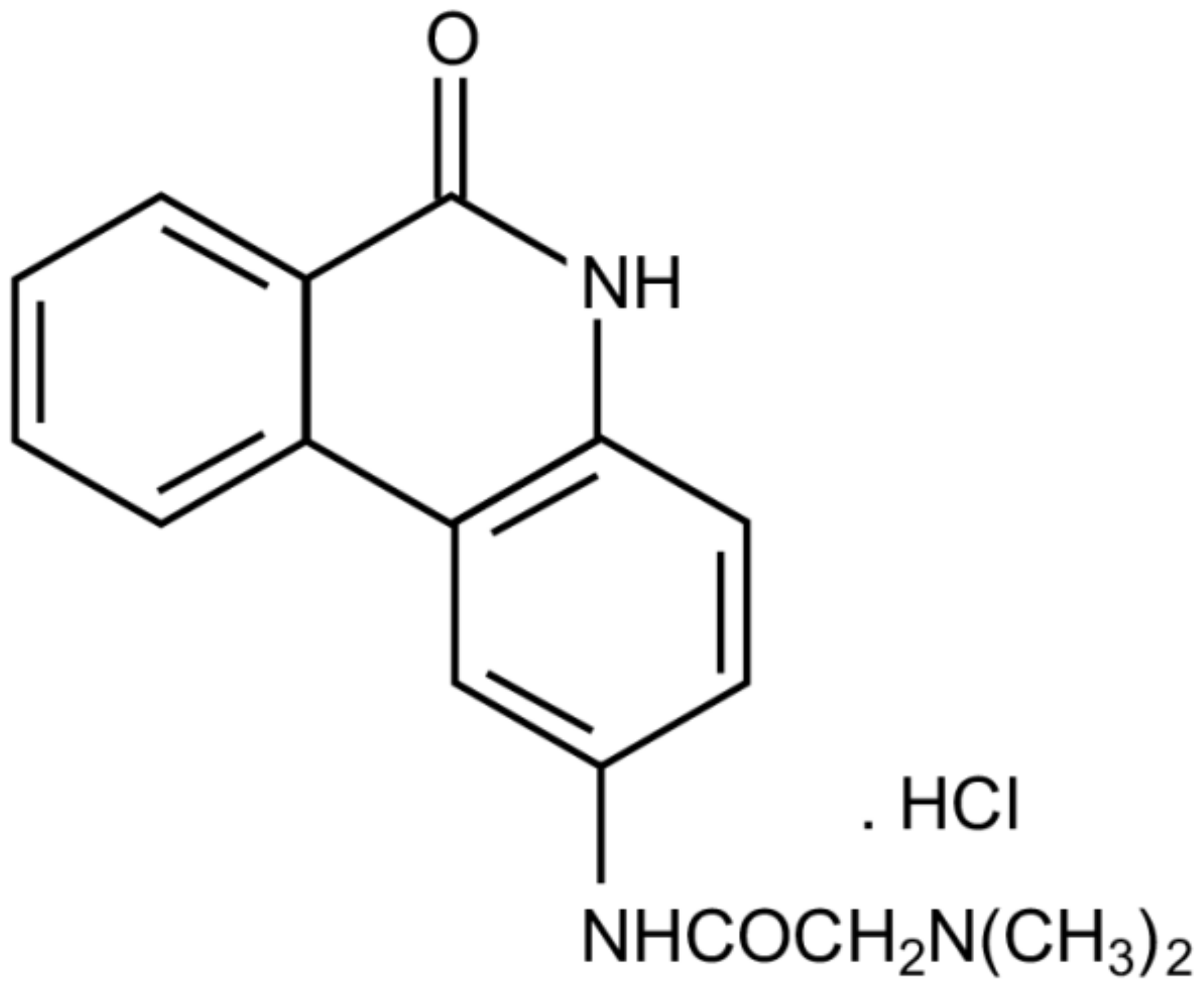
The Modified Phenanthridine PJ34 Unveils an Exclusive Cell-Death Mechanism in Human Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
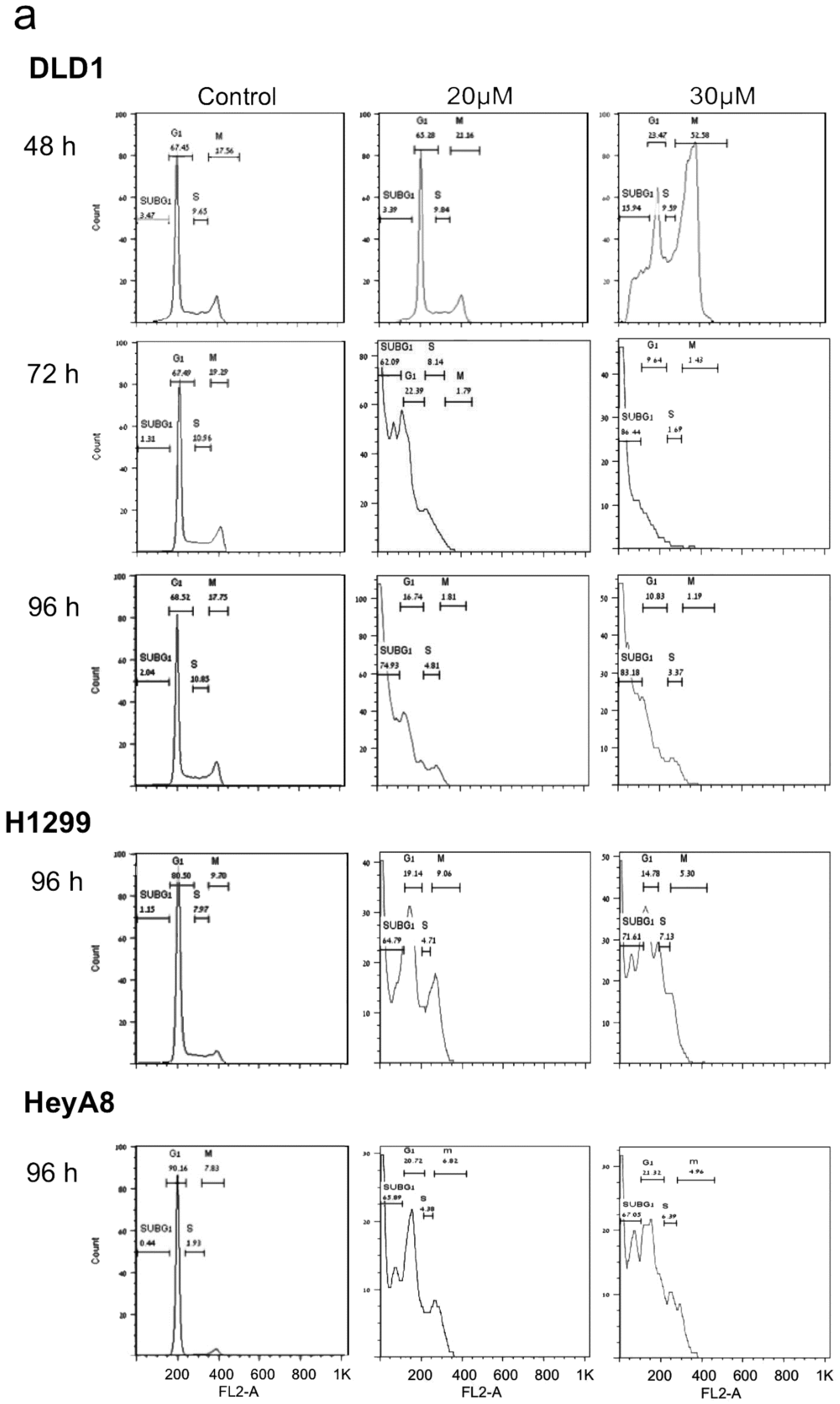
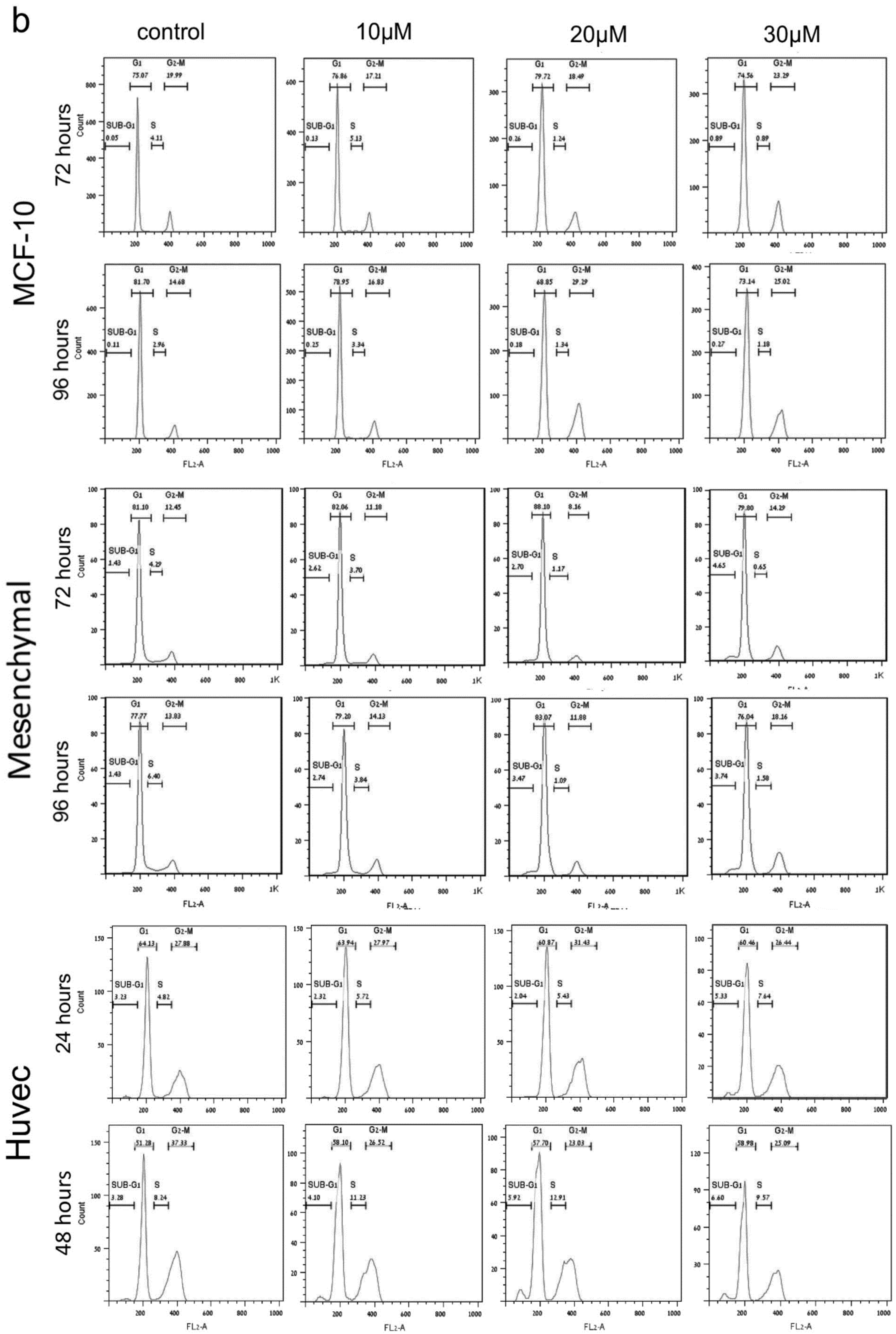
2. PJ34 Efficiently Eradicates a Variety of Human Cancer Cells in Tissue Cultures
3. PJ34 Causes Eradication of Human Cancer Cells in Xenografts
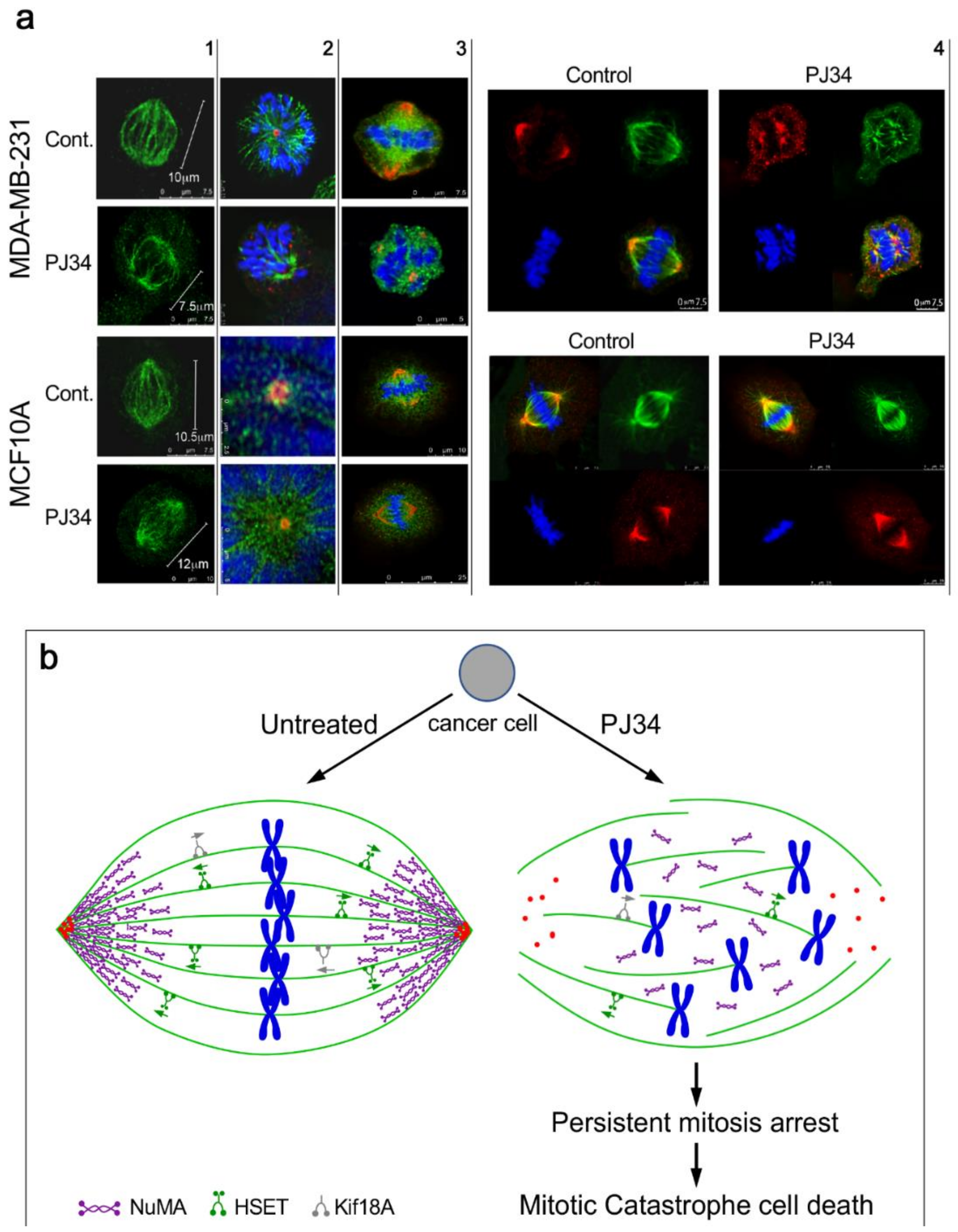
4. The Mechanism of Action of PJ34 in Human Cancer Cells
5. The Potency of PJ34 and Other PARP Inhibitors in Preventing Metastases
6. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Abdelkarim, G.E.; Gertz, K.; Harm, C.; Katchanov, J.; Dirnagi, U.; Szabo, C.; Enders, M. Protective effects of PJ34, a novel potent inhibitor of poly(ADP-ribose)polymerase (PARP) in in-vitro and in-vivo models of stroke. Int. J. Mol. Med. 2001, 7, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose)polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Gene Dev. 2020, 34, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Carden, C.P.; Yap, T.A.; Kaye, S.B. PARP inhibition: Targeting the Achilles heelof DNA repair to treat germline and sporadic ovarian cancers. Curr. Opin. Oncol. 2010, 22, 473–480. [Google Scholar] [CrossRef]
- Plummer, R. Perspective on the pipeline of drugs being developed with modulationof DNA damage as a target. Clin. Cancer Res. 2010, 16, 4527–4531. [Google Scholar] [CrossRef]
- Mangerich, A.; Burkle, A. How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int. J. Cancer 2011, 128, 251–265. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPs, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef]
- Wahlberg, E.; Karlberg, T.; Kouznetsova, E.; Markova, N.; Macchiarulo, A.; Torsell, A.-G.; Pol, E.; Frostell, A.; Ekblad, T.; Öncü, D.; et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotech. 2012, 30, 283–289. [Google Scholar] [CrossRef]
- Citarelli, M.; Teotia, S.; Lamb, R.S. Evolutionary history of the poly(ADP-ribose)polymerase gene family in eukaryotes. BMC Evol. Biol. 2010, 10, 308. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Swanson, R.A. The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience 2007, 145, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Strosznajder, R.P.; Czubowicz, K.; Jesko, H.; Strosznajder, J.B. Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol. Neurobiol. 2010, 41, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Ba, X.; Garg, N.J. Signaling mechanism of poly(ADP-ribose)polymerase-1 (PARP-1) in inflammatory diseases. Am. J. Pathol. 2011, 178, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Jesper, J.Q. The cellular response to transcription blocking DNA damage. Trend Biochem. Sci. 2018, 43, 327–341. [Google Scholar] [CrossRef]
- Lesueur, P.; Chevalier, F.; Austry, J.-B.; Waissi, W.; Burckel, H.; Noel, G.; Habrand, J.-L.; Saintigny, Y.; Joly, F. PolyADP-ribose) polymerase inhibitors as radiosensitizers: A systematic review of preclinical human studies. Oncotarget 2017, 8, 69105–69124. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.-S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sassaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, C.K.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Paddock, M.N.; Bauman, A.T.; Higdon, R.; Kolker, E.; Takeda, S.; Scharenberg, A.M. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair 2011, 10, 338–343. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.P.Y.; Ma, H.T.; Poon, R.Y.C. Synergism between ATM and PARP1 inhibition involves DNA damage and abrogating the G2 DNA damage checkpoint. Mol. Cancer Ther. 2020, 19, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hong, J.-J.; Yang, Q.; Ong, C.-T.; Li, B.-A.; Liou, Y.-C. Poly(ADP- ribosyl)ation of OVOL2 regulates aneuploidy and cell death in cancer cells. Oncogene 2019, 38, 2750–2766. [Google Scholar] [CrossRef]
- Zhou, J.X.; Feng, L.J.; Zhang, X. Risk of severe hematologic toxicities in cancer patients treated with PARP inhibitors: A meta-analysis of randomized controlled trials. Drug Des. Dev. Ther. 2017, 11, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Yu, D.-S.; Liang, Y.-C.; Huang, K.-F.; Chou, S.-J.; Chen, T.-C.; Lee, C.-C.; Chen, C.-L.; Chiou, S.-H.; Huang, H.-S. New approaches of PARP1 inhibitors in human lung cancer cells and cancer stem-like cells by some selected anthraquinone-derived small molecules. PLoS ONE 2013, 8, e56284. [Google Scholar] [CrossRef]
- Kishi, Y.; Fujibara, H.; Kawaguchi, K.; Yamada, H.; Nakayaima, R.; Yamamoto, N.; Fujihara, Y.; Hamada, Y.; Satomura, K.; Masutani, M. PARP inhibitor PJ34 supresses osteogenic differentiation in mouse mesenchymal stem cells by modulating BMP-2 signaling pathway. Int. J. Mol. Sci. 2015, 16, 24820–24838. [Google Scholar] [CrossRef] [PubMed]
- Antolin, A.A.; Ameratunga, M.; Banerji, U.; Clarke, P.A.; Workman, P.; Al-Lazikani, B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci. Rep. 2020, 10, 2585. [Google Scholar] [CrossRef] [PubMed]
- Inbar-Rozensal, D.; Visochek, L.; Castel, D.; Castiel, A.; Izraeli, S.; Dantzer, F.; Cohen-Armon, M. A selective eradication of human nonhereditary breast cancer cells by phenanthridine-derived polyADP-ribose polymerase inhibitors. Breast Cancer Res. 2009, 11, R78. [Google Scholar] [CrossRef]
- Castiel, A.; Visochek, L.; Mittelman, L.; Dantzer, F.; Izraeli, S.; Cohen-Armon, M. A phenanthrene derived PARP inhibitor is an extra-centrosomes de-clustering agent exclusively eradicating human cancer cells. BMC Cancer 2011, 11, 412. [Google Scholar] [CrossRef]
- Castiel, A.; Visochek, L.; Mittelman, L.; Zilberstein, Y.; Dantzer, F.; Izraeli, S.; Cohen-Armon, M. Cell death associated with abnormal mitosis observed by confocal imaging in live cancer cells. J. Vis. Exp. 2013, 78, e50568. [Google Scholar] [CrossRef]
- Visochek, L.; Castiel, A.; Mittelman, L.; Elkin, M.; Atias, D.; Golan, T.; Izraeli, S.; Peretz, T.; Cohen-Armon, M. Exclusive destruction of mitotic spindles in human cancer cells. Oncotarget 2017, 8, 20813–20824. [Google Scholar] [CrossRef]
- Visochek, L.; Atias, D.; Spektor, I.; Castiel, A.; Golan, T.; Cohen-Armon, M. The phenanthrene derivative PJ34 exclusively eradicates human pancreatic cancer cells in xenografts. Oncotarget 2019, 10, 6269–6282. [Google Scholar] [CrossRef] [PubMed]
- Okuda, A.; Kurokawa, S.; Takehashi, M.; Maeda, A.; Fukuda, K.; Kubo, Y.; Nogusa, H.; Takatani-Nakase, T.; Okuda, S.; Ueda, K.; et al. Poly(ADP-ribose) polymerase inhibitors activate the p53 signaling pathway in neural stem/progenitor cells. BMC Neurosci. 2017, 18, 2–18. [Google Scholar] [CrossRef]
- Visochek, L.; Grigoryan, G.; Kalal, A.; Milshtein-Parush, H.; Gazit, N.; Slutsky, I.; Yeheskel, A.; Shainberg, A.; Castiel, A.; Seger, R.; et al. A PARP1-ERK2 synergism is required for the induction of LTP. Sci. Rep. 2016, 6, 24950. [Google Scholar] [CrossRef] [PubMed]
- Madison, D.L.; Stauffer, D.; Lundblad, J.R. The PARP inhibitor PJ34 causes a PARP1-independent, p21 dependent mitotic arrest. DNA Repair 2011, 10, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Lamoral-Theys, D.; Andolfi, A.; Van Goietsenoven, G.; Cimmino, A.; Le Calv, B.; Wauthoz, N.; Mégalizzi, V.; Gras, T.; Bruyère, C.; Dubois, J.; et al. Lycorine, the Main Phenanthridine Amaryllidaceae Alkaloid, Exhibits Significant Antitumor Activity in Cancer Cells That Display Resistance to Proapoptotic Stimuli: An Investigation of Structure-Activity Relationship and Mechanistic Insight. J. Med. Chem. 2009, 52, 6244–6256. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Taniguchi, T.; Baba, T.; Li, Y.-Y.; Isibashi, H.; Nukaida, N. Identification of a phenanthrene derivative as a potent anticancer drug with pim kinase inhibitory activity. Cancer Sci. 2011, 103, 107–115. [Google Scholar] [CrossRef]
- Mariappan, A.; Soni, K.; Schorpp, K.; Zhao, F.; Minakar, A.; Zheng, X.; Mandad, S.; Macheleidt, I.; Ramani, A.; Kubelka, T.; et al. Inhibition of CPAP-tubulin interaction prevents proliferation of centrosome-amplified cancer cells. EMBO J. 2019, 38, e99876. [Google Scholar] [CrossRef]
- Huang, S.H.; Xiong, M.; Chen, X.P.; Xiao, Z.Y.; Zhao, Y.F.; Huang, Z.Y. PJ34 an inhibitor of PARP1, suppresses cell growth and enhances the suppressive effects of cisplatin in liver cancer cells. Oncol. Rep. 2008, 20, 567–572. [Google Scholar]
- Pyriochou, A.; Olah, G.; Deitch, O.; Szabo, C.; Papaetropoulos, A. Inhibition of angiogenesis by the poly(ADP-ribose) polymerase inhibitor. J. Mol. Med. 2008, 22, 113–118. [Google Scholar] [CrossRef]
- Gangopadhyay, N.N.; Luketich, J.D.; Opest, A.; Meyer, E.M.; Landreneau, R.; Schuchert, M.J. Inhibition of Poly(ADP-Ribose) Polymerase (PARP) Induces Apoptosis in Lung Cancer Cell Lines. Cancer Investig. 2011, 29, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Xiong, M.; Ji, G.; Zhang, E.; Zhang, Z.; Dong, K.; Chen, X.; Huang, Z.-Y. Synergistic suppressive effect of PARP-1 inhibitor PJ34 and HDAC inhibitor SAHA on proliferation of liver cancer cells. J. Huazhong Univ. Sci. Technol. 2015, 35, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.; Chen, X.; Wei, H.; Xiao, H. Influence of PJ34 on the genotoxicity induced by melphalan in human multiple myeloma cells. Arch. Med. Sci. 2015, 11, 301–306. [Google Scholar] [CrossRef]
- Bai, X.T.; Moles, R.; Chaib-Mezrag, H.; Nicot, C. Small PARP inhibitor PJ34 induces cell-cycle arrest and apoptosis of adult T cell leukemia cells. J. Hematol. Oncol. 2015, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Lv, S.; Tian, Y. Inhibition of proliferation and invasiveness of ovarian cancer C13 cells by a poly(ADP-ribose)polymerase inhibitor and the role of nuclear factor-kappaB. J. Int. Med. Res. 2013, 41, 1577–1585. [Google Scholar] [CrossRef]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of breast cancer cells to PARP inhibitors is independent of BRCA status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef]
- Chevanne, M.; Zampieri, M.; Rizzo, A.C.R.; Ciccarone, F.; Catizone, A.; D’Angelo, C.; Guastafierro, T.; Biroccio, A.; Raele, A.; Zupi, G.; et al. Inhibition of PARP activity by PJ34 leads to growth impairment and cell death associated with aberrant mitotic pattern and nucleolar actin accumulation in M14 melanoma cell line. J. Cell Physiol. 2010, 222, 401–410. [Google Scholar] [CrossRef]
- Lavarone, E.; Puppin, C.; Passon, N.; Filetti, S.; Russo, D.; Damante, G. The PARP inhibitor PJ34 modifies proliferation, NIS expression and epigenetic marks in thyroid cancer cell lines. Mol. Cell Endocrinol. 2013, 365, 1–10. [Google Scholar] [CrossRef]
- Magan, N.; Isaacs, R.J.; Stowell, K.M. Treatment with the PARP-inhibitor PJ34 causes enhanced doxorubicin-mediated cell death in HeLa cells. Anticancer Drugs 2012, 23, 627–637. [Google Scholar] [CrossRef]
- Majuelos-Melguizo, J.; Rodríguez, M.I.; López-Jiménez, L.; Jose, M.; Rodríguez-Vargas, J.; Martín-Consuegra, M.; Serrano-Sáenz, S.; Gavard, J.; Ruiz de Almodóvar, J.M.; Oliver, F.J. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget 2015, 6, 4790–4803. [Google Scholar] [CrossRef]
- Toller, I.M.; Altmeyer, M.; Kohler, E.; Hottiger, M.O.; Muller, A. Inhibition of ADP-ribosylation prevents and cures Helicobacter-induced Gastric Preneoplasia. Cancer Res. 2010, 70, 5912–5922. [Google Scholar] [CrossRef] [PubMed]
- Horvat, L.; Grubar, M.; Madunic, J.; Antica, M.; Matulic, M. Inhibition of PARP activity does not affect the differentiation processes caused by retinoic acid in SHSY5Y cells. Mol. Exp. Biol. Med. 2019, 1, 38–43. [Google Scholar] [CrossRef]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.-J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Press, J.Z.; Kenyon, J.A.; Xue, H.; Miller, M.A.; DeLuca, A.; Miller, D.M.; Huntsman, D.G.; Gilk, C.B.; McAlpine, J.N.; Wang, Y.Z. Xenografts of primary human gynecological tumors grown under the renal capsule of NOD/SCID mice show genetic stability during serial transplantation and respond to cytotoxic chemotherapy. Gyn. Oncol. 2008, 109, 256–264. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Pareja, F.; Aime, P.; Shu, C.; Chau, L.; Westhoff, A.; Halatsch, M.-E.; Crary, J.F.; Canoll, P.; Siegelin, M. PARP inhibition restores extrinsic apoptotic sensitivity in Glioblastoma. PLoS ONE 2014, 9, e114583. [Google Scholar] [CrossRef]
- Yuan, K.; Sun, Y.; Zhou, T.; McDonald, J.; Chen, Y. PARP1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin. Cancer Res. 2013, 19, 4750–4759. [Google Scholar] [CrossRef]
- Stepnik, M.; Spryszynska, S.; Gorzkiewicz, A.; Ferlinska, M. Cytotoxicity of anticancer drugs and PJ-34 (polyADP-ribose)polymerase-1(PARP-1) inhibitor) on HL-60 Jurkat cells. Adv. Clin. Exp. Med. 2017, 26, 379–385. [Google Scholar] [CrossRef]
- Cseh, M.; Zsolt, F.; Quintana-Cabrera, R.; Szabo, A.; Eros, K.; Eugenia Soriano, M.; Gallyas, F.; Scorrano, L.; Sumegi, B. PARP Inhibitor PJ34 Protects mitochondria and induces DNA-damage mediated apoptosis in combination with Cisplatin or Temozolomide in B16F10 Melanoma. Cells Front. Physiol. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Heng Wei, X.; Chen, X.; Xiao, H. PJ34, a poly(ADP-ribose) polymerase (PARP) inhibitor, reverses melphalan-resistance and inhibits repair of DNA double-strand breaks by targeting the FA/BRCA pathway in multidrug resistant multiple myeloma cell line RPMI8226/R TING. Int. J. Oncol. 2015, 46, 223. [Google Scholar]
- Meng, W.; Koh, B.D.; Jin-San Zhang, J.-S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) Polymerase Inhibitors Sensitize Cancer Cells to Death Receptor-mediated Apoptosis by Enhancing Death Receptor Expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef]
- Passeri, D.; Camaioni, E.; Lisco, P.; Sabbatini, P.; Ferri, M.; Carroti, A.; Giacche, N.; Pellicciari, R.; Gioiello, A.; Macchiarulo, A. Concepts and molecular aspects in the polypharmacology of PARP-1 inhibitors. ChemMedChem 2016, 11, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Quintyne, N.J.; Reing, J.E.; Hoffelder, D.R.; Gollin, S.M.; Saunders, W.S. Spindle multipolarity is prevented by centrosomal clustering. Science 2005, 307, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Anderhub, S.; Maier, B. Mechanisms and Consequences of centrosomes clustering in cancer cells. In The Centrosome: Cell and Molecular Mechanisms of Functions and Disfunctions in Disease; Schatten, E., Ed.; Humana Press Springer: Totowa, NJ, USA, 2012; pp. 255–277. [Google Scholar]
- Rieder, C.L.; Maiato, H. Stuck in Division or Passing through: What Happens When Cells Cannot Satisfy the Spindle Assembly Checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef]
- Etemad, B.; Kops, J.P.L.G. Kinetochore transformations and spindle checkpoint silencing. Curr. Opin. Cell Biol. 2016, 39, 101–108. [Google Scholar] [CrossRef]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef]
- Galimberti, F.; Sarah, L.; Thompson, S.L.; Ravi, S.; Compton, D.A.; Dmitrovsky, E. Anaphase Catastrophe is a Target for Cancer Therapy. Clin. Cancer Res. 2011, 17, 1218–1222. [Google Scholar] [CrossRef]
- La Terra, S.; English, C.N.; Hergert, P.; McEwen, B.F.; Sluder, G.; Khodjakov, A. The de novo centriole assembly pathway in HeLa cells: Cell cycle progression and centriole assembly/maturation. J. Cell Biol. 2005, 168, 713–722. [Google Scholar] [CrossRef]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130467. [Google Scholar] [CrossRef]
- Cohen-Armon, M.; Yeheskel, A.; Pascal, J.M. Signal-induced PARP1-Erk synergism mediates IEG expression. Sig. Transduct. Target. Ther. 2019, 4, 1–8. [Google Scholar] [CrossRef]
- Miki, H.; Okada, Y.; Hirokawa, N. Analysis of the kinesin superfamily: Insights into structure and function. Trends Cell Biol. 2005, 15, 467–476. [Google Scholar] [CrossRef]
- Simeonov, D.R.; Kenny, K.; Seo, L.; Moyer, A.; Allen, J.; Paluh, J. Distinct kinesin-14 mitotic mechanisms in spindle polarity. Cell Cycl. 2009, 8, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Goshima, G.; Nedelec, F.; Vale, R.D. Mechanisms for focusing mitotic spindle poles by minus end directed motor proteins. J. Cell Biol. 2005, 171, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Verhey, K.; Hammond, J.W. Traffic control: Regulation of kinesin motors. Nat. Rev. Mol. Cell Biol. 2009, 10, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.I.; Hummer, S.; Bormann, J.; Gruner, T.; Adio, S.; Woehlke, G.; Mayer, T.U. The human kinesin Kif18A is a motile microtubule depolymerase essential for chromosome congression. Curr. Biol. 2007, 17, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.D.; Holland, A.J.; Cleveland, D.W. Requirement for NuMA in maintenance and establishment of mammalian spindle poles. J. Cell Biol. 2009, 184, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Kleylein-Sohn, J.; Pollinger, B.; Ohmer, M.; Hofmann, F.; Nigg, E.A.; Hemmings, B.A.; Wartmann, M. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J. Cell Sci. 2012, 125, 5391–5402. [Google Scholar] [CrossRef]
- Cai, S.; Weaver, L.N.; Ems-McClung, S.C.; Walczak, E. Kinesin-14 Family Proteins HSET/XCTK2 Control Spindle Length by Cross-Linking and Sliding Microtubules. Mol. Biol. Cell 2009, 20, 1348–1359. [Google Scholar] [CrossRef]
- Gordon, M.B.; Howard, L.; Compton, D.A. Chromosome Movement in Mitosis Require Microtubule Anchorage at Spindle Poles. J. Cell Biol. 2001, 152, 425–434. [Google Scholar] [CrossRef]
- Cai, S.; Weaver, L.N.; Ems-McClung, S.C.; Walczak, C.E. Proper Organization of Microtubule Minus-Ends is needed for Midzone Stability and Cytokinesis. Curr. Biol. 2010, 20, 880–885. [Google Scholar] [CrossRef]
- Haren, L.; Gnadt, N.; Wright, M.; Merdes, A. NuMA is required for proper spindle assembly and chromosome alignment in prometaphase. BMC Res. Notes 2009, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Haren, L.; Andreas -Merdes, A. Direct binding of NuMA to tubulin is mediated by a novel sequence motif in the tail domain that bundles and stabilizes microtubules. J. Cell Sci. 2002, 115, 1815–1823. [Google Scholar] [PubMed]
- Sukhai, M.A.; Wu, X.; Xuan, Y.; Zhang, T.P.P.; Dubé, K.; Rego, E.M.; Bhaumik, M.; Bailey, D.J.; Wells, R.A.; Kamel-Reid, S.; et al. Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG–NuMA–RARα. Oncogene 2004, 23, 665–678. [Google Scholar] [CrossRef]
- Zink, D.; Fischer, A.H.; Nickerson, J.A. Nuclear structure in cancer cells. Nat. Rev. Cancer 2004, 4, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, A.; Xie, Y.; Purinton, S.C.; Capell, K.M.; Swanik, J.T.; Larson, B.; Girard, L.; Schorge, J.O.; White, M.A. Tumor antigen Acrosin binding protein normalizes mitotic spindle function to promote cancer cell proliferation. Cancer Res. 2010, 70, 7652–7661. [Google Scholar] [CrossRef]
- Compton, D.A.; Luo, C. Mutations in the predicted p34cdc2 phosphorylation sites in NuMA impair the assembly of the mitotic spindle and block mitosis. J. Cell Sci. 1995, 108, 621–633. [Google Scholar] [PubMed]
- Chang, W.; Dynek, J.N.; Smith, S. NuMA is a major acceptor of polyADP-ribosylation by tankyrase1 in mitosis. Biochem. J. 2005, 391, 177–184. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Wang, Z.; Davitt, C.; McKenzie, I.F.; Xing, P.X.; Magnuson, N.S. Pim-1 associates with protein complexes necessary for mitosis. Chromosoma 2002, 111, 80–95. [Google Scholar] [CrossRef]
- Haikarainen, T.; Narwal, M.; Joensu, P.; Lehtiö, L. Evaluation and structural basis for the inhibition of tankyrases by PARP inhibitors. ACS Med. Chem. Lett. 2014, 5, 18–22. [Google Scholar] [CrossRef]
- Kirby, C.A.; Cheung, A.; Fazal, A.; Shultz, M.D.; Stam, T. Structure of Human tankyrase1 in complex with small-molecule inhibitors PJ34 and XAV939. Acta Cryst. 2012, 68, 115–118. [Google Scholar]
- Antolín, A.A.; Jalencas, X.; Yelamos, J.; Mestres, J. Identification of Pim Kinases as Novel Targets for PJ34 with Confounding Effects in PARP Biology. ACS Chem. Biol. 2012, 7, 1962–1967. [Google Scholar] [CrossRef] [PubMed]
- Haikarainen, T.; Krauss, S.; Lehtiö, L. Tankyrases: Structure, Function and Therapeutic Implications in Cancer. Curr. Pharm. Des. 2014, 20, 6472–6488. [Google Scholar] [CrossRef] [PubMed]
- Paul Chang, P.; Margaret Coughlin, M.; Mitchison, T.J. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat. Cell Biol. 2005, 7, 1133–1139. [Google Scholar]
- Tian, X.H.; Hou, W.J.; Fang, Y.; Fan, J.; Tong, H.; Bai, S.-L.; Chen, Q.; Xu, H.; Li, Y. XAV939, a tankyrase 1 inhibitior, promotes cell apoptosis in neuroblastoma cell lines by inhibiting Wnt/β-catenin signaling pathway. J. Exp. Clin. Cancer Res. 2013, 32, 100. [Google Scholar] [CrossRef]
- Leber, B.; Maler, B.; Fuchs, F.; Chi, J.; Riffel, P.; Anderhub, S.; Wagner, L.; Ho, A.D.; Salisbury, J.L.; Boutros, M.; et al. Proteins required for chromosome clustering in cancer cells. Sci. Trans. Med. 2010, 2, 1–11. [Google Scholar] [CrossRef]
- Nicolescu, A.C.; Holt, A.; Kandasamy, A.D.; Pacher, P.; Schultz, R. Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochim. Biophys. Res. Commun. 2009, 374, 646–650. [Google Scholar] [CrossRef]
- Chambers, A.F.; Matrisian, L.M. Changing views on the role of metalloproteinases in metastasis. J. Natl. Cancer Inst. 1997, 89, 1260–1270. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef]
- Inbar, D.; Cohen-Armon, M.; Neumann, D. Erythropoietin-driven signaling and cell migration mediated by polyADP-ribosylation. Br. J. Cancer 2012, 107, 1317–1326. [Google Scholar] [CrossRef]
- Rodriguez, M.I.; Peralta-Leal, A.; OValle, F.; Rodriguez-Vagas, J.M.; Gonzales-Flores, A.; Majuelos-Melquizo, J.; Lopez, L.; Serrano, S.; Garcia de Herreros, A.; Rodrigues-Manzaneque, J.C.; et al. PARP1 regulates metastatic melanoma through modulation of vimentin–induced malignant transformation. PLoS Genet. 2013, 9, e1003531. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen-Armon, M. The Modified Phenanthridine PJ34 Unveils an Exclusive Cell-Death Mechanism in Human Cancer Cells. Cancers 2020, 12, 1628. https://doi.org/10.3390/cancers12061628
Cohen-Armon M. The Modified Phenanthridine PJ34 Unveils an Exclusive Cell-Death Mechanism in Human Cancer Cells. Cancers. 2020; 12(6):1628. https://doi.org/10.3390/cancers12061628
Chicago/Turabian StyleCohen-Armon, Malka. 2020. "The Modified Phenanthridine PJ34 Unveils an Exclusive Cell-Death Mechanism in Human Cancer Cells" Cancers 12, no. 6: 1628. https://doi.org/10.3390/cancers12061628
APA StyleCohen-Armon, M. (2020). The Modified Phenanthridine PJ34 Unveils an Exclusive Cell-Death Mechanism in Human Cancer Cells. Cancers, 12(6), 1628. https://doi.org/10.3390/cancers12061628