Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors



Abstract
1. Introduction
2. DNA Damage and PARP Inhibition
2.1. Role of PARP in DNA Damage Response
2.2. The Lethal Synthetic Effect of PARP Inhibitors
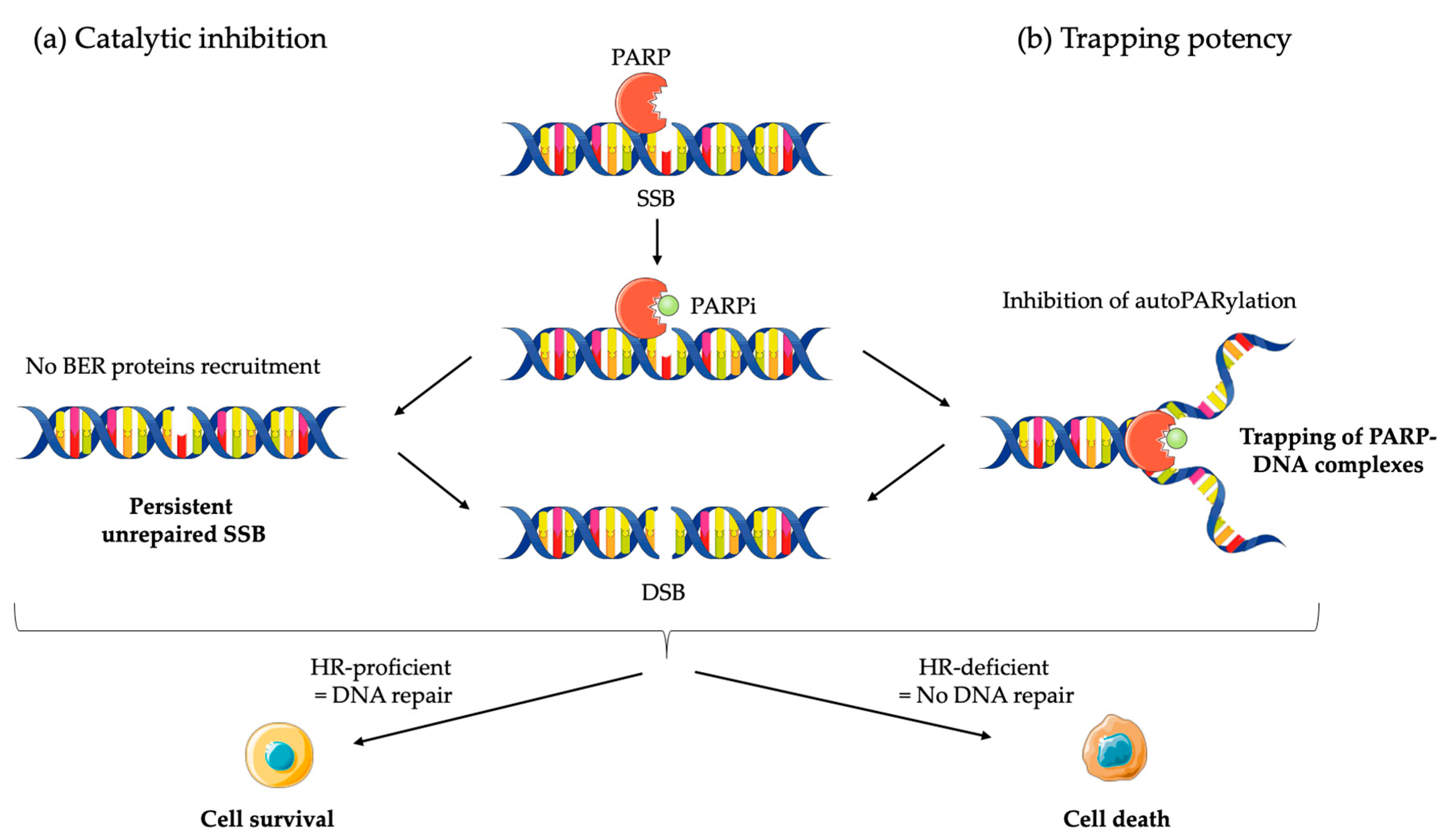
2.2.1. Mechanism of Action of PARPi
2.2.2. Clinical Applications of PARPi
3. The Revolution of Cancer Immunotherapy and Immune Checkpoint Inhibitors
3.1. CD80/86-CTLA-4 Signaling Pathway
3.2. PD-1/PD-L1 Signaling Pathway
3.3. Clinical Application of ICI
4. Combination of PARPi and ICI Therapy
4.1. A Rational to Combine PARPi and ICI
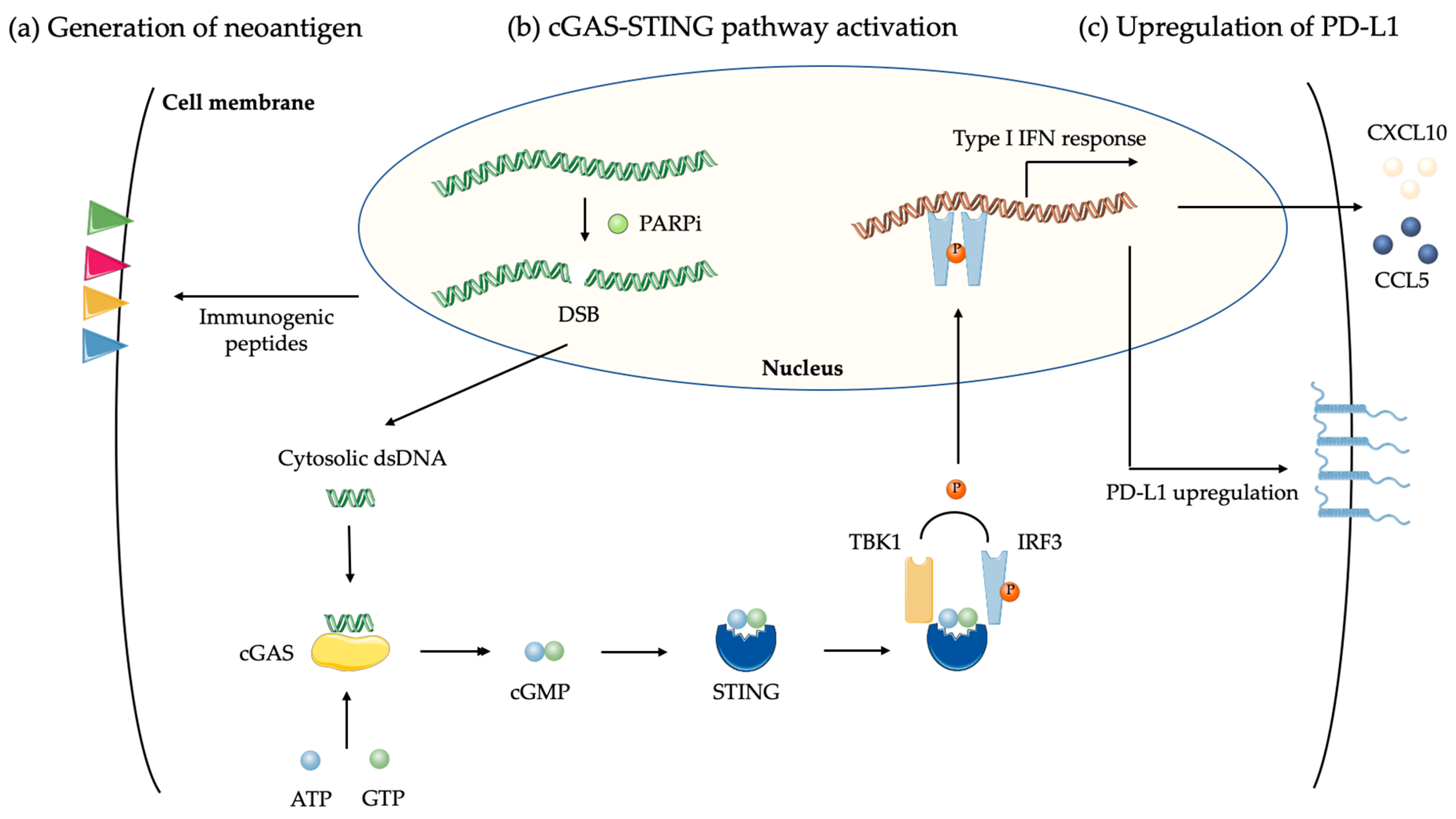
4.1.1. Tumor Mutation Burden and Neoantigen
4.1.2. DNA Damages and cGAS-STING Pathway
4.1.3. PD-L1 Upregulation by PARPi
4.1.4. Reprogramming of Immune Microenvironnement by PARPi
4.2. Preclinical Data and Clinical Studies
4.2.1. Combination of PARPi with Anti-PD1/PD-L1 ICIs
4.2.2. Combination of PARPi with Anti-CTLA-4 ICIs
4.2.3. Combination of ICI with Others DDR Inhibitors: Moving beyond PARP in Targeting the DDR
4.3. Future Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA. Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA. -Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Nowsheen, S.; Maraboyina, S.; Xia, F. The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci. 2020, 10, 35. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Lee, C.-H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2017, 2, e000213. [Google Scholar] [CrossRef]
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Fucá, G.; Reppel, L.; Landoni, E.; Savoldo, B.; Dotti, G. Enhancing Chimeric Antigen Receptor T cell Efficacy in Solid Tumors. Clin. Cancer Res. 2020, 26, 2444–2451. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Stewart, R.A.; Pilié, P.G.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes: Regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Im, S.-A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Thompson, L.H.; Schild, D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001, 477, 131–153. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Ström, C.E.; Johansson, F.; Uhlén, M.; Szigyarto, C.A.-K.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Wang, P.-Y.; Wang, Y.-T.; Yang, G.-F.; Zhang, A.; Miao, Z.-H. An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. J. Med. Chem. 2016, 59, 9575–9598. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Dhawan, M.S.; Bartelink, I.H.; Aggarwal, R.R.; Leng, J.; Zhang, J.Z.; Pawlowska, N.; Terranova-Barberio, M.; Grabowsky, J.A.; Gewitz, A.; Chien, A.J.; et al. Differential Toxicity in Patients with and without DNA Repair Mutations: Phase I Study of Carboplatin and Talazoparib in Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 6400–6410. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Diéras, V.C.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Phase III study of veliparib with carboplatin and paclitaxel in HER2-negative advanced/metastatic gBRCA-associated breast cancer. Ann. Oncol. 2019, 30, v857–v858. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Lord, C.J. Dissecting PARP inhibitor resistance with functional genomics. Curr. Opin. Genet. Dev. 2019, 54, 55–63. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell. 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef]
- Parish, C.R. Cancer immunotherapy: The past, the present and the future. Immunol. Cell. Biol. 2003, 81, 106–113. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Germain, C.; de Reyniès, A.; Laurent-Puig, P.; Zucman-Rossi, J.; Dieu-Nosjean, M.-C.; Sautès-Fridman, C.; Fridman, W.H. Immune Contexture, Immunoscore, and Malignant Cell Molecular Subgroups for Prognostic and Theranostic Classifications of Cancers. Adv. Immunol. 2016, 130, 95–190. [Google Scholar] [CrossRef]
- Bretscher, P.A. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc. Natl. Acad. Sci. USA 1999, 96, 185–190. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Boussiotis, V.A.; Lorsbach, R.B.; Abbas, A.K.; Sharpe, A.H. CTLA-4 Regulates Induction of Anergy In Vivo. Immunity 2001, 14, 145–155. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Catenacci Daniel, V.; Wainberg, Z.; Fuchs Charles, S.; Garrido, M.; Bang, Y.-J.; Muro, K.; Savage, M.; Wang, J.; Koshiji, M.; Dalal Rita, P.; et al. KEYNOTE-059 cohort 3: Safety and efficacy of pembrolizumab monotherapy for first-line treatment of patients (pts) with PD-L1-positive advanced gastric/gastroesophageal (G/GEJ) cancer. Ann. Oncol. 2017, 28 (Suppl. 3), iii153. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. JCO 2018, 36, 1428–1439. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. of. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit from PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Honjo, T. Combination therapy strategies for improving PD-1 blockade efficacy: A new era in cancer immunotherapy. J. Intern. Med. 2018, 283, 110–120. [Google Scholar] [CrossRef]
- Seliger, B. Combinatorial Approaches with Checkpoint Inhibitors to Enhance Anti-tumor Immunity. Front. Immunol. 2019, 10, 999. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e4. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Legrand, F.A.; Gandara, D.R.; Mariathasan, S.; Powles, T.; He, X.; Zhang, W.; Jhunjhunwala, S.; Nickles, D.; Bourgon, R.; Schleifman, E.; et al. Association of high tissue TMB and atezolizumab efficacy across multiple tumor types. JCO 2018, 36, 12000. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome. Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA. -Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Damania, B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host. Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Rieke, D.T.; Ochsenreither, S.; Klinghammer, K.; Seiwert, T.Y.; Klauschen, F.; Tinhofer, I.; Keilholz, U. Methylation of RAD51B, XRCC3 and other homologous recombination genes is associated with expression of immune checkpoints and an inflammatory signature in squamous cell carcinoma of the head and neck, lung and cervix. Oncotarget 2016, 7, 75379–75393. [Google Scholar] [CrossRef]
- Green, A.R.; Aleskandarany, M.A.; Ali, R.; Hodgson, E.G.; Atabani, S.; De Souza, K.; Rakha, E.A.; Ellis, I.O.; Madhusudan, S. Clinical Impact of Tumor DNA Repair Expression and T-cell Infiltration in Breast Cancers. Cancer Immunol. Res. 2017, 5, 292–299. [Google Scholar] [CrossRef]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.-M.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, C.E.; Mills, A.M.; Cross, J.V.; Ring, K.L. Tumor-associated macrophage expression of PD-L1 in implants of high grade serous ovarian carcinoma: A comparison of matched primary and metastatic tumors. Gynecol. Oncol. 2017, 144, 607–612. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- LaFargue, C.J.; Dal Molin, G.Z.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Karzai, F.; Madan, R.A.; Owens, H.; Couvillon, A.; Hankin, A.; Williams, M.; Bilusic, M.; Cordes, L.M.; Trepel, J.B.; Killian, K.; et al. A phase 2 study of olaparib and durvalumab in metastatic castrate-resistant prostate cancer (mCRPC) in an unselected population. JCO 2018, 36, 163. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Lee, J.-M.; Annunziata, C.M.; Houston, N.; Kohn, E.C.; Lipkowitz, S.; Minasian, L.; Nichols, E.; Trepel, J.; Trewhitt, K.; Zia, F.; et al. A phase II study of durvalumab, a PD-L1 inhibitor and olaparib in recurrent ovarian cancer (OvCa). Ann. Oncol. 2018, 29, viii334. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.S.; Padget, M.R.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: A proof-of-concept phase 2 study. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef]
- Drew, Y.; de Jonge, M.; Hong, S.H.; Park, Y.H.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.E.; Alvarez, R.H.; Gresty, C.; et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Domchek, S.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Phase II study of olaparib (O) and durvalumab (D) (MEDIOLA): Updated results in patients (pts) with germline BRCA-mutated (gBRCAm) metastatic breast cancer (MBC). Ann. Oncol. 2019, 30, v477. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Kaufman, B.; Geva, R.; Stemmer, S.M.; Hong, S.-H.; Lee, J.-S.; Domchek, S.M.; Lanasa, M.C.; Tang, M.; Gresty, C.; et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in patients with relapsed gastric cancer. J. Clin. Oncol 2019. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132. [Google Scholar] [CrossRef]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.; Norris, C.; Mu, S.; et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: Results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1306–1315. [Google Scholar] [CrossRef]
- Yap, T.A.; Konstantinopoulos, P.; Telli, M.L.; Saraykar, S.; Beck, J.T.; Galsky, M.D.; Abraham, J.; Wise, D.R.; Khasraw, M.; Rubovszky, G.; et al. Abstract P1-19-03: JAVELIN PARP Medley, a phase 1b/2 study of avelumab plus talazoparib: Results from advanced breast cancer cohorts. Cancer Res. 2020, 80, P1-P1-19–03. [Google Scholar] [CrossRef]
- Rodriguez-Moreno, J.F.; de Velasco, G.; Bravo Fernandez, I.; Alvarez-Fernandez, C.; Fernandez, R.; Vazquez-Estevez, S.; Virizuela, J.A.; Gajate, P.; Font, A.; Lainez, N.; et al. Impact of the combination of durvalumab (MEDI4736) plus olaparib (AZD2281) administered prior to surgery in the molecular profile of resectable urothelial bladder cancer: NEODURVARIB Trial. JCO 2020, 38, 542. [Google Scholar] [CrossRef]
- Clarke, B.; Tinker, A.V.; Lee, C.-H.; Subramanian, S.; van de Rijn, M.; Turbin, D.; Kalloger, S.; Han, G.; Ceballos, K.; Cadungog, M.G.; et al. Intraepithelial T cells and prognosis in ovarian carcinoma: Novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 2009, 22, 393–402. [Google Scholar] [CrossRef]
- McAlpine, J.N.; Porter, H.; Köbel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef]
- Wen, W.X.; Leong, C.-O. Association of BRCA1- and BRCA2-deficiency with mutation burden, expression of PD-L1/PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer. PLoS ONE 2019, 14, e0215381. [Google Scholar] [CrossRef]
- Adams, S.F.; Rixe, O.; Lee, J.-H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I study combining olaparib and tremelimumab for the treatment of women with BRCA-deficient recurrent ovarian cancer. JCO 2017, 35, e17052. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM–Chk2 and ATR–Chk1 Pathways in DNA Damage Signaling and Cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherland, 2010; Volume 108, pp. 73–112. ISBN 978-0-12-380888-2. [Google Scholar]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3348–3353. [Google Scholar] [CrossRef]
- Alimzhanov, M.; Soulard, P.; Zimmermann, A.; Schroeder, A.; Mehr, K.T.; Amendt, C.; Sim, G.C.; Blaukat, A.; Halle, J.-P.; Zenke, F.T. Abstract 2269: ATR inhibitor M6620 enhances anti-tumor efficacy of the combination of the anti-PD-L1 antibody avelumab with platinum-based chemotherapy. Cancer Res. 2019, 79, 2269. [Google Scholar] [CrossRef]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef]
- Carr, M.; Zimmermann, A.; Guo, Y.; Liu, X.; Steiner, P.; Hahn, S.; Zenke, F.; Blaukat, A.; Vassilev, L.T. Abstract 2923: DNA-PK inhibitor, M3814, is a potent inducer of inflammatory micronucleation in irradiated p53-deficient cancer cells: Implications for combination radio-immunotherapy. Cancer Res. 2019, 79, 2923. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; Bang, Y.J.; El-Khoueiry, A.; Abida, W.; Harrington, K.; Sundar, R.; Carter, L.; Castanon-Alvarez, E.; et al. Phase I modular study of AZD6738, a novel oral, potent and selective ataxia telangiectasia Rad3-related (ATR) inhibitor in combination (combo) with carboplatin, olaparib or durvalumab in patients (pts) with advanced cancers. Eur. J. Cancer 2016, 69, S2. [Google Scholar] [CrossRef]
- Powles, T.; Kilgour, E.; Mather, R.; Galer, A.; Arkenau, H.-T.; Farnsworth, A.; Wilde, J.; Ratnayake, J.; Landers, D. BISCAY, a phase Ib, biomarker-directed multidrug umbrella study in patients with metastatic bladder cancer. JCO 2016, 34, TPS4577. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. In American Society of Clinical Oncology Educational Book; ASCO: Alexandria, VA, USA, 2019; pp. 185–195. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tymor Type | Study Identifier | Setting | ICI Agent | PARPi Agent | Design | Patients | Primary Endpoint | Outcome |
|---|---|---|---|---|---|---|---|---|
| Ovarian | NCT02571725 | Phase I | Tremelimumab (10 mg/kg Q4W) | Olaparib (300 mg BID) | gBRCAm recurrent ovarian cancer | 3 | Safety and RP2D | No DLT or grade 3 AE ORR 100% with 3 PRs |
| NCT02484404 | Phase II | Durvalumab (1500 mg Q4W) | Olaparib (300 mg BID) | Platinum-resistant recurrent ovarian cancer | 35 | Clinical activity (ORR) | ORR 14% with 5 PRs (irrespective of BRCA status), DCR 71%, mPFS 3.9 months Acceptable toxicity | |
| NCT02657889 (TOPACIO/KEYNOTE-162) | Phase II | Pembrolizumab (200 mg Q3W) | Niraparib (200 mg QD) | Platinum-resistant recurrent ovarian cancer | 60 | Clinical activity (ORR) | ORR 18% with 3 CRs and 8 PRs (irrespective of BRCA and HRD status), DCR 65% mPFS 3.4 months Acceptable toxicity | |
| NCT02734004 (MEDIOLA) | Phase II | Durvalumab (1500 mg Q4W) | Olaparib (300 mg BID) | gBRCAm platinum-sensitive ovarian cancer | 32 | Clinical activity (DCR) | 12-week DCR 81%, ORR 63% with 6 CRs and 14 PRs Acceptable toxicity | |
| Breast | NCT02657889 (TOPACIO/KEYNOTE-162) | Phase II | Pembrolizumab (200 mg Q3W) | Niraparib (200 mg QD) | Advanced/Metastatic TNBC | 55 | Clinical activity (ORR) | ORR 21% with 5 CRs and 5 PRs (stronger activity in BRCA-mutated tumors), DCR 49% Acceptable toxicity |
| NCT02734004 (MEDIOLA) | Phase II | Durvalumab (1500 mg Q4W) | Olaparib (300 mg BID) | gBRCAm HER2 negative mBC | 30 for clinical activity and 34 for safety | Clinical activity (DCR) and safety | 12-week DCR 80%, 28-week DCR 80%, ORR 63%, mPFS 8.2 months, mOS 20.5 months (especially in chemotherapy-free patients) Acceptable toxicity | |
| Prostate | NCT02484404 | Phase II | Durvalumab (1500 mg Q4W) | Olaparib (300 mg BID) | Previously treated mCRPC | 17 | Clinical activity (rPFS) and safety | rPFS 16.1 months with 12-month rPFS 51.5% (especially in men with DDR abnomalities) Acceptable toxicity |
| SCLC | NCT02484404 | Phase II | Durvalumab (1500 mg Q4W) | Olaparib (300 mg BID) | Relapsed SCLC | 19 | Clinical activity (ORR) | ORR 10.5% with 1 PRs and 1 CRs, clinical benefit 21.1% (preexisting TIL predictive of response) Acceptable toxicity |
| Bladder | NCT03534492 (NEODURVARIB) | Phase II | Durvalumab (1500 mg Q4W) | Olaparib (300 mg BID) | Resectable muscle-invasive bladder cancer | 28 | Clinical activity and safety | Pathological CR 44.5% Acceptable toxicity (grade 3 or higher AEs 8.3%) |
| Gastric | NCT02734004 (MEDIOLA) | Phase II | Durvalumab (1500 mg Q4W) | Olaparib (300 mg BID) | Platinum-resistant relapsed gastric cancer | 39 for clinical activity and 40 for safety | Clinical activity (DCR) and safety | 12-week DCR 26%, ORR 10% with 2 CRs and 2 PRs Unacceptable toxicity (grade 3 or higher AEs 48%) |
| Solid tumors | NCT02660034 | Phase Ia/b | Tislelizumab (2 mk/kg Q2W) | Pamiparib (20, 40 or 60 mg BID) | Previously treated advanced solid tumors | 49 patients | Safety and RP2D | DLT 8% with 23 immune-related AEs RP2D tislelizumab 200 mg and pamiparib 40 mg ORR 20% with 2CRs and 8 PRs |
| Solid tumors | NCT03330405 (JAVELIN PARP Medley) | Phase Ib/II | Avelumab (800 mg Q2W) | Talazoparib (1 mg QD) | Previously treated advanced solid tumors | 34 patients | Safety and clinical activity (ORR) | First-cycle DLT 25% ORR 8% with 1 PR, SD 50% |
| PARPi | ICI | Study Identifier | Phase | Tumor Type and Conditions | Status | |
|---|---|---|---|---|---|---|
| Type | Drug | |||||
| Olaparib | Anti-CTLA-4 | Tremelimumab | NCT02571725 | I/II | gBRCAm recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma | Recruiting |
| Tremelimumab | NCT02485990 | I/II | Recurrent or persistent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma | Not recruiting | ||
| Tremelimumab | NCT04034927 | II | Platinum-sensitive advanced epithelial ovarian, fallopian tube, or primary peritoneal carcinoma | Recruiting | ||
| Anti-PD1 | Pembrolizumab | NCT04209686 | II | Locally advanced or metastatic gastric carcinoma | Not yet recruiting | |
| Pembrolizumab | NCT04306367 | II | Locally advanced or metastatic cholangiocarcinoma | Recruiting | ||
| Pembrolizumab | NCT02861573 (KEYNOTE-365) | I | Metastatic castration-resistant prostate cancer | Recruiting | ||
| Pembrolizumab | NCT03740165 (KEYLYNK-001 | III | BRCA-non-mutated advanced epithelial ovarian, fallopian tube, or primary peritoneal carcinoma | Recruiting | ||
| Pembrolizumab | NCT03976323 (KEYLINK-006) | III | Metastatic non-squamous cell lung cancer | Recruiting | ||
| Pembrolizumab | NCT04123366 (KEYLYNK-007) | II | HRR-mutated or HRD positive advanced or metastatic solid tumors | Recruiting | ||
| Pembrolizumab | NCT03976362 (KEYLYNK-008) | III | Metastatic squamous non-small cell lung cancer | Recruiting | ||
| Pembrolizumab | NCT04191135 (KEYLYNK-009) | II/III | Locally advanced triple negative breast cancer | Recruiting | ||
| Pembrolizumab | NCT03834519 (KEYLYNK-010) | III | Metastatic castration-resistant prostate cancer | Recruiting | ||
| Anti-PD-L1 | Atezolizumab | NCT02849496 | II | Locally advanced unresectable and or metastatic HER negative breast cancer | Recruiting | |
| Durvalumab | NCT03594396 | I/II | Resectable stage II/III triple negative breast cancer | Recruiting | ||
| Durvalumab | NCT03534492 (NEODURVARIB) | II | Resectable urothelial carcinoma | Recruiting | ||
| Durvalumab | NCT03459846 (BAYOU) | II | Advanced or metastatic platinum-ineligible urothelial carcinoma | Not recruiting | ||
| Durvalumab | NCT03951415 (DOMEC) | II | Recurrent, refractory or metastatic endometrial cancer or carcinosarcoma of the endometrium | Recruiting | ||
| Durvalumab | NCT03851614 (DAPPER) | II | Locally advanced or metastatic mismatch repair proficient colorectal cancer, pancreatic adenocarcinoma or leiomyosarcoma | Recruiting | ||
| Durvalumab | NCT03167619 (DORA) | II | Locally advanced or metastatic platinum-treated advanced triple negative breast cancer | Recruiting | ||
| Durvalumab | NCT03544125 | I | Metastatic triple negative breast cancer | Not recruiting | ||
| Durvalumab | NCT03801369 | II | Metastatic triple negative breast cancer | Recruiting | ||
| Durvalumab | NCT04053322 (DOLAF) | II | Locally advanced or metastatic ER positive HER2 negative breast cancer | Recruiting | ||
| Durvalumab | NCT03810105 | II | DDR-mutated castration sensitive biochemically recurrent non-metastatic prostate cancer | Recruiting | ||
| Durvalumab | NCT02882308 (OPHELIA) | II | Resectable squamous cell carcinoma of the head and neck | Completed | ||
| Durvalumab | NCT03991832 | II | IDH-mutated solid tumors (glioma, cholangiocarcinoma, and solid tumors) | Not yet recruiting | ||
| Durvalumab | NCT03772561 (MEDIPAC) | I | Advanced or metastatic solid tumors | Recruiting | ||
| Durvalumab | NCT02484404 | I/II | Advanced, recurrent or metastatic ovarian, triple negative breast, lung, prostate, colorectal carcinoma or solid tumors | Recruiting | ||
| Durvalumab | NCT03579784 | II | Unresectable or recurrent gastric carcinoma | Recruiting | ||
| Durvalumab | NCT03737643 (DUO-O) | III | Newly diagnosed advanced ovarian, fallopian tube or primary peritoneal carcinoma or carcinosarcoma | Recruiting | ||
| Durvalumab | NCT04269200 (DUO-E) | III | Newly diagnosed advanced or recurrent endometrial carcinoma | Not yet recruiting | ||
| Durvalumab | NCT03801369 | II | Metastatic triple negative breast cancer | Recruiting | ||
| Durvalumab | NCT03775486 | II | Metastatic non-squamous cell lung cancer | Recruiting | ||
| Durvalumab | NCT02734004 (MEDIOLA) | I/II | Advanced or metastatic solid tumors | Recruiting | ||
| Durvalumab | NCT03842228 | I | DDR-mutated unresectable, advanced or metastatic solid tumors | Recruiting | ||
| Anti-PD-L1 + Anti-CTLA-4 | Durvalumab + Tremelimumab | NCT04169841 (GUIDE2REPAIR) | II | HRR-mutated advanced or metastatic solid tumors (breast, lung, head and neck, clear cell renal, endometrial, ovarian, urothelial and prostate cancer | Not yet recruiting | |
| Durvalumab + Tremelimumab | NCT03923270 | I | Extensive small cell lung cancer | Recruiting | ||
| Durvalumab + Tremelimumab | NCT02953457 | I/II | DDR-mutated recurrent or refractory ovarian, fallopian tube or primary peritoneal carcinoma | Recruiting | ||
| Niraparib | Anti-CTLA-4 | Ipilimumab | NCT03404960 (Parpvax) | I/II | Advanced pancreatic cancer | Recruiting |
| Anti-PD1 | Cetrelimab | NCT03431350 (QUEST) | I/II | Metastatic castration-resistant prostate cancer | Recruiting | |
| Dostarlimab | NCT04068753 (STAR) | II | Recurrent or progressive cervix cancer | Recruiting | ||
| Dostarlimab | NCT04313504 | II | Reccurent or metastatic head and nead squamous carcinoma | Not yet recruiting | ||
| Dostarlimab | NCT03016338 | II | Recurrent or advanced endometrial cancer | Recruiting | ||
| Dostarlimab | NCT03602859 (FIRST) | III | Stage III or IV non-mucinous epithelial ovarian, fallopian tube or primary peritoneal cancer | Recruiting | ||
| Dostarlimab | NCT03955471 (MOONSTONE) | II | Advanced platinum-resistant ovarian, fallopian tube or primary peritoneal carcinoma | Recruiting | ||
| Dostarlimab | NCT03806049 | III | Advanced or recurrent platinum-sensitive ovarian, fallopian tube or primary peritoneal carcinoma | Not yet recruiting | ||
| Dostarlimab | NCT03307785 | I | Advanced or metastatic solid tumors | Not recruiting | ||
| Dostarlimab | NCT03651206 (ROCSAN) | II/III | Reccurent or progressive uterine or ovarian carcinosarcoma | Not yet recruiting | ||
| Nivolumab | NCT03404960 (Parpvax) | I/II | Advanced pancreatic cancer | Recruiting | ||
| Pembrolizumab | NCT02657889 (TOPACIO) | I/II | Advanced or metastatic triple negative breast or ovarian cancer | Not recruiting | ||
| PD-1 inhibitor | NCT03308942 | II | Locally advanced or metastatic non-small cell lung carcinoma | Not recruiting | ||
| Anti-PD-L1 | Atezolizumab | NCT03695380 | I | Advanced ovarian, fallopian tube or primary peritoneal carcinoma | Recruiting | |
| Atezolizumab | NCT03598270 (ANITA) | III | Recurrent ovarian, fallopian tube or primary peritoneal carcinoma | Recruiting | ||
| Atezolizumab | NCT04185831 (MEGALiT) | II | Advanced or metastatic solid tumors | Not yet recruiting | ||
| Anti-PD-1 | Nivolumab | NCT04187833 | II | BRCA- or BRCAness-mutated resectable or metastatic melanoma | Recruiting | |
| Pembrolizumab | NCT04158336 | I/II | Solid tumors | Recruiting | ||
| Anti-PD-L1 | Avelumab | NCT02912572 | II | Recurrent or persistent endometrial cancer | Recruiting | |
| Avelumab | NCT03964532 (TALAVE) | I/II | Advanced breast cancer | Recruiting | ||
| Avelumab | NCT03637491 | II | Locally advanced or metastatic RAS-mutant solid tumors | Recruiting | ||
| Avelumab | NCT04173507 (A LUNG-MAP) | II | STK11-mutated recurrent or metastatic non-squamous non-small cell lung cancer | Recruiting | ||
| Avelumab | NCT03565991 (JAVELIN BRCA/ATM) | II | BRCA or ATM-mutated locally advanced or metastatic solid tumors | Recruiting | ||
| Avelumab | NCT04068831 | II | Locally advanced or metastatic clear-cell renal cell carcinoma | Recruiting | ||
| Avelumab | NCT04052204 | II | Locally advanced or metastatic head and neck squamous carcinoma or CRPC | Recruiting | ||
| Avelumab | NCT03330405 (JAVELIN PARP MEDLEY) | II | Locally advanced or metastatic solid tumors | Recruiting | ||
| Avelumab | NCT03642132 (JAVELIN Ovarian PARP 100) | III | Locally advanced or metastatic ovarian cancer (NSCLC, triple negative breast cancer, HR+ breast cancer, recurrent platinum-sensitive ovarian cancer, urothelial cancer, CRPC) | Not recruiting | ||
| Veliparib | Anti-PD-1 | Nivolumab | NCT02944396 | I | Advanced or metastatic NSCLC | Completed |
| Nivolumab | NCT03061188 | I | Advanced, recurrent, refractory or metastatic solid tumors | Not recruiting | ||
| Rucaparib | Anti-PD-1 | Nivolumab | NCT03572478 | I/II | Metastatic CRPC or recurrent endometrial cancer | Recruiting |
| Nivolumab | NCT03639935 | II | Advanced or metastatic cholangiocarcinoma | Recruiting | ||
| Nivolumab | NCT02873962 | II | Relapsed ovarian, fallopian tube or peritoneal cancer | Recruiting | ||
| Nivolumab | NCT03338790 (CheckMate 9KD) | II | Metastatic castration-resistant prostate cancer | Recruiting | ||
| Nivolumab | NCT03522246 (ATHENA) | III | Newly diagnosed advanced ovarian, fallopian tube or primary peritoneal carcinoma or carcinosarcoma | Recruiting | ||
| Nivolumab | NCT03824704 (ARIES) | II | Platinum-treated advanced ovarian, fallopian tube or primary peritoneal carcinoma or carcinosarcoma | Not recruiting | ||
| Nivolumab | NCT03958045 | II | Platinum-sensitive small cell lung carcinoma | Recruiting | ||
| Nivolumab | NCT03995017 (RiME) | I/II | Unresectable or metastatic gastric or esophageal adenocarcinoma | Recruiting | ||
| Pembrolizumab | NCT03559049 | I/II | Metastatic NSCLC | Recruiting | ||
| Anti-PD-L1 | Atezolizumab | NCT03101280 | I | Advanced or metastatic platinum-sensitive ovarian or endometrial cancer or triple negative breast cancer | Not recruiting | |
| Atezolizumab | NCT04276376 (ARIANES) | II | DDR-deficient or platinum sensitive solid tumors | Recruiting | ||
| Atezolizumab | NCT03694262 (EndoBARR) | II | Recurrent progressive endometrial carcinoma | Recruiting | ||
| Pamiparib | Anti-PD-L1 | Tislelizumab | NCT02660034 | I | Advanced or metastatic solid tumors | Recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. https://doi.org/10.3390/cancers12061502
Peyraud F, Italiano A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers. 2020; 12(6):1502. https://doi.org/10.3390/cancers12061502
Chicago/Turabian StylePeyraud, Florent, and Antoine Italiano. 2020. "Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors" Cancers 12, no. 6: 1502. https://doi.org/10.3390/cancers12061502
APA StylePeyraud, F., & Italiano, A. (2020). Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers, 12(6), 1502. https://doi.org/10.3390/cancers12061502