. [Google Scholar]
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar] [PubMed]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer. Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Duesberg, P.; Li, R.; Fabarius, A.; Hehlmann, R. The chromosomal basis of cancer. Cell. Oncol. 2005, 27, 293–318. [Google Scholar]
- Alpermann, T.; Haferlach, C.; Eder, C.; Nadarajah, N.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Schnittger, S. AML with gain of chromosome 8 as the sole chromosomal abnormality (+8sole) is associated with a specific molecular mutation pattern including ASXL1 mutations in 46.8% of the patients. Leuk. Res. 2015, 39, 265–272. [Google Scholar] [CrossRef]
- Klein, A.; Li, N.; Nicholson, J.M.; McCormack, A.A.; Graessmann, A.; Duesberg, P. Transgenic oncogenes induce oncogene-independent cancers with individual karyotypes and phenotypes. Cancer Genet. Cytogenet. 2010, 200, 79–99. [Google Scholar] [CrossRef]
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Cancer 2004, 4, 197–205. [Google Scholar] [CrossRef]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Li, R.; Sachs, R.; Fabarius, A.; Upender, M.B.; Hehlmann, R. Cancer drug resistance: The central role of the karyotype. Drug Resist. Updat. 2007, 10, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rondon, N.; Villegas, V.E.; Rondon-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Lee, N.C.; Kouprina, N.; Kim, J.H.; Kagansky, A.; Bates, S.; Trepel, J.B.; Pommier, Y.; Sackett, D.; Larionov, V. Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res. 2016, 76, 902–911. [Google Scholar] [CrossRef]
- Stanta, G.; Bonin, S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front. Med. 2018, 5, 85. [Google Scholar] [CrossRef]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef]
- Sotillo, R.; Schvartzman, J.M.; Benezra, R. Very CIN-ful: Whole chromosome instability promotes tumor suppressor loss of heterozygosity. Cancer Cell 2009, 16, 451–452. [Google Scholar] [CrossRef]
- Duesberg, P.; Mandrioli, D.; McCormack, A.; Nicholson, J.M. Is carcinogenesis a form of speciation? Cell Cycle 2011, 10, 2100–2114. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Amon, A. Aneuploidy: Cancer’s fatal flaw? Cancer Res. 2009, 69, 5289–5291. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Danilova, O.V.; Kaur, P.; Levy, N.B.; Compton, D.A. Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin. Cancer Res. 2011, 17, 7704–7711. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kobunai, T.; Yamamoto, Y.; Matsuda, K.; Ishihara, S.; Nozawa, K.; Yamada, H.; Hayama, T.; Inoue, E.; Tamura, J.; et al. Chromosomal instability (CIN) phenotype, CIN high or CIN low, predicts survival for colorectal cancer. J. Clin. Oncol. 2012, 30, 2256–2264. [Google Scholar] [CrossRef]
- Hahn, P.J. Molecular biology of double-minute chromosomes. Bioessays 1993, 15, 477–484. [Google Scholar] [CrossRef]
- Favero, F.; McGranahan, N.; Salm, M.; Birkbak, N.J.; Sanborn, J.Z.; Benz, S.C.; Becq, J.; Peden, J.F.; Kingsbury, Z.; Grocok, R.J.; et al. Glioblastoma adaptation traced through decline of an IDH1 clonal driver and macro-evolution of a double-minute chromosome. Ann. Oncol. 2015, 26, 880–887. [Google Scholar] [CrossRef]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ‘Go or grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65. [Google Scholar] [CrossRef]
- Gallaher, J.A.; Brown, J.S.; Anderson, A.R.A. The impact of proliferation-migration tradeoffs on phenotypic evolution in cancer. Sci. Rep. 2019, 9, 2425. [Google Scholar] [CrossRef]
- Hu, Y.; Ru, N.; Xiao, H.; Chaturbedi, A.; Hoa, N.T.; Tian, X.J.; Zhang, H.; Ke, C.; Yan, F.; Nelson, J.; et al. Tumor-specific chromosome mis-segregation controls cancer plasticity by maintaining tumor heterogeneity. PLoS ONE 2013, 8, e80898. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Chen, Y.; HU, Y.; YU, L.; Tran, K.; Giedzinski, E.; Ru, N.; Gau, A.; Pan, F.; Qiao, J.; et al. The role of EGFR double minutes in modulating the response of malignant gliomas to radiotherapy. Oncotarget 2017, 8, 80853–80868. [Google Scholar] [CrossRef]
- Kondo, T. Brain cancer stem-like cells. Eur. J. Cancer 2006, 42, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Trinh, A.L.; Chen, H.; Chen, Y.; Hu, Y.; Li, Z.; Siegel, E.R.; Linskey, M.E.; Wang, P.H.; Digman, M.A.; Zhou, Y.H. Tracking Functional Tumor Cell Subpopulations of Malignant Glioma by Phasor Fluorescence Lifetime Imaging Microscopy of NADH. Cancers 2017, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer. 2020, 20, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Warburg, O.; Posener, K.; Negelein, E. On Metabolism of Tumors; Constable: London, UK, 1930. [Google Scholar]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Padma, M.V.; Said, S.; Jacobs, M.; Hwang, D.R.; Dunigan, K.; Satter, M.; Christian, B.; Ruppert, J.; Bernstein, T.; Kraus, G.; et al. Prediction of pathology and survival by FDG PET in gliomas. J. Neurooncol. 2003, 64, 227–237. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Kaye, S.B. New antimetabolites in cancer chemotherapy and their clinical impact. Br. J. Cancer 1998, 78 (Suppl. 3), 1–7. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef]
- Elf, S.E.; Chen, J. Targeting glucose metabolism in patients with cancer. Cancer 2014, 120, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Clem, B.F.; O’Neal, J.; Klarer, A.C.; Telang, S.; Chesney, J. Clinical development of cancer therapeutics that target metabolism. QJM 2016, 109, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G. Exploiting tumor metabolism: Challenges for clinical translation. J. Clin. Investig. 2013, 123, 3648–3651. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Troncone, M.; Cargnelli, S.M.; Villani, L.A.; Isfahanian, N.; Broadfield, L.A.; Zychla, L.; Wright, J.; Pond, G.; Steinberg, G.R.; Tsakiridis, T. Targeting metabolism and AMP-activated kinase with metformin to sensitize non-small cell lung cancer (NSCLC) to cytotoxic therapy: Translational biology and rationale for current clinical trials. Oncotarget 2017, 8, 57733–57754. [Google Scholar] [CrossRef] [PubMed]
- Neugent, M.L.; Goodwin, J.; Sankaranarayanan, I.; Yetkin, C.E.; Hsieh, M.H.; Kim, J.W. A New Perspective on the Heterogeneity of Cancer Glycolysis. Biomol. Ther. 2018, 26, 10–18. [Google Scholar] [CrossRef]
- Narula, J.; Williams, C.J.; Tiwari, A.; Marks-Bluth, J.; Pimanda, J.E.; Igoshin, O.A. Mathematical model of a gene regulatory network reconciles effects of genetic perturbations on hematopoietic stem cell emergence. Dev. Biol. 2013, 379, 258–269. [Google Scholar] [CrossRef]
- Seo, J.; Jin, D.; Choi, C.H.; Lee, H. Integration of MicroRNA, mRNA, and Protein Expression Data for the Identification of Cancer-Related MicroRNAs. PLoS ONE 2017, 12, e0168412. [Google Scholar] [CrossRef]
- Rangel, M.C.; Bertolette, D.; Castro, N.P.; Klauzinska, M.; Cuttitta, F.; Salomon, D.S. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res. Treat. 2016, 156, 211–226. [Google Scholar] [CrossRef]
- Sneddon, J.B.; Werb, Z. Location, location, location: The cancer stem cell niche. Cell Stem Cell 2007, 1, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Afrasiabi, K. Fundamentals of Living and Non-Living Universes; Page Publishing, Inc.: New York, NY, USA, 2018. [Google Scholar]
- Fittall, M.W.; Van Loo, P. Translating insights into tumor evolution to clinical practice: Promises and challenges. Genome Med. 2019, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Swanton, C. The Role of Aneuploidy in Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a028373. [Google Scholar] [CrossRef]
- Janssen, A.; Kops, G.J.; Medema, R.H. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19108–19113. [Google Scholar] [CrossRef]
- Galimberti, F.; Thompson, S.L.; Ravi, S.; Compton, D.A.; Dmitrovsky, E. Anaphase catastrophe is a target for cancer therapy. Clin. Cancer Res. 2011, 17, 1218–1222. [Google Scholar] [CrossRef]
- Tovar, C.; Higgins, B.; Deo, D.; Kolinsky, K.; Liu, J.J.; Heimbrook, D.C.; Vassilev, L.T. Small-molecule inducer of cancer cell polyploidy promotes apoptosis or senescence: Implications for therapy. Cell Cycle 2010, 9, 3364–3375. [Google Scholar] [CrossRef][Green Version]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Kabeche, L.; Wood, M.D.; Laucius, C.D.; Qu, D.; Laughney, A.M.; Reynolds, G.E.; Louie, R.J.; Phillips, J.; Chan, D.A.; et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat. Commun. 2015, 6, 5990. [Google Scholar] [CrossRef]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651. [Google Scholar] [CrossRef]
- Mason, J.M.; Wei, X.; Fletcher, G.C.; Kiarash, R.; Brokx, R.; Hodgson, R.; Beletskaya, I.; Bray, M.R.; Mak, T.W. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Patterson, J.O.; Dewhurst, S.; Lopez-Garcia, C.; Koch, A.; McGranahan, N.; Chao, W.C.H.; Barry, D.J.; Rowan, A.; Instrell, R.; et al. APC/C Dysfunction Limits Excessive Cancer Chromosomal Instability. Cancer Discov. 2017, 7, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.H.; Afrasiabi, K.; Linskey, M.E. Extracellular control of chromosomal instability and maintenance of intra-tumoral heterogeneity. J. Cancer Metastasis Treat. 2018, 4, 15. [Google Scholar] [CrossRef]
- Hu, Y.; Ke, C.; Ru, N.; Chen, Y.; Yu, L.; Siegel, E.R.; Linskey, M.E.; Wang, P.; Zhou, Y.H. Cell context-dependent dual effects of EFEMP1 stabilizes subpopulation equilibrium in responding to changes of in vivo growth environment. Oncotarget 2015, 6, 30762–30772. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Hu, Y.; Yu, L.; Ke, C.; Vo, C.; Hsu, H.; Li, Z.; Di Donato, A.T.; Chaturbedi, A.; Hwang, J.W.; et al. Weaponizing human EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1) for 21st century cancer therapeutics. Oncoscience 2016, 3, 208–219. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 5387041. [Google Scholar] [CrossRef]
- Morash, M.; Mitchell, H.; Beltran, H.; Elemento, O.; Pathak, J. The Role of Next-Generation Sequencing in Precision Medicine: A Review of Outcomes in Oncology. J. Pers. Med. 2018, 8, 30. [Google Scholar] [CrossRef]
- Beck, S.; Ng, T. C2c: Turning cancer into chronic disease. Genome Med. 2014, 6, 38. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, Z.; Wang, S.; Lu, S.; Song, Y.; Cheng, Y.; Ying, J.; Liu, W.; Hou, Y.; Li, Y.; et al. Application of next-generation sequencing technology to precision medicine in cancer: Joint consensus of the Tumor Biomarker Committee of the Chinese Society of Clinical Oncology. Cancer Biol. Med. 2019, 16, 189–204. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afrasiabi, K.; Linskey, M.E.; Zhou, Y.-H. Exploiting Cancer’s Tactics to Make Cancer a Manageable Chronic Disease. Cancers 2020, 12, 1649. https://doi.org/10.3390/cancers12061649
Afrasiabi K, Linskey ME, Zhou Y-H. Exploiting Cancer’s Tactics to Make Cancer a Manageable Chronic Disease. Cancers. 2020; 12(6):1649. https://doi.org/10.3390/cancers12061649
Chicago/Turabian StyleAfrasiabi, Kambiz, Mark E. Linskey, and Yi-Hong Zhou. 2020. "Exploiting Cancer’s Tactics to Make Cancer a Manageable Chronic Disease" Cancers 12, no. 6: 1649. https://doi.org/10.3390/cancers12061649
APA StyleAfrasiabi, K., Linskey, M. E., & Zhou, Y.-H. (2020). Exploiting Cancer’s Tactics to Make Cancer a Manageable Chronic Disease. Cancers, 12(6), 1649. https://doi.org/10.3390/cancers12061649