Well-Differentiated Papillary Mesothelioma of the Peritoneum Is Genetically Distinct from Malignant Mesothelioma

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
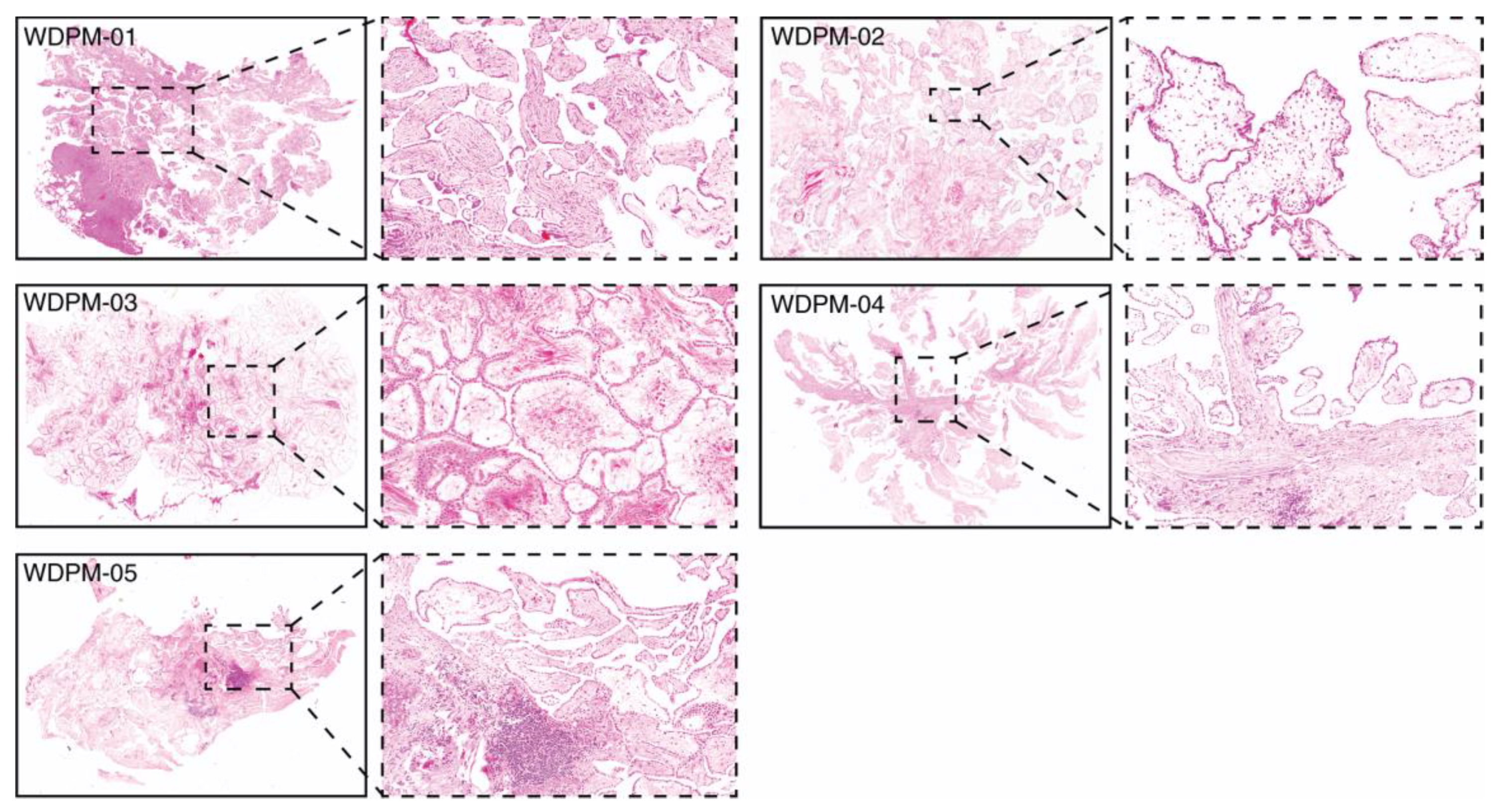
2.1. Histopathological Features of WDPM
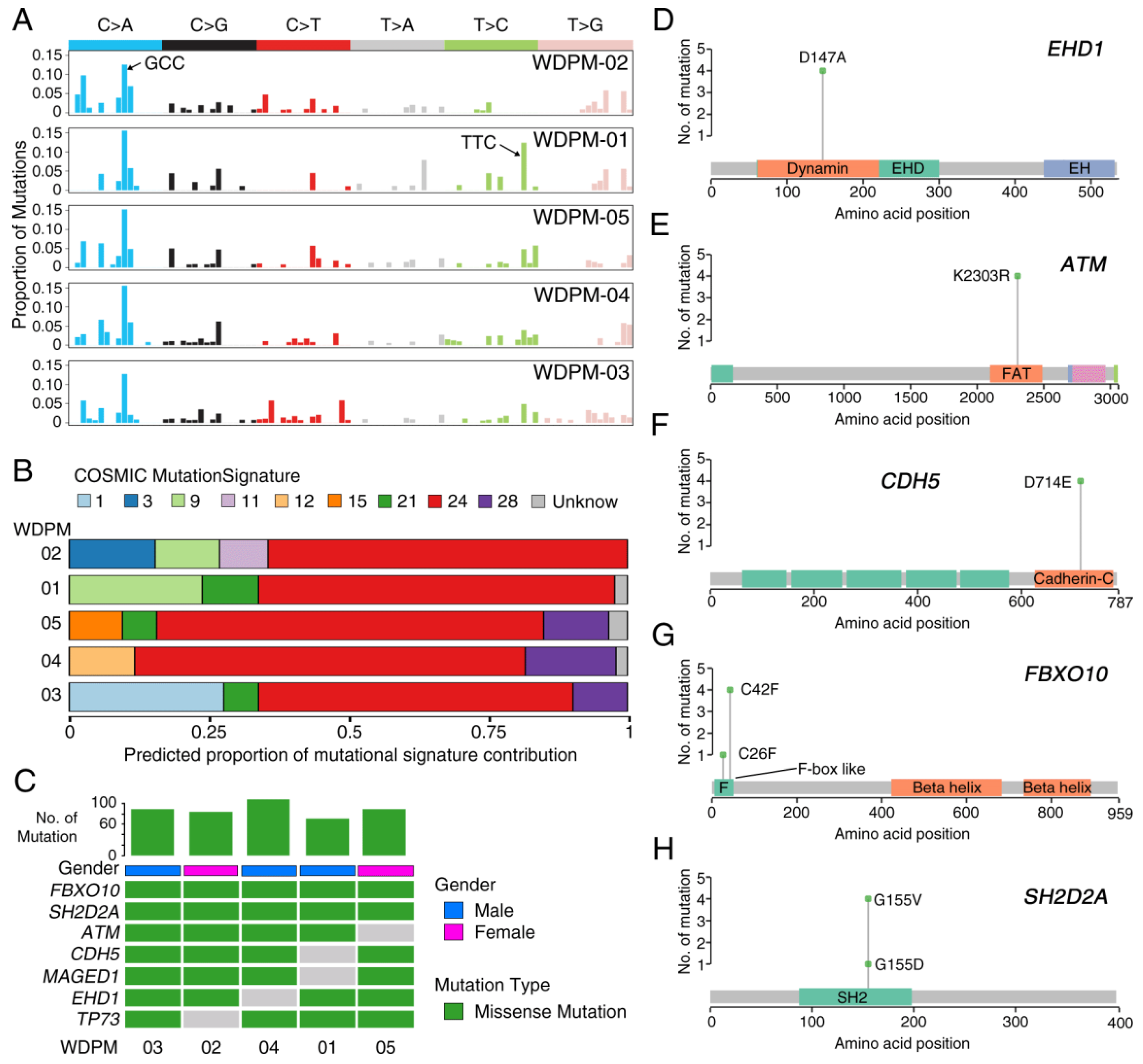
2.2. Mutational Landscape of WDPM
2.3. Copy Number Landscape of WDPM
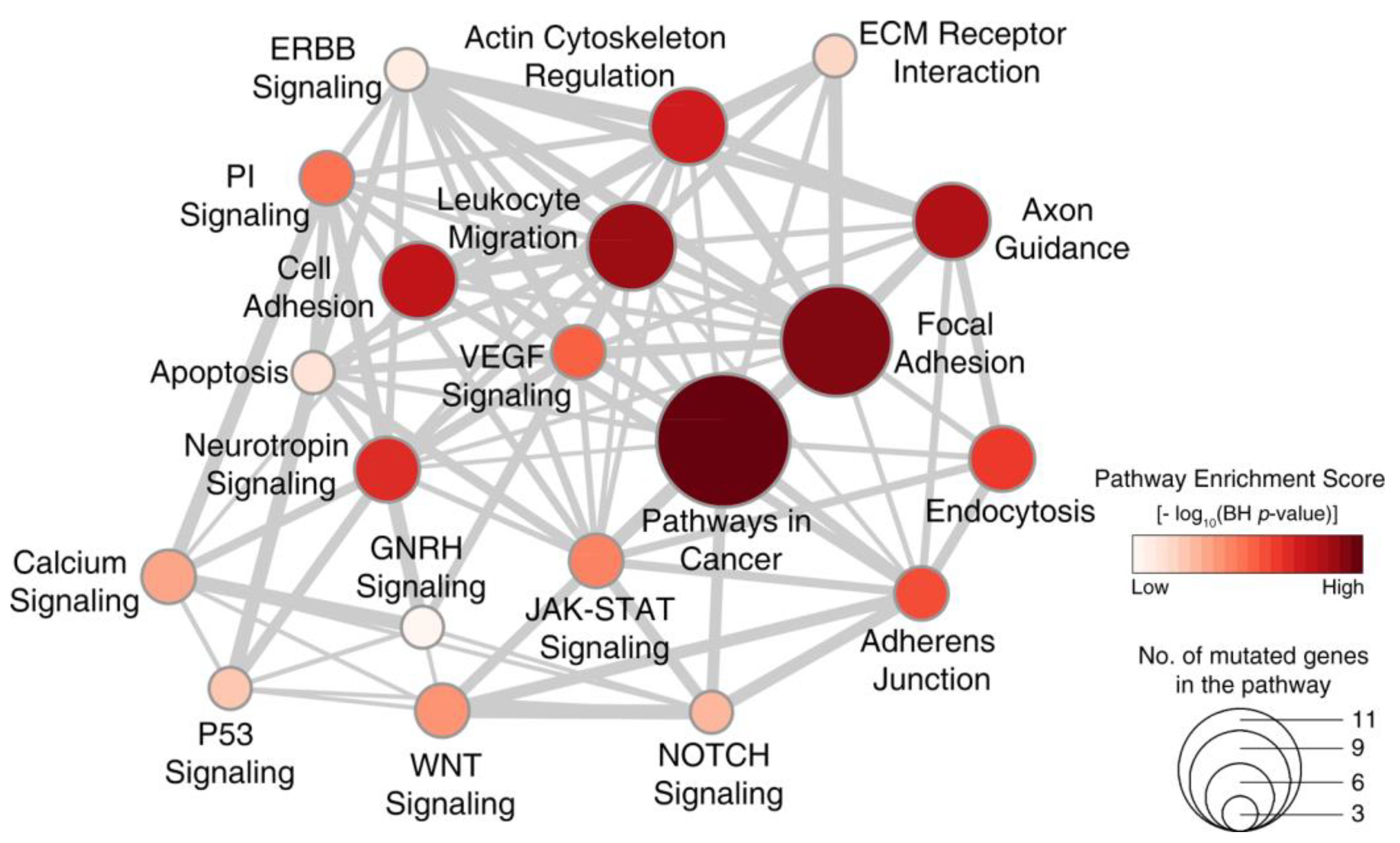
2.4. Signaling Pathways Dysregulated in WDPM
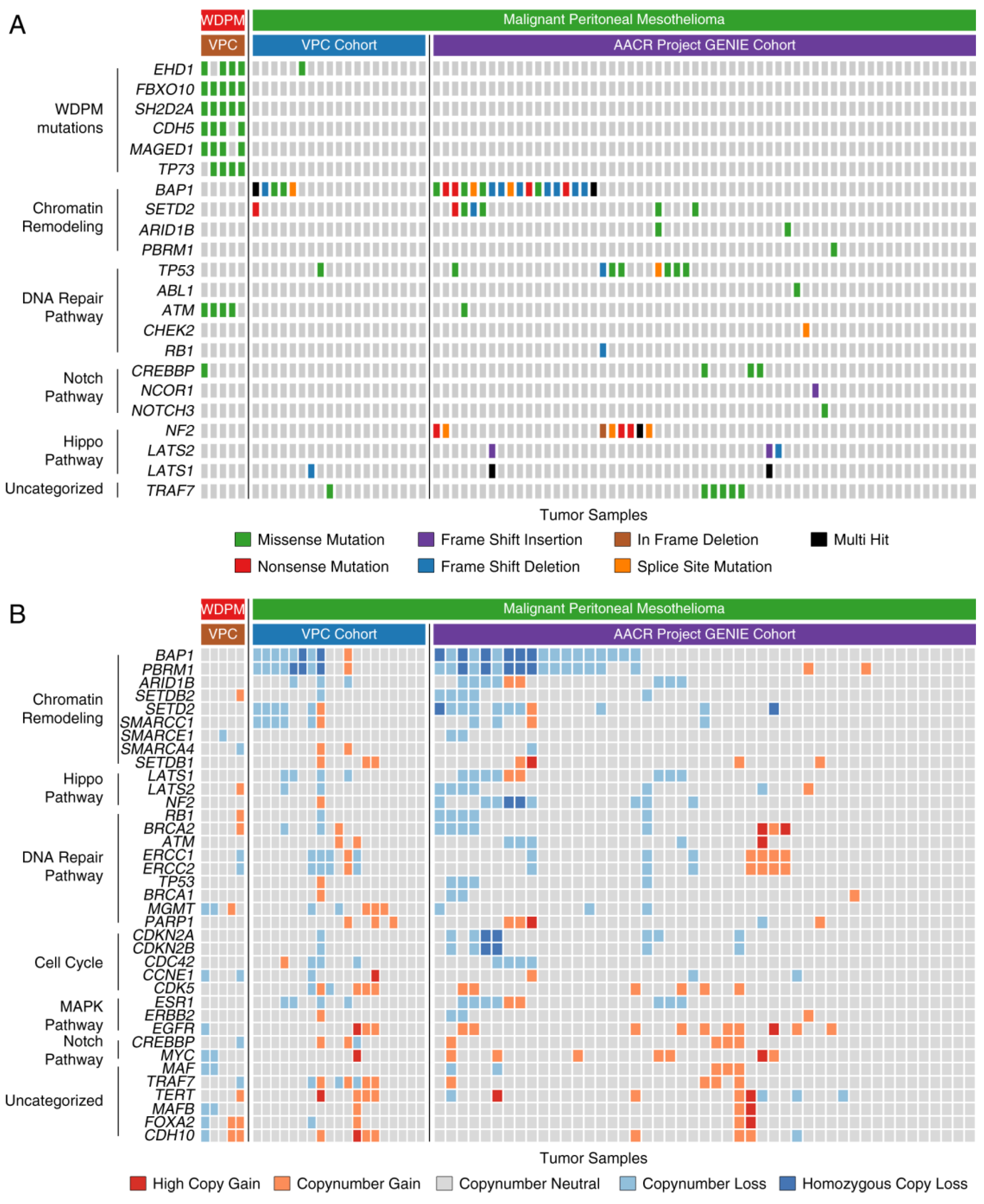
2.5. WDPM is Genetically Distinct from Malignant Mesothelioma
3. Discussion
4. Materials and Methods
4.1. Patient Cohort Description and Tissue Procurement
4.2. Whole Exome Sequencing
4.3. Single Nucleotide Variant Calling
4.4. Copy Number Aberration (CNA) Calls
4.5. Mutational Signature Analysis
4.6. Pathway Enrichment Analysis
4.7. Peritoneal Mesothelioma Datasets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Daya, D.; McCaughey, W.T. Well-differentiated papillary mesothelioma of the peritoneum. A clinicopathologic study of 22 cases. Cancer 1990, 65, 292–296. [Google Scholar] [CrossRef]
- Butnor, K.J.; Sporn, T.A.; Hammar, S.P.; Roggli, V.L. Well-differentiated papillary mesothelioma. Am. J. Surg. Pathol. 2001, 25, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Deraco, M.; Nizri, E.; Glehen, O.; Baratti, D.; Tuech, J.-J.; Bereder, J.-M.; Kepenekian, V.; Kusamura, S.; Goere, D. Well differentiated papillary peritoneal mesothelioma treated by cytoreduction and hyperthermic intraperitoneal chemotherapy-the experience of the PSOGI registry. Eur. J. Surg. Oncol. 2019, 45, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Nabavi, N.; Lin, Y.-Y.; Mo, F.; Anderson, S.; Volik, S.; Adomat, H.H.; Lin, D.; Xue, H.; Dong, X.; et al. BAP1 haploinsufficiency predicts a distinct immunogenic class of malignant peritoneal mesothelioma. Genome Med. 2019, 11, 8. [Google Scholar] [CrossRef]
- Yu, W.; Chan-On, W.; Teo, M.; Ong, C.K.; Cutcutache, I.; Allen, G.E.; Wong, B.; Myint, S.S.; Lim, K.H.; Voorhoeve, P.M.; et al. First somatic mutation of E2F1 in a critical DNA binding residue discovered in well-differentiated papillary mesothelioma of the peritoneum. Genome Biol. 2011, 12, R96. [Google Scholar] [CrossRef]
- Nemoto, H.; Tate, G.; Kishimoto, K.; Saito, M.; Shirahata, A.; Umemoto, T.; Matsubara, T.; Goto, T.; Mizukami, H.; Kigawa, G.; et al. Heterozygous loss of NF2 is an early molecular alteration in well-differentiated papillary mesothelioma of the peritoneum. Cancer Genet. 2012, 205, 594–598. [Google Scholar] [CrossRef]
- Ribeiro, C.; Campelos, S.; Moura, C.S.; Machado, J.C.; Justino, A.; Parente, B. Well-differentiated papillary mesothelioma: Clustering in a Portuguese family with a germline BAP1 mutation. Ann. Oncol. 2013, 24, 2147–2150. [Google Scholar] [CrossRef]
- Lee, H.E.; Molina, J.R.; Sukov, W.R.; Roden, A.C.; Yi, E.S. BAP1 loss is unusual in well-differentiated papillary mesothelioma and may predict development of malignant mesothelioma. Hum. Pathol. 2018, 79, 168–176. [Google Scholar] [CrossRef]
- Stevers, M.; Rabban, J.T.; Garg, K.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Zaloudek, C.; Solomon, D.A. Well-differentiated papillary mesothelioma of the peritoneum is genetically defined by mutually exclusive mutations in TRAF7 and CDC42. Mod. Pathol. 2019, 32, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Müller-Hermelink, H.K.; Harris, C.C. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart; Travis, W.D., Brambilla, E., Müller-Hermelink, H.K., Harris, C.C., Eds.; WHO Press: Lyon, France, 2004; ISBN 92 832 2418 3. [Google Scholar]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [CrossRef]
- Rufini, A.; Agostini, M.; Grespi, F.; Tomasini, R.; Sayan, B.S.; Niklison-Chirou, M.V.; Conforti, F.; Velletri, T.; Mastino, A.; Mak, T.W.; et al. p73 in Cancer. Genes Cancer 2011, 2, 491–502. [Google Scholar] [CrossRef]
- Wang, X.; Gao, X.; Xu, Y. MAGED1: Molecular insights and clinical implications. Ann. Med. 2011, 43, 347–355. [Google Scholar] [CrossRef]
- Laitman, Y.; Boker-Keinan, L.; Berkenstadt, M.; Liphsitz, I.; Weissglas-Volkov, D.; Ries-Levavi, L.; Sarouk, I.; Pras, E.; Friedman, E. The risk for developing cancer in Israeli ATM, BLM, and FANCC heterozygous mutation carriers. Cancer Genet. 2016, 209, 70–74. [Google Scholar] [CrossRef]
- Huang, M.N.; Yu, W.; Teoh, W.W.; Ardin, M.; Jusakul, A.; Ng, A.W.T.; Boot, A.; Abedi-Ardekani, B.; Villar, S.; Myint, S.S.; et al. Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors. Genome Res. 2017, 27, 1475–1486. [Google Scholar] [CrossRef]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Chirac, P.; Maillet, D.; Leprêtre, F.; Isaac, S.; Glehen, O.; Figeac, M.; Villeneuve, L.; Péron, J.; Gibson, F.; Galateau-Sallé, F.; et al. Genomic copy number alterations in 33 malignant peritoneal mesothelioma analyzed by comparative genomic hybridization array. Hum. Pathol. 2016, 55, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F. Beyond E-cadherin: Roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yin, H.; Zhang, H.; Hu, J.; Lu, H.; Li, C.; Cao, M.; Yan, S.; Cai, L. NF-κB-driven improvement of EHD1 contributes to erlotinib resistance in EGFR-mutant lung cancers. Cell Death Dis. 2018, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, M.; Rui, L.; Yang, Y.; Ceribelli, M.; Tishbi, N.; Maurer, C.W.; Ranuncolo, S.M.; Zhao, H.; Xu, W.; Chan, W.-C.C.; et al. Related F-box proteins control cell death in Caenorhabditis elegans and human lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3943–3948. [Google Scholar] [CrossRef]
- Acuto, O.; Bartolo, V.D.; Michel, F. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat. Rev. Immunol. 2008, 8, 699–712. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Emi, M.; Hashimoto-Tamaoki, T.; Ohmuraya, M.; Sato, A.; Tsujimura, T.; Hasegawa, S.; Nakano, T.; Nasu, M.; Pastorino, S.; et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc. Natl. Acad. Sci. USA 2016, 113, 13432–13437. [Google Scholar] [CrossRef]
- Goode, B.; Joseph, N.M.; Stevers, M.; Van Ziffle, J.; Onodera, C.; Talevich, E.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Phillips, J.J.; et al. Adenomatoid tumors of the male and female genital tract are defined by TRAF7 mutations that drive aberrant NF-kB pathway activation. Mod. Pathol. 2018, 31, 660–673. [Google Scholar] [CrossRef]
- Sneddon, S.; Dick, I.; Lee, Y.C.G.; Musk, A.W. (Bill.; Patch, A.M.; Pearson, J.V.; Waddell, N.; Allcock, R.J.N.; Holt, R.A.; Robinson, B.W.S.; et al. Malignant cells from pleural fluids in malignant mesothelioma patients reveal novel mutations. Lung Cancer 2018, 119, 64–70. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, R.; Nabavi, N.; Volik, S.; Anderson, S.; Haegert, A.; McConeghy, B.; Sar, F.; Brahmbhatt, S.; Bell, R.; Le Bihan, S.; et al. Well-Differentiated Papillary Mesothelioma of the Peritoneum Is Genetically Distinct from Malignant Mesothelioma. Cancers 2020, 12, 1568. https://doi.org/10.3390/cancers12061568
Shrestha R, Nabavi N, Volik S, Anderson S, Haegert A, McConeghy B, Sar F, Brahmbhatt S, Bell R, Le Bihan S, et al. Well-Differentiated Papillary Mesothelioma of the Peritoneum Is Genetically Distinct from Malignant Mesothelioma. Cancers. 2020; 12(6):1568. https://doi.org/10.3390/cancers12061568
Chicago/Turabian StyleShrestha, Raunak, Noushin Nabavi, Stanislav Volik, Shawn Anderson, Anne Haegert, Brian McConeghy, Funda Sar, Sonal Brahmbhatt, Robert Bell, Stephane Le Bihan, and et al. 2020. "Well-Differentiated Papillary Mesothelioma of the Peritoneum Is Genetically Distinct from Malignant Mesothelioma" Cancers 12, no. 6: 1568. https://doi.org/10.3390/cancers12061568
APA StyleShrestha, R., Nabavi, N., Volik, S., Anderson, S., Haegert, A., McConeghy, B., Sar, F., Brahmbhatt, S., Bell, R., Le Bihan, S., Wang, Y., Collins, C., & Churg, A. (2020). Well-Differentiated Papillary Mesothelioma of the Peritoneum Is Genetically Distinct from Malignant Mesothelioma. Cancers, 12(6), 1568. https://doi.org/10.3390/cancers12061568