Safety of BRAF+MEK Inhibitor Combinations: Severe Adverse Event Evaluation


Abstract
1. Introduction
2. Methods
2.1. Study Design and Data Sources
2.2. Procedures
2.3. Statistical Analysis
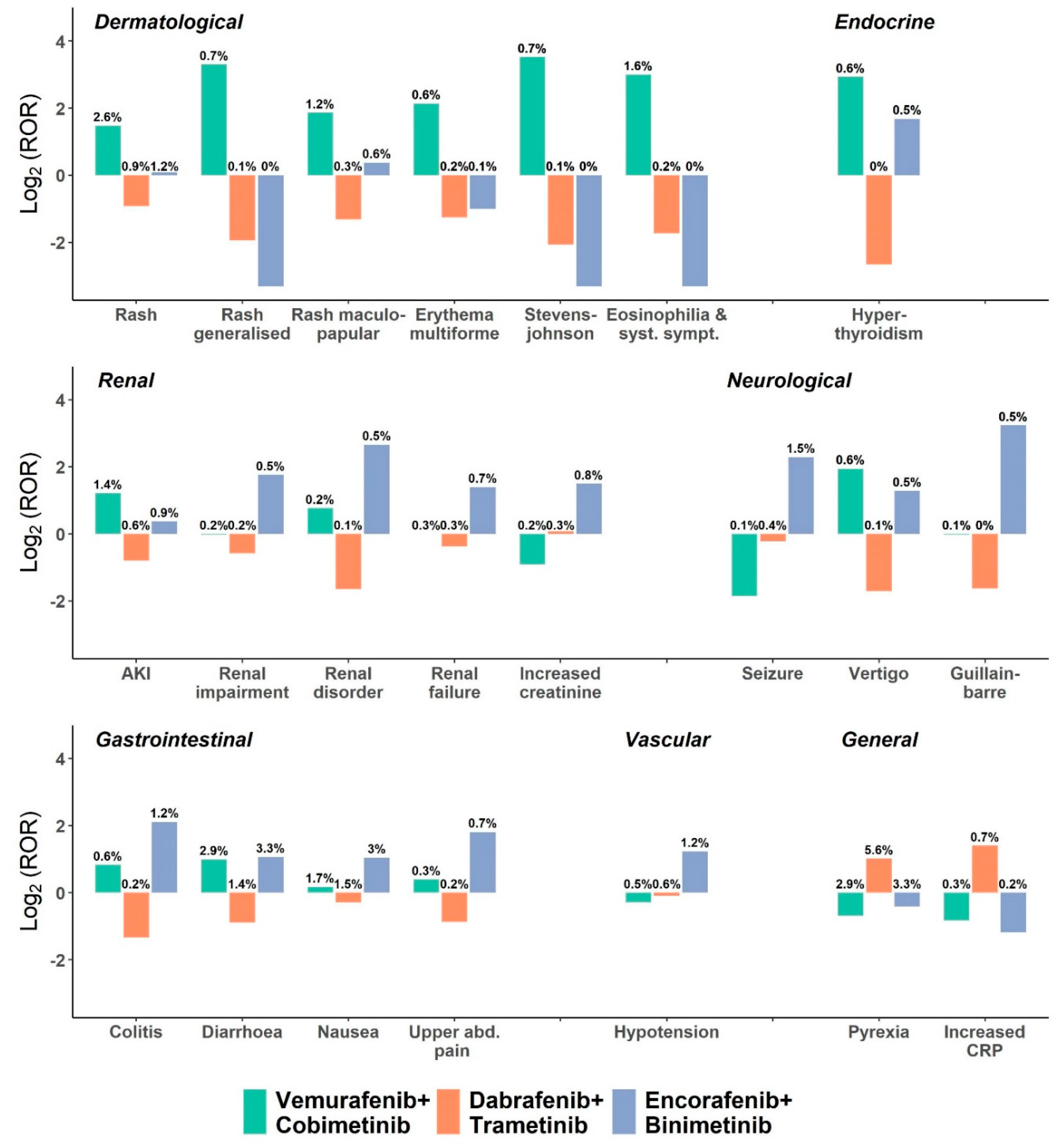
3. Results
3.1. Patient Characteristics
3.2. Toxicity Profile of BRAFi and MEKi Compared to the Full Database
3.3. Toxicity Profile of BRAFi and MEKi Combinations
3.4. Clinical Characteristics Using BRAFi and MEKi Combinations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.V.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Dreno, B.; Ascierto, P.A.; McArthur, G.A.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.V.; Stroyakovskiy, D.; Thomas, L.; et al. Efficacy and safety of cobimetinib (C) combined with vemurafenib (V) in patients (pts) with BRAF V600 mutation–positive metastatic melanoma: Analysis from the 4-year extended follow-up of the phase 3 coBRIM study. J. Clin. Oncol. 2018, 36, 9522. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.; Arance, A.; Mandalà, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsová, I.; Gutzmer, R.; et al. Overall survival in COLUMBUS: A phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) vs. vemurafenib (VEM) or enco in BRAF-mutant melanoma. J. Clin. Oncol. 2018, 36, 9504. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroyakovskiy, D.; Dummer, R.; Grange, F.; Mortier, L.; Chiarion-Sileni, V.; et al. Three-year estimate of overall survival in COMBI-v, a randomized phase 3 study evaluating first-line dabrafenib (D)+ trametinib (T) in patients (pts) with unresectable or metastatic BRAF V600E/K–mutant cutaneous melanoma. Ann. Oncol. 2016, 27, vi575. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Sileni, V.C.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage IIIBRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Flaherty, K.T.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Update on tolerability and overall survival in COLUMBUS: Landmark analysis of a randomised phase 3 trial of encorafenib plus binimetinib vs. vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. Eur. J. Cancer 2020, 126, 33–44. [Google Scholar] [CrossRef]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.-J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies–update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; Van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Rosen, L.S.; LoRusso, P.; Ma, W.W. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Kefford, R.; Pavlick, A.C. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013, 31, 482. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbé, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.; Grob, J.-J.; et al. Phase II Trial (BREAK-2) of the BRAF Inhibitor Dabrafenib (GSK2118436) in Patients with Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Delord, J.-P.; Robert, C.; Nyakas, M.; McArthur, G.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF -Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef]
- Heinzerling, L.; Eigentler, T.K.; Fluck, M.; Hassel, J.C.; Heller-Schenck, D.; Leipe, J.; Pauschinger, M.; Vogel, A.; Zimmer, L.; Gutzmer, R. Tolerability of BRAF/MEK inhibitor combinations: Adverse event evaluation and management. ESMO Open 2019, 4, e000491. [Google Scholar] [CrossRef]
- Zhang, W. BRAF inhibitors: The current and the future. Curr. Opin. Pharmacol. 2015, 23, 68–73. [Google Scholar] [CrossRef]
- Boussemart, L.; Routier, E.; Mateus, C.; Opletalova, K.; Sebille, G.; Kamsu-Kom, N.; Thomas, M.; Vagner, S.; Favre, M.; Tomasic, G.; et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: A study of 42 patients. Ann. Oncol. 2013, 24, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Indini, A.; Tondini, C.A.; Mandalà, M. Cobimetinib in malignant melanoma: How to MEK an impact on long-term survival. Futur. Oncol. 2019, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.H.C.; Duits, D.E.M.; Versluis, M.; Luyten, G.P.M.; Bergen, A.A.B.; Kapiteijn, E.; De Lange, M.J.; Boon, C.J.F.; Van Der Velden, P. Loss of MAPK Pathway Activation in Post-Mitotic Retinal Cells as Mechanism in MEK Inhibition-Related Retinopathy in Cancer Patients. Medicine 2016, 95, e3457. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef]
- Daud, A.N.A.; Tsai, K. Management of Treatment-Related Adverse Events with Agents Targeting the MAPK Pathway in Patients with Metastatic Melanoma. Oncologist 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Jhaveri, K.D.; Sakhiya, V.; Fishbane, S.; Sakhiya, M.V. Nephrotoxicity of the BRAF Inhibitors Vemurafenib and Dabrafenib. JAMA Oncol. 2015, 1, 1133. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, J.P.; Borges, F.C.; Murteira, R.; Ramos, C.; Gouveia, E.; Passos, M.J.; Miranda, A.; Da Costa, F.A. Using a cancer registry to capture signals of adverse events following immune and targeted therapy for melanoma. Int. J. Clin. Pharm. 2018, 40, 852–861. [Google Scholar] [CrossRef]
- Dumas, M.; Laly, P.; Gottlieb, J.; Vercellino, L.; Paycha, F.; Bagot, M.; Baroudjian, B.; Madelaine, I.; Basset-Séguin, N.; Eftekhari, P.; et al. Osteopenia and fractures associated with long-term therapy with MEK inhibitors. Melanoma Res. 2018, 28, 641–644. [Google Scholar] [CrossRef]
- Cornet, L.; Khouri, C.; Roustit, M.; Guignabert, C.; Chaumais, M.-C.; Humbert, M.; Revol, B.; Despas, F.; Montani, D.; Cracowski, J.-L. Pulmonary arterial hypertension associated with protein kinase inhibitors: A pharmacovigilance–pharmacodynamic study. Eur. Respir. J. 2019, 53, 1802472. [Google Scholar] [CrossRef]
- Sanlorenzo, M.; Choudhry, A.; Vujic, I.; Posch, C.; Chong, K.; Johnston, K.; Meier, M.; Osella-Abate, S.; Quaglino, P.; Daud, A.N.A.; et al. Comparative profile of cutaneous adverse events: BRAF/MEK inhibitor combination therapy versus BRAF monotherapy in melanoma. J. Am. Acad. Dermatol. 2014, 71, 1102–1109.e1. [Google Scholar] [CrossRef]
- Alves, C.; Ribeiro, I.; Penedones, A.; Mendes, D.; Batel-Marques, F. Risk of Ophthalmic Adverse Effects in Patients Treated with MEK Inhibitors: A Systematic Review and Meta-Analysis. Ophthalmic Res. 2016, 57, 60–69. [Google Scholar] [CrossRef]
- Mackin, A.G.; Pecen, P.E.; Dinsmore, A.L.; Patnaik, J.L.; Gonzalez, R.; Robinson, W.A.; Palestine, A.G. Inflammatory side effects of BRAF and MEK inhibitors. Melanoma Res. 2019, 29, 522–526. [Google Scholar] [CrossRef]
- Bronte, E.; Bronte, G.; Novo, G., Jr.; Rinaldi, G.; Bronte, F.; Passiglia, F.; Russo, A. Cardiotoxicity mechanisms of the combination of BRAF-inhibitors and MEK-inhibitors. Pharmacol. Ther. 2018, 192, 65–73. [Google Scholar] [CrossRef]
- Mourad, N.; Lourenço, N.; Delyon, J.; Eftekhari, P.; Bertheau, P.; Allayous, C.; Ballon, A.; Pagès, C.; Allez, M.; Lebbé, C.; et al. Severe gastrointestinal toxicity of MEK inhibitors. Melanoma Res. 2019, 29, 556–559. [Google Scholar] [CrossRef]
- Goldman, S.A. Limitations and strengths of spontaneous reports data. Clin. Ther. 1998, 20, C40–C44. [Google Scholar] [CrossRef]
- Kessler, D.A. Introducing MEDWatch. A new approach to reporting medication and device adverse effects and product problems. JAMA 1993, 269, 2765–2768. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Adverse Event Reporting System (FAERS) Public Dashboard; US Food Drug Administration, Silver Spring: Maryland, MD, USA, 2018.
- Mozzicato, P. MedDRA. Pharm. Med. 2009, 23, 65–75. [Google Scholar] [CrossRef]
- Moore, N.; Kreft-Jais, C.; Haramburu, F.; Noblet, C.; Andrejak, M.; Ollagnier, M.; Bégaud, B. Reports of hypoglycaemia associated with the use of ACE inhibitors and other drugs: A case/non-case study in the French pharmacovigilance system database. Br. J. Clin. Pharmacol. 1997, 44, 513–518. [Google Scholar] [CrossRef]
- Wilson, A.; Thabane, L.; Holbrook, A. Application of data mining techniques in pharmacovigilance. Br. J. Clin. Pharmacol. 2004, 57, 127–134. [Google Scholar] [CrossRef]
- Bate, A.; Lindquist, M.; Edwards, I.R.; Olsson, S.; Orre, R.; Lansner, A.; De Freitas, R.M. A Bayesian neural network method for adverse drug reaction signal generation. Eur. J. Clin. Pharmacol. 1998, 54, 315–321. [Google Scholar] [CrossRef]
- Rothman, K.J.; Lanes, S.; Sacks, S.T. The reporting odds ratio and its advantages over the proportional reporting ratio. Pharmacoepidemiol. Drug Saf. 2004, 13, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, P.G.M.; Van Buuren, S.; Van Der Hofstede, J.W.; Van Puijenbroek, E.P. On the assessment of adverse drug reactions from spontaneous reporting systems: The influence of under-reporting on odds ratios. Stat. Med. 2002, 21, 2027–2044. [Google Scholar] [CrossRef] [PubMed]
- Sakaeda, T.; Tamon, A.; Kadoyama, K.; Okuno, Y. Data Mining of the Public Version of the FDA Adverse Event Reporting System. Int. J. Med. Sci. 2013, 10, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Star, K.; Sandberg, L.; Bergvall, T.; Choonara, I.; Caduff-Janosa, P.; Edwards, I.R. Paediatric safety signals identified in VigiBase: Methods and results from Uppsala Monitoring Centre. Pharmacoepidemiol. Drug Saf. 2019, 28, 680–689. [Google Scholar] [CrossRef]
- Ahmed, I.; Haramburu, F.; Fourrier-Réglat, A.; Thiessard, F.; Kreft-Jais, C.; Miremont-Salamé, G.; Begaud, B.; Tubert-Bitter, P. Bayesian pharmacovigilance signal detection methods revisited in a multiple comparison setting. Stat. Med. 2009, 28, 1774–1792. [Google Scholar] [CrossRef]
- Ahmed, I.; Dalmasso, C.; Haramburu, F.; Thiessard, F.; Broët, P.; Tubert-Bitter, P. False Discovery Rate Estimation for Frequentist Pharmacovigilance Signal Detection Methods. Biometrics 2009, 66, 301–309. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ahmed, I.; Poncet, A. PhViD: An R Package for Pharmacovigilance Signal Detection, R Package Version; 2013. Available online: https://cran.r-project.org/web/packages/PhViD (accessed on 28 July 2019).
- Stevenson, M.; Nunes, T.; Sanchez, J. EpiR: An R Package for the Analysis of Epidemiological Data; R Package Version 0.9-48; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- ArrayExpress—A Database of Functional Genomics Experiments. Available online: http://www.ebi.ac.uk/arrayexpress/ (accessed on 12 November 2012).
- Oneal, P.A.; Kwitkowski, V.; Luo, L.; Shen, Y.L.; Subramaniam, S.; Shord, S.; Goldberg, K.B.; McKee, A.E.; Kaminskas, E.; Farrell, A.; et al. FDA Approval Summary: Vemurafenib for the Treatment of Patients with Erdheim-Chester Disease with the BRAF V600 Mutation. Oncologist 2018, 23, 1520–1524. [Google Scholar] [CrossRef]
- Falchook, G.S.; Millward, M.; Hong, D.S.; Naing, A.; Piha-Paul, S.A.; Waguespack, S.; Cabanillas, M.E.; I Sherman, S.; Ma, B.; Curtis, M.; et al. BRAF Inhibitor Dabrafenib in Patients with Metastatic BRAF-Mutant Thyroid Cancer. Thyroid 2015, 25, 71–77. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.-J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef]
- Pirmohamed, M.; Darbyshire, J. Collecting and sharing information about harms. BMJ 2004, 329, 6–7. [Google Scholar] [CrossRef]
- Talbot, J.C.; Nilsson, B.S. Pharmacovigilance in the pharmaceutical industry. Br. J. Clin. Pharmacol. 1998, 45, 427–431. [Google Scholar] [CrossRef]
- Salem, J.-E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.-P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- Launay-Vacher, V.; Zimner-Rapuch, S.; Poulalhon, N.; Fraisse, T.; Garrigue, V.; Gosselin, M.; Amet, S.; Janus, N.; Deray, G. Acute renal failure associated with the new BRAF inhibitor vemurafenib: A case series of 8 patients. Cancer 2014, 120, 2158–2163. [Google Scholar] [CrossRef]
- Perico, L.; Mandalà, M.; Schieppati, A.; Carrara, C.; Rizzo, P.; Conti, S.; Longaretti, L.; Benigni, A.; Remuzzi, G. BRAF Signaling Pathway Inhibition, Podocyte Injury, and Nephrotic Syndrome. Am. J. Kidney Dis. 2017, 70, 145–150. [Google Scholar] [CrossRef]
- Maanaoui, M.; Saint-Jacques, C.; Gnemmi, V.; Frimat, M.; Lionet, A.; Hazzan, M.; Noël, C.; Provot, F. Glomerulonephritis and granulomatous vasculitis in kidney as a complication of the use of BRAF and MEK inhibitors in the treatment of metastatic melanoma. Medicine 2017, 96, e7196. [Google Scholar] [CrossRef]
- Nussbaum, E.Z.; Perazella, M.A. Update on the nephrotoxicity of novel anticancer agents. Clin. Nephrol. 2018, 89, 149–165. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.V.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Harrisingh, M.C.; Pérez-Nadales, E.; Parkinson, D.B.; Malcolm, D.S.; Mudge, A.W.; Lloyd, A.C. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004, 23, 3061–3071. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chuang, T.-J.; Lyu, R.-K.; Ro, L.-S.; Wu, Y.-R.; Chang, H.-S.; Huang, C.-C.; Kuo, H.-C.; Hsu, W.-C.; Chu, C.-C.; et al. Identification of Gene Networks and Pathways Associated with Guillain-Barré Syndrome. PLoS ONE 2012, 7, e29506. [Google Scholar] [CrossRef]
- Urner-Bloch, U.; Urner, M.; Stieger, P.; Galliker, N.; Winterton, N.; Zubel, A.; Parseval, L.M.-D.; Dummer, R.; Goldinger, S.M. Transient MEK inhibitor-associated retinopathy in metastatic melanoma. Ann. Oncol. 2014, 25, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Stjepanovic, N.; Velazquez-Martin, J.P.; Bedard, P.L. Ocular toxicities of MEK inhibitors and other targeted therapies. Ann. Oncol. 2016, 27, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Niro, A.; Strippoli, S.; Alessio, G.; Sborgia, L.; Recchimurzo, N.; Guida, M. Ocular Toxicity in Metastatic Melanoma Patients Treated with Mitogen-Activated Protein Kinase Kinase Inhibitors: A Case Series. Am. J. Ophthalmol. 2015, 160, 959–967.e1. [Google Scholar] [CrossRef]
- Libenciuc, C.; Mateus, C.; Routier, E.; Reigneau, M.; Fahmy, J.; Ghoufi, L.; Boutros, C.; Cauquil, C.; Robert, C. Neuropathies sensitives sous la combinaison inhibiteurs de BRAF et de MEK: Dabrafénib et tramétinib. Ann. Dermatol. Vénéréol. 2016, 143, S207. [Google Scholar] [CrossRef]
- Compter, A.; Boogerd, W.; Van Thienen, J.V.; Brandsma, D. Acute polyneuropathy in a metastatic melanoma patient treated with vemurafenib and cobimetinib. Neurol. Clin. Pr. 2017, 7, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schwartz, G.K.; DeAngelis, L.M.; Kaley, T.J.; Carvajal, R.D. Dropped head syndrome: Report of three cases during treatment with a MEK inhibitor. Neurology 2012, 79, 1929–1931. [Google Scholar] [CrossRef]
- Boasberg, P.; Redfern, C.H.; Daniels, G.A.; Bodkin, D.; Garrett, C.R.; Ricart, A.D. Pilot study of PD-0325901 in previously treated patients with advanced melanoma, breast cancer, and colon cancer. Cancer Chemother. Pharmacol. 2011, 68, 547–552. [Google Scholar] [CrossRef]
- Taha, T.; Tzuk-Shina, T.; Forschner, I.; Bar-Sela, G. Acute motor and sensory axonal neuropathy related to treatment with MEK inhibitors in a patient with advanced melanoma. Melanoma Res. 2017, 27, 632–634. [Google Scholar] [CrossRef]
- Maurice, C.; Marcus, B.; Mason, W. Guillain-Barre Syndrome after Treatment with Dabrafenib for Metastatic Recurrent Melaloma. (P4. 232); Neurology: Alphen aan den Rijn, The Netherlands, 2015. [Google Scholar]
- Greco, A.; Gallo, A.; Fusconi, M.; Marinelli, C.; Macri, G.; De Vincentiis, M. Bell’s palsy and autoimmunity. Autoimmun. Rev. 2012, 12, 323–328. [Google Scholar] [CrossRef]
- Grampp, G.; Felix, T. Pharmacovigilance Considerations for Biosimilars in the USA. BioDrugs 2015, 29, 309–321. [Google Scholar] [CrossRef]
- Wang, H.-W.; Hochberg, A.M.; Pearson, R.K.; Hauben, M.; Hauben, M. An Experimental Investigation of Masking in the US FDA Adverse Event Reporting System Database. Drug Saf. 2010, 33, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Pariente, A.; Avillach, P.; Salvo, F. Effect of competition bias in safety signal generation. Drug Saf. 2012, 35, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Morganstein, D.L.; Lai, Z.; Spain, L.; Diem, S.; Levine, D.; Mace, C.; Gore, M.; Larkin, J. Thyroid abnormalities following the use of cytotoxic T-lymphocyte antigen-4 and programmed death receptor protein-1 inhibitors in the treatment of melanoma. Clin. Endocrinol. (Oxf.) 2017, 86, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Robert, E.; Rosa, F. Valproate and Birth Defects. Lancet 1983, 322, 1142. [Google Scholar] [CrossRef]
- Stricker, B.H.; Tijssen, J.G. Serum sickness-like reactions to cefaclor. J. Clin. Epidemiol. 1992, 45, 1177–1184. [Google Scholar] [CrossRef]
- Sommet, A.; Grolleau, S.; Bagheri, H.; Lapeyre-Mestre, M.; Montastruc, J.L.; French Network of Regional Pharmacovigilance Centres. Was the thrombotic risk of rofecoxib predictible from the French Pharmacovigilance Database before 30 September 2004? Eur. J. Clin. Pharmacol. 2008, 64, 829–834. [Google Scholar] [CrossRef]
- Souyri, C.; Olivier, P.; Grolleau, S.; Lapeyre-Mestre, M.; Centres, T.F.N.O.P. Severe necrotizing soft-tissue infections and nonsteroidal anti-inflammatory drugs. Clin. Exp. Dermatol. 2008, 33, 249–255. [Google Scholar] [CrossRef]
- Montastruc, J.L.; Sommet, A.; Bagheri, H.; Lapeyre-Mestre, M. Benefits and strengths of the disproportionality analysis for identification of adverse drug reactions in a pharmacovigilance database. Br. J. Clin. Pharmacol. 2011, 72, 905–908. [Google Scholar] [CrossRef]
- Hauben, M.; Madigan, D.; Gerrits, C.M.; Walsh, L.; Van Puijenbroek, E.P. The role of data mining in pharmacovigilance. Expert Opin. Drug Saf. 2005, 4, 929–948. [Google Scholar] [CrossRef]


{kind=link}
| Vemurafenib+Cobimetinib | Dabrafenib+Trametinib | Encorafenib+Binimetinib | ||
|---|---|---|---|---|
| Patients | 584 | 1893 | 350 | |
| Reports, No. (%) | 2345 (20) | 8411 (71.8) | 965 (8.2) | |
| Age, Mean (SD), y | 58.8 (14.05) | 59.4 (16) | 62.93 (12.96) | |
| Sex, No. (%) | ||||
| Female | 276 (47.26) | 789 (41.68) | 24 (6.86) | |
| Male | 275 (47.09) | 943 (49.82) | 26 (7.43) | |
| Not reported | 33 (5.65) | 161 (8.51) | 300 (85.71) | |
| Indications, No. (%) | ||||
| Melanoma | 480 (80.9) | 1219 (62.6) | 204 (57.8) | |
| Lung cancer | 3 (0.5) | 111 (5.7) | 4 (1.1) | |
| Gastrointestinal cancer | 4 (0.7) | 29 (1.5) | 48 (13.6) | |
| Thyroid cancer | 20 (3.4) | 34 (1.7) | 2 (0.6) | |
| Hematological malignancy | 18 (3) | 17 (0.9) | 1 (0.3) | |
| Other/Unspecified | 71 (12) | 542 (27.8) | 96 (27.2) | |
| Country, No. (%) | ||||
| Americas | 389 (16.59) | 1695 (20.15) | 600 (62.18) | |
| Europe | 1570 (66.95) | 3389 (40.29) | 233 (24.15) | |
| Australia | 33 (1.41) | 178 (2.12) | 19 (1.97) | |
| Asia | 70 (2.99) | 1097 (13.04) | 41 (4.25) | |
| Africa | 19 (0.81) | 10 (0.12) | 0 (0) | |
| Other | 264 (11.26) | 2032 (24.16) | 72 (7.46) |
| V/C Reports (n = 2345) | D/T Reports (n = 8411) | E/B Reports (n = 965) | Reports in Full Database; (n = 3,285,265) | Adj. ROR 1 (95% CI) V+C vs. D+T and E+B | Adj. ROR 1 (95% CI) D+T vs. V+C and E+B | Adj. ROR 1 (95% CI) E+B vs. V+C and D+T | |
|---|---|---|---|---|---|---|---|
| Epidermal and dermal conditions | 311 (13.3%) | 355 (4.2%) | 47 (4.9%) | 303188 (9.2%) | 3.4 (2.9–4.0) † | 0.4 (0.3–0.4) † | 0.8 (0.6–1.1) |
| Stevens–Johnson syndrome | 17 (0.7%) | 6 (0.1%) | 0 (0%) | 7099 (0.2%) | 10.4 (4–26.9) † | 0.1 (0.1–0.3) † | |
| Pyrexia | 67 (2.9%) | 472 (5.6%) | 32 (3.3%) | 75,155 (2.3%) | 0.5 (0.4–0.6) † | 1.9 (1.5–2.4) † | 0.7 (0.5–1.1) |
| Increased CRP | 7 (0.3%) | 58 (0.7%) | 2 (0.2%) | 13,829 (0.4%) | 0.4 (0.2–0.9) | 2.3 (1.2–4.8) † | 0.6 (0.1–2.6) |
| Renal disorders NEC | 5 (0.2%) | 8 (0.1%) | 6 (0.6%) | 20,466 (0.6%) | 1.7 (0.6–4.7) | 0.3 (0.1–0.8) † | 4.1 (1.3–12.5) † |
| Renal disorders (excl. nephropathies) | 61 (2.6%) | 115 (1.4%) | 28 (2.9%) | 335,796 (10.2%) | 1.8 (1.3–2.4) † | 0.5 (0.4–0.7) † | 1.8 (1.2–2.9) † |
| GI motility and defecation conditions | 91 (3.9%) | 155 (1.8%) | 38 (3.9%) | 151,750 (4.6%) | 2 (1.6–2.6) † | 0.5 (0.4–0.6) † | 1.8 (1.2–2.6) † |
| Colitis | 13 (0.6%) | 19 (0.2%) | 12 (1.2%) | 8681 (0.3%) | 1.9 (1–3.7) | 0.3 (0.2–0.6) † | 3.3 (1.5–7.1) † |
| Nausea | 40 (1.7%) | 127 (1.5%) | 29 (3%) | 86,674 (2.6%) | 1 (0.7–1.5) | 0.7 (0.5–1.0) | 2.3 (1.4–3.7) † |
| Seizure | 2 (0.1%) | 31 (0.4%) | 14 (1.5%) | 25,004 (0.8%) | 0.2 (0–0.8) † | 0.9 (0.5–1.6) | 3.8 (1.8–8.0) † |
| Peripheral neuropathies | 16 (0.7%) | 35 (0.4%) | 9 (0.9%) | 35,676 (1.1%) | 1.3 (0.8–2.4) | 0.6 (0.3–0.9) | 2.7 (1.2–6.1) † |
| Guillain–Barre syndrome | 2 (0.1%) | 4 (0%) | 5 (0.5%) | 1374 (0%) | 0.9 (0.2–4.2) | 0.2 (0.1–0.8) | 8.5 (2.1–35.0) † |
| Hypotension | 11 (0.5%) | 49 (0.6%) | 12 (1.2%) | 57,946 (1.8%) | 0.7 (0.4–1.3) | 0.9 (0.5–1.4) | 2.5 (1.2–5.1) † |
| Dermal Conditions | Body Temp. Conditions | Renal Disorders * | GI Motility and Defac. Conditions | Seizures (Incl. Subtypes) | Periph. Neurop. | Vascular Hypoten. Disorders | ||
|---|---|---|---|---|---|---|---|---|
| Drug dosing | ||||||||
| V/C | ||||||||
| <960/<60 mg | 32/212 (15%) | 5/69 (7%) | 7/53 (13%) | 12/71 (17%) | 3/14 (21%) | 1/9 (11%) | 2/16 (12%) | |
| 960/60 mg | 67/212 (32%) | 25/69 (36%) | 18/53 (34%) | 19/71 (27%) | 3/14 (21%) | 2/9 (22%) | 6/16 (38%) | |
| >960/>60 mg | 26/212 (12%) | 9/69 (13%) | 6/53 (11%) | 9/71 (13%) | 1/14 (7%) | 3/9 (33%) | 2/16 (12%) | |
| Unreported | 87/212 (41%) | 30/69 (43%) | 22/53 (42%) | 31/71 (44%) | 7/14 (50%) | 3/9 (33%) | 6/16 (38%) | |
| D/T | ||||||||
| <150/<2 mg | 154/238 (65%) | 281/485 (58%) | 58/109 (53%) | 80/141 (57%) | 27/58 (47%) | 11/32 (34%) | 29/55 (53%) | |
| 150/2 mg | 10/238 (4%) | 56/485 (12%) | 9/109 (8%) | 8/141 (6%) | 5/58 (9%) | 2/32 (6%) | 9/55 (16%) | |
| >150/>2 mg | 7/238 (3%) | 31/485 (6%) | 12/109 (11%) | 8/141 (6%) | 2/58 (3%) | 1/32 (3%) | 3/55 (5%) | |
| Unreported | 67/238 (28%) | 117/485 (24%) | 30/109 (28%) | 45/141 (32%) | 24/58 (41%) | 18/32 (56%) | 14/55 (25%) | |
| E/B | ||||||||
| <450/<45 mg | 1/29 (3%) | 2/33 (6%) | 1/28 (4%) | 1/37 (3%) | 0/15 (0%) | 0/8 (0%) | 0/12 (0%) | |
| 450/45 mg | 9/29 (31%) | 14/33 (42%) | 15/28 (54%) | 16/37 (43%) | 6/15 (40%) | 6/8 (75%) | 7/12 (58%) | |
| >450/>45 mg | 3/29 (10%) | 7/33 (21%) | 2/28 (7%) | 7/37 (19%) | 1/15 (7%) | 0/8 (0%) | 1/12 (8%) | |
| Unreported | 16/29 (55%) | 10/33 (30%) | 10/28 (36%) | 13/37 (35%) | 8/15 (53%) | 2/8 (25%) | 4/12 (33%) | |
| Time to AE onset, days (IQR) | ||||||||
| V/C | 9 (7–14); n = 138 | 9 (8–22); n = 42 | 14 (7–80); n = 35 | 9 (5–27); n = 39 | 37 (11–185); n = 9 | 237 (154–320); n = 2 | 9 (7–13); n = 8 | |
| D/T | 30 (13–50); n = 20 | 28 (16–52); n = 88 | 70.5 (54–138); n = 24 | 23 (10–50); n = 19 | 141.5 (22–303); n = 8 | 107 (44–206); n = 7 | 11 (6–76); n = 12 | |
| E/B | 12 (2–32); n = 5 | 34.5 (12–122); n = 14 | 17.5 (5–54); n = 12 | 21 (12–27); n = 13 | 124 (64–128); n = 3 | 54 (41–57); n = 3 | 54 (14–131); n = 5 | |
| Dechallenge | ||||||||
| V/C | 123/134 (92%) | 38/44 (86%) | 34/36 (94%) | 25/27 (93%) | 2/2 (100%) | 1/1 (100%) | 11/11 (100%) | |
| D/T | 116/120 (97%) | 239/251 (95%) | 60/64 (94%) | 57/60 (95%) | 15/16 (94%) | 10/12 (83%) | 32/33 (97%) | |
| E/B | 7/8 (88%) | 17/17 (100%) | 10/11 (91%) | 14/16 (88%) | 0/1 (0%) | 1/2 (50%) | 4/5 (80%) | |
| Rechallenge | ||||||||
| V/C | 4/9 (44%) | 0/2 (0%) | 2/2 (100%) | 1/2 (50%) | ||||
| D/T | 1/2 (50%) | 8/19 (42%) | 3/3 (100%) | 0/1 (0%) | 1/1 (100%) | |||
| E/B | 2/2 (100%) | 2/3 (67%) | 1/3 (33%) | 2/3 (67%) | 0/1 (0%) | |||
| Outcome | ||||||||
| V/C | ||||||||
| Death | 19/212 (9%) | 10/69 (14%) | 7/53 (13%) | 16/71 (23%) | 6/14 (43%) | 1/9 (11%) | 0/16 (0%) | |
| Life-threatening | 18/212 (8%) | 7/69 (10%) | 5/53 (9%) | 3/71 (4%) | 3/14 (21%) | 2/9 (22%) | 2/16 (12%) | |
| Hospitalization | 202/212 (95%) | 68/69 (99%) | 49/53 (92%) | 65/71 (92%) | 13/14 (93%) | 7/9 (78%) | 16/16 (100%) | |
| Other | 58/212 (27%) | 28/69 (41%) | 18/53 (34%) | 37/71 (52%) | 10/14 (71%) | 8/9 (89%) | 6/16 (38%) | |
| D/T | ||||||||
| Death | 57/238 (24%) | 119/485 (25%) | 22/109 (20%) | 43/141 (30%) | 31/58 (53%) | 2/32 (6%) | 9/55 (16%) | |
| Life-threatening | 22/238 (9%) | 48/485 (10%) | 18/109 (17%) | 6/141 (4%) | 2/58 (3%) | 5/32 (16%) | 7/55 (13%) | |
| Hospitalization | 204/238 (86%) | 441/485 (91%) | 103/109 (94%) | 132/141 (94%) | 52/58 (90%) | 32/32 (100%) | 53/55 (96%) | |
| Other | 140/238 (59%) | 269/485 (55%) | 65/109 (60%) | 89/141 (63%) | 43/58 (74%) | 20/32 (62%) | 31/55 (56%) | |
| E/B | ||||||||
| Death | 4/29 (14%) | 2/33 (6%) | 6/28 (21%) | 2/37 (5%) | 2/15 (13%) | 1/8 (12%) | 4/12 (33%) | |
| Life-threatening | 5/29 (17%) | 3/33 (9%) | 5/28 (18%) | 3/37 (8%) | 4/15 (27%) | 0/8 (0%) | 1/12 (8%) | |
| Hospitalization | 26/29 (90%) | 30/33 (91%) | 22/28 (79%) | 33/37 (89%) | 14/15 (93%) | 6/8 (75%) | 11/12 (92%) | |
| Other | 12/29 (41%) | 12/33 (36%) | 12/28 (43%) | 12/37 (32%) | 9/15 (60%) | 3/8 (38%) | 5/12 (42%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meirson, T.; Asher, N.; Bomze, D.; Markel, G. Safety of BRAF+MEK Inhibitor Combinations: Severe Adverse Event Evaluation. Cancers 2020, 12, 1650. https://doi.org/10.3390/cancers12061650
Meirson T, Asher N, Bomze D, Markel G. Safety of BRAF+MEK Inhibitor Combinations: Severe Adverse Event Evaluation. Cancers. 2020; 12(6):1650. https://doi.org/10.3390/cancers12061650
Chicago/Turabian StyleMeirson, Tomer, Nethanel Asher, David Bomze, and Gal Markel. 2020. "Safety of BRAF+MEK Inhibitor Combinations: Severe Adverse Event Evaluation" Cancers 12, no. 6: 1650. https://doi.org/10.3390/cancers12061650
APA StyleMeirson, T., Asher, N., Bomze, D., & Markel, G. (2020). Safety of BRAF+MEK Inhibitor Combinations: Severe Adverse Event Evaluation. Cancers, 12(6), 1650. https://doi.org/10.3390/cancers12061650