A Driver Never Works Alone—Interplay Networks of Mutant p53, MYC, RAS, and Other Universal Oncogenic Drivers in Human Cancer

 ,
,
Abstract
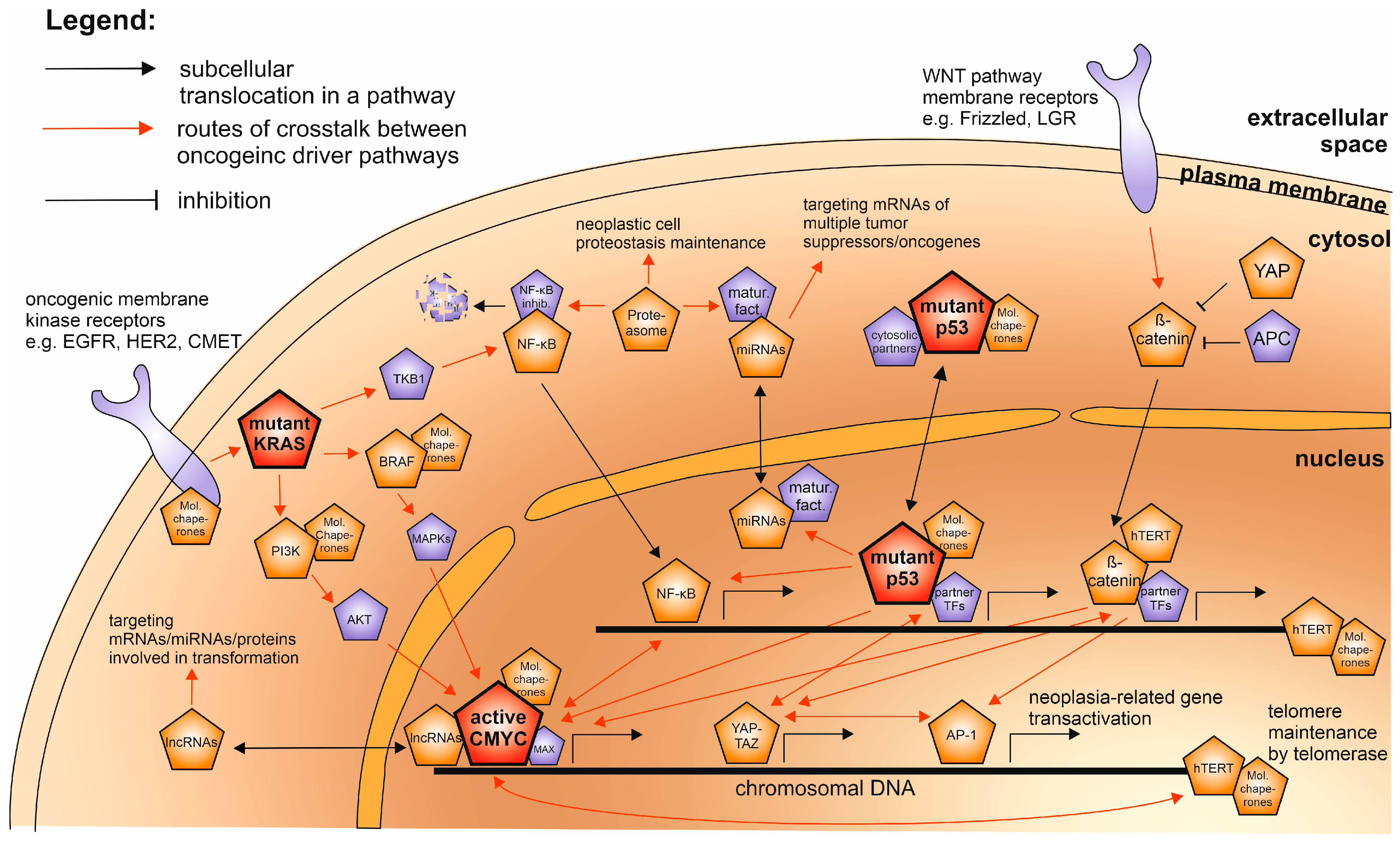
1. Oncogene Cooperation in a Game of Multistep Carcinogenesis
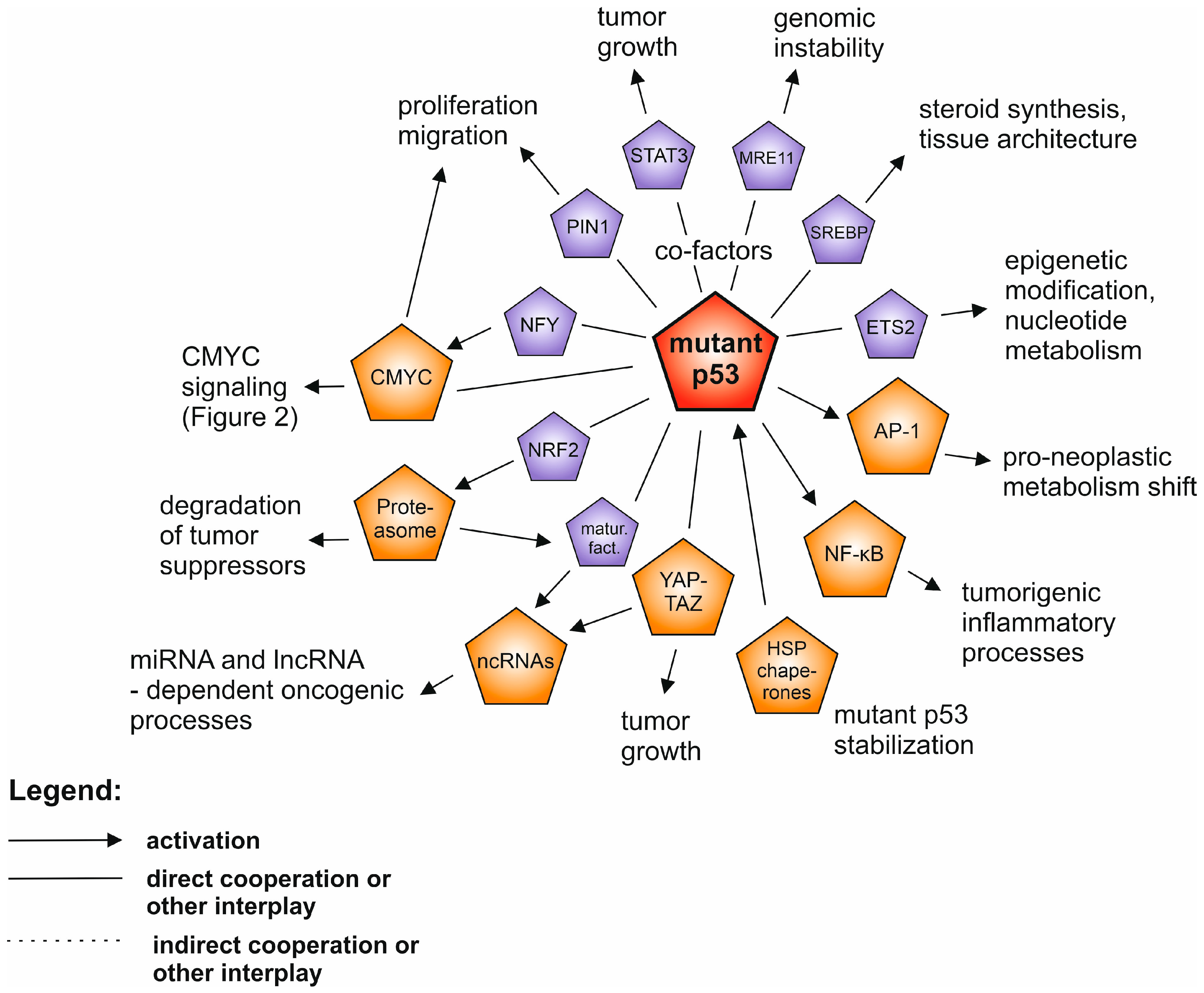
2. Mutant p53 Gain-of-Function
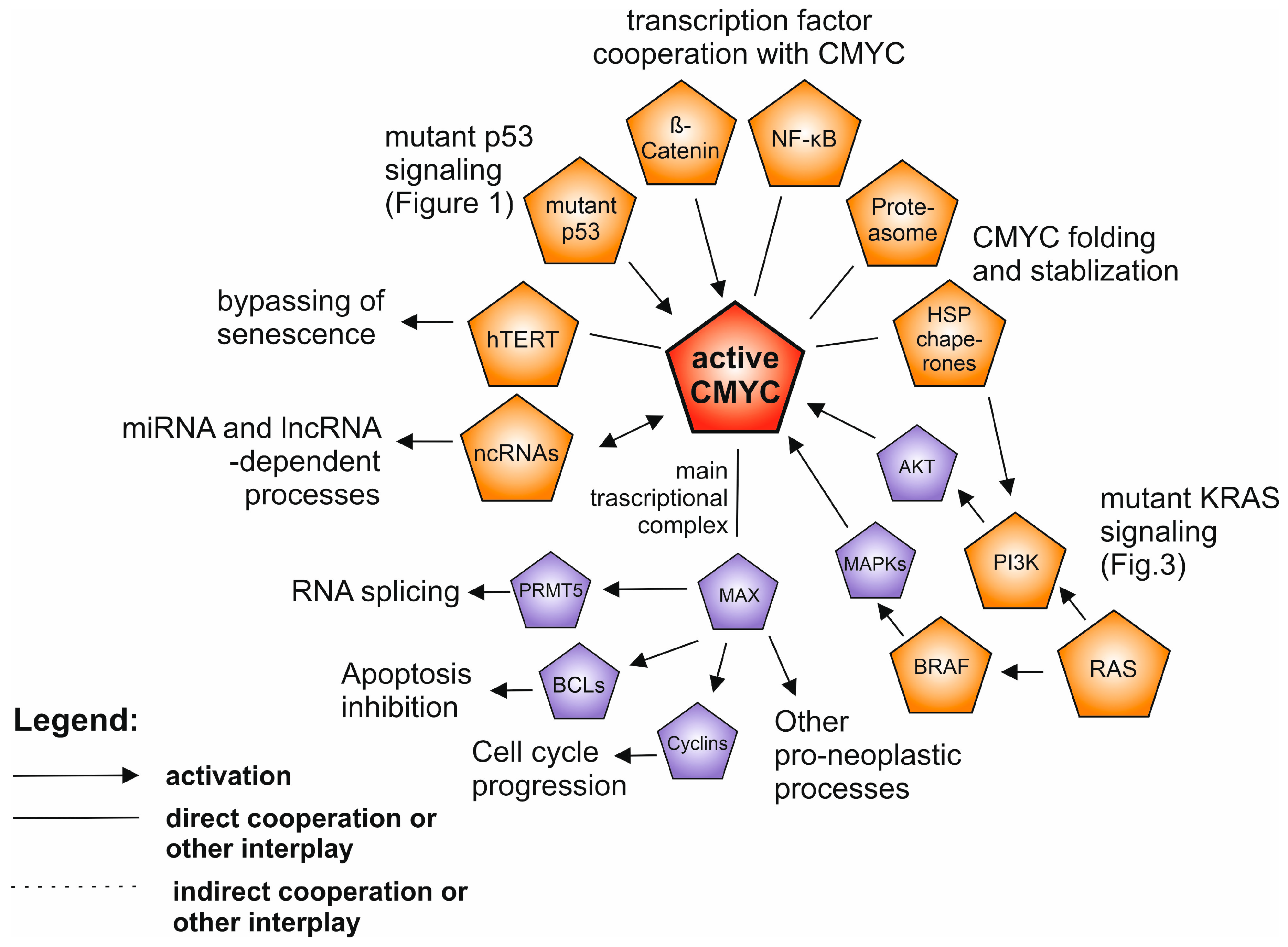
3. CMYC and MYC Family of Proteins
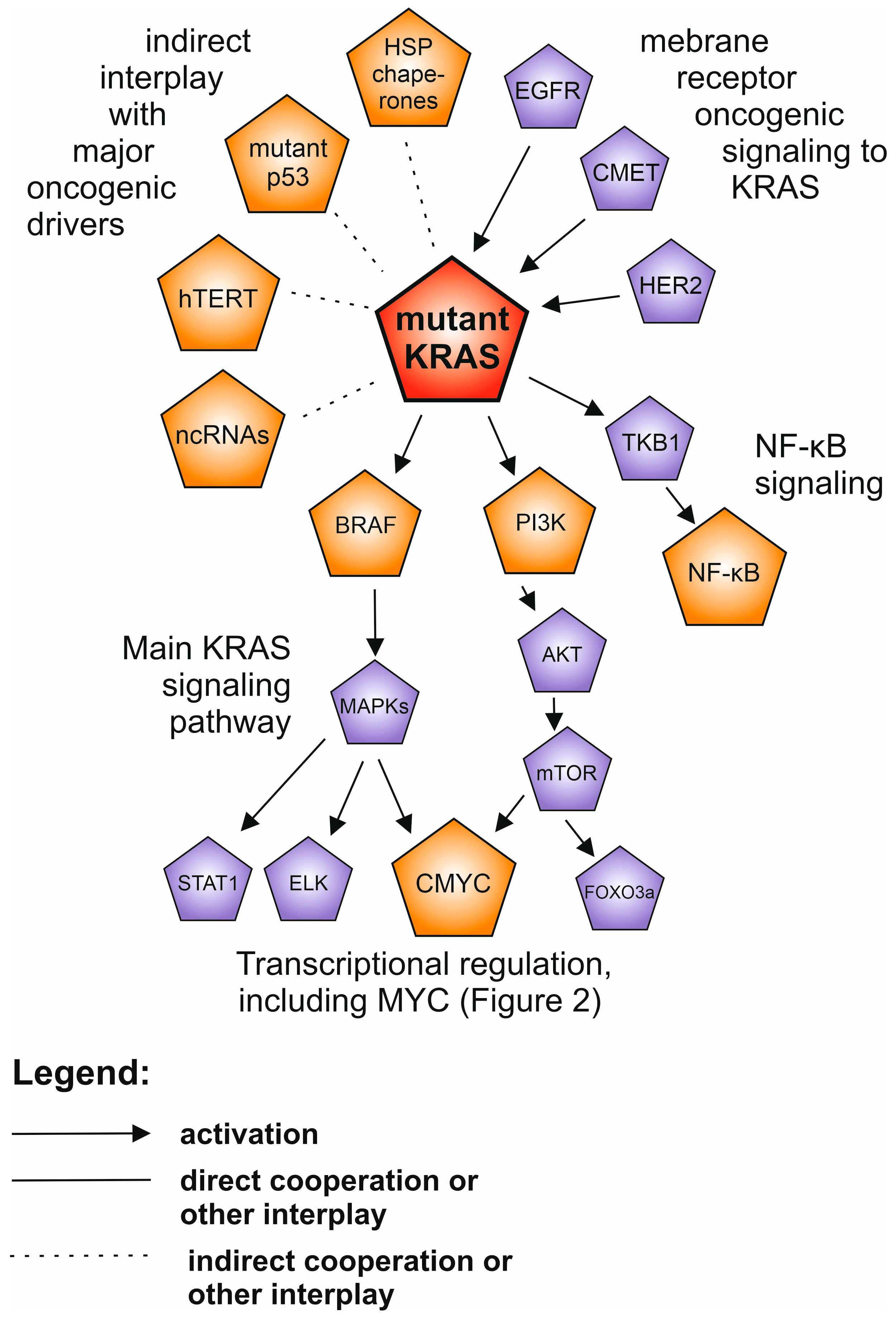
4. KRAS and RAS Family of Proteins
5. PI3K and BRAF
6. Telomerase
7. Cellular Proteasome Machinery
8. HSP Molecular Chaperones
9. NF-κB
10. AP-1—FOS/JUN
11. β-catenin and the WNT Pathway
12. YAP/TAZ
13. Non-coding RNAs (Micro RNAs and Long Non-codong RNAs)
14. Exploitation of the Oncogene Cooperation in Clinics
15. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- ICGC Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Weinberg, R.A. Rules for making human tumor cells. N. Engl. J. Med. 2002, 347, 1593–1603. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, A.; Hong, S.J.; Gifford, A.; Weinberg, R.A. Species—And cell type-specific requirements for cellular transformation. Cancer Cell 2004, 6, 171–183. [Google Scholar] [CrossRef]
- Zhang, F.; Zakaria, S.M.; Hogqvist Tabor, V.; Singh, M.; Tronnersjo, S.; Goodwin, J.; Selivanova, G.; Bartek, J.; Castell, A.; Larsson, L.G. MYC and RAS are unable to cooperate in overcoming cellular senescence and apoptosis in normal human fibroblasts. Cell Cycle 2018, 17, 2697–2715. [Google Scholar] [CrossRef]
- Kress, T.R.; Sabo, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC deregulation in primary human cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Narisawa-Saito, M.; Yoshimatsu, Y.; Ohno, S.; Yugawa, T.; Egawa, N.; Fujita, M.; Hirohashi, S.; Kiyono, T. An in vitro multistep carcinogenesis model for human cervical cancer. Cancer Res. 2008, 68, 5699–5705. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Pruszko, M.; Zyla, L.; Wezyk, M.; Gaweda-Walerych, K.; Zylicz, A. Wild-type p53 oligomerizes more efficiently than p53 hot-spot mutants and overcomes mutant p53 gain-of-function via a “dominant-positive” mechanism. Oncotarget 2018, 9, 32063–32080. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-function mutant p53: All the roads lead to tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef]
- Seger, Y.R.; Garcia-Cao, M.; Piccinin, S.; Cunsolo, C.L.; Doglioni, C.; Blasco, M.A.; Hannon, G.J.; Maestro, R. Transformation of normal human cells in the absence of telomerase activation. Cancer Cell 2002, 2, 401–413. [Google Scholar] [CrossRef]
- Boehm, J.S.; Hession, M.T.; Bulmer, S.E.; Hahn, W.C. Transformation of human and murine fibroblasts without viral oncoproteins. Mol. Cell. Biol. 2005, 25, 6464–6474. [Google Scholar] [CrossRef]
- Kendall, S.D.; Linardic, C.M.; Adam, S.J.; Counter, C.M. A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res. 2005, 65, 9824–9828. [Google Scholar] [CrossRef]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. MCR 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Olszewski, M.B.; Gutkowska, M.; Helwak, A.; Zylicz, M.; Zylicz, A. Hsp70 molecular chaperones are required to support p53 tumor suppressor activity under stress conditions. Oncogene 2009, 28, 4284–4294. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell. Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef]
- Stindt, M.H.; Muller, P.A.; Ludwig, R.L.; Kehrloesser, S.; Dotsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015, 34, 4300–4310. [Google Scholar] [CrossRef]
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant p53 gains its function via c-Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Mol. Cell 2017, 68, 1134–1146. [Google Scholar] [CrossRef]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. Embo. Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, Y.; Tang, Y.; Butler, N.; Kim, J.; Guessous, F.; Schiff, D.; Mandell, J.; Abounader, R. A novel PTEN/mutant p53/c-Myc/Bcl-XL axis mediates context-dependent oncogenic effects of PTEN with implications for cancer prognosis and therapy. Neoplasia 2013, 15, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Matas, D.; Sigal, A.; Stambolsky, P.; Milyavsky, M.; Weisz, L.; Schwartz, D.; Goldfinger, N.; Rotter, V. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. Embo. J. 2001, 20, 4163–4172. [Google Scholar] [CrossRef]
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015, 6, 7389. [Google Scholar] [CrossRef]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef]
- Weisz, L.; Damalas, A.; Liontos, M.; Karakaidos, P.; Fontemaggi, G.; Maor-Aloni, R.; Kalis, M.; Levrero, M.; Strano, S.; Gorgoulis, V.G.; et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007, 67, 2396–2401. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits STAT3-mediated tumor growth and invasion. Cancer Cell 2018, 34, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Lisek, K.; Del Sal, G. Mutant p53: One, no one, and one hundred thousand. Front. Oncol. 2015, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hollstein, M.; Xu, Y. P53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi, F.; Falcone, E.; Trisciuoglio, D.; Colombo, T.; Lisek, K.; Walerych, D.; Del Sal, G.; Paci, P.; Bossi, G.; Piaggio, G.; et al. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex. Oncogene 2016, 35, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 2014, 56, 617–629. [Google Scholar] [CrossRef]
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Sal, G.; et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 2019, 38, 6184–6195. [Google Scholar] [CrossRef]
- Hashimoto, S.; Furukawa, S.; Hashimoto, A.; Tsutaho, A.; Fukao, A.; Sakamura, Y.; Parajuli, G.; Onodera, Y.; Otsuka, Y.; Handa, H.; et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 17450–17459. [Google Scholar] [CrossRef]
- Bailey, J.M.; Hendley, A.M.; Lafaro, K.J.; Pruski, M.A.; Jones, N.C.; Alsina, J.; Younes, M.; Maitra, A.; McAllister, F.; Iacobuzio-Donahue, C.A.; et al. P53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 2016, 35, 4282–4288. [Google Scholar] [CrossRef]
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. P53 loss in breast cancer leads to Myc activation, increased cell plasticity, and expression of a mitotic signature with prognostic value. Cell Rep. 2019, 26, 624–638. [Google Scholar] [CrossRef]
- Munoz, D.M.; Tung, T.; Agnihotri, S.; Singh, S.; Guha, A.; Zadeh, G.; Hawkins, C. Loss of p53 cooperates with K-ras activation to induce glioma formation in a region-independent manner. Glia 2013, 61, 1862–1872. [Google Scholar] [CrossRef]
- Stampfer, M.R.; Garbe, J.; Nijjar, T.; Wigington, D.; Swisshelm, K.; Yaswen, P. Loss of p53 function accelerates acquisition of telomerase activity in indefinite lifespan human mammary epithelial cell lines. Oncogene 2003, 22, 5238–5251. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Groudine, M. Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 1991, 56, 1–48. [Google Scholar] [PubMed]
- Sears, R.C. The life cycle of C-myc: From synthesis to degradation. Cell Cycle 2004, 3, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Xu-Monette, Z.Y.; Deng, Q.; Manyam, G.C.; Tzankov, A.; Li, L.; Xia, Y.; Wang, X.X.; Zou, D.; Visco, C.; Dybkaer, K.; et al. Clinical and biologic significance of MYC genetic mutations in de novo diffuse large B-cell lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3593–3605. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer alterations of the MYC oncogene and its proximal network across the cancer genome atlas. Cell Syst. 2018, 6, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef]
- Yagi, K.; Furuhashi, M.; Aoki, H.; Goto, D.; Kuwano, H.; Sugamura, K.; Miyazono, K.; Kato, M. C-MYC is a downstream target of the Smad pathway. J. Biol. Chem. 2002, 277, 854–861. [Google Scholar] [CrossRef]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Lim, C.; Chu, G.; Hua, S.; Ding, Z.; Collins, M.; Hu, J.; Jiang, S.; Fletcher-Sananikone, E.; Zhuang, L.; et al. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell 2010, 18, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Notari, M.; Neviani, P.; Santhanam, R.; Blaser, B.W.; Chang, J.S.; Galietta, A.; Willis, A.E.; Roy, D.C.; Caligiuri, M.A.; Marcucci, G.; et al. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood 2006, 107, 2507–2516. [Google Scholar] [CrossRef]
- Tsai, W.B.; Aiba, I.; Long, Y.; Lin, H.K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, I. Lncrnas and MYC: An intricate relationship. Int. J. Mol. Sci. 2017, 18, 1497. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Yu, Q.; Ciemerych, M.A.; Sicinski, P. Ras and Myc can drive oncogenic cell proliferation through individual D-cyclins. Oncogene 2005, 24, 7114–7119. [Google Scholar] [CrossRef] [PubMed]
- He, T.L.; Zhang, Y.J.; Jiang, H.; Li, X.H.; Zhu, H.; Zheng, K.L. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med. Oncol. 2015, 32, 187. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cetinkaya, C.; Munoz-Alonso, M.J.; von der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; Leon, J.; Larsson, L.G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Woo, D.; Roussel, M.F.; Cleveland, J.L. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 2001, 21, 5063–5070. [Google Scholar] [CrossRef]
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabo, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-MYC. Oncogene 2008, 27, 6462–6472. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Pellanda, P.; Pellegrinet, L.; Bianchi, V.; Nicoli, P.; Doni, M.; Recordati, C.; Bianchi, S.; Rotta, L.; Capra, T.; et al. Identification of MYC-dependent transcriptional programs in oncogene-addicted liver tumors. Cancer Res. 2016, 76, 3463–3472. [Google Scholar] [CrossRef]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.; Matallanas, D.; Kolch, W. The spatiotemporal regulation of RAS signalling. Biochem. Soc. Trans. 2016, 44, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Grieco, M.; Tsuchida, N. RAS mutations in human cancers: Roles in precision medicine. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the "undruggable". Biochim. Et Biophys. Acta Mol. Cell Res. 2019, 118570. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. RAS interaction with PI3K: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yin, Y.; Wang, J.; Shi, B.; Zhang, L.; Qian, D.; Li, C.; Zhang, H.; Wang, S.; Zhu, J.; et al. Kras mutations increase telomerase activity and targeting telomerase is a promising therapeutic strategy for Kras-mutant NSCLC. Oncotarget 2017, 8, 179–190. [Google Scholar] [CrossRef]
- Frezzetti, D.; De Menna, M.; Zoppoli, P.; Guerra, C.; Ferraro, A.; Bello, A.M.; De Luca, P.; Calabrese, C.; Fusco, A.; Ceccarelli, M.; et al. Upregulation of miR-21 by Ras in vivo and its role in tumor growth. Oncogene 2011, 30, 275–286. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, F.; Rong, W.; Zeng, Q.; Sun, W. Positive feedback between oncogenic KRAS and HIF-1alpha confers drug resistance in colorectal cancer. OncoTargets Ther. 2015, 8, 1229–1237. [Google Scholar]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc cooperates with ras by programming inflammation and immune suppression. Cell 2017, 171, 1301–1315. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS suppression-induced degradation of MYC is antagonized by a MEK5-ERK5 compensatory mechanism. Cancer Cell 2018, 34, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C inhibitor, MRTX849, provides insight toward therapeutic susceptibility of KRAS mutant cancers in mouse models and patients. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Drilon, A. KRAS-mutant lung cancers in the era of targeted therapy. Adv. Exp. Med. Biol. 2016, 893, 155–178. [Google Scholar]
- Arafeh, R.; Samuels, Y. PIK3CA in cancer: The past 30 years. Semin. Cancer Biol. 2019, 59, 36–49. [Google Scholar] [CrossRef]
- Kang, S.; Bader, A.G.; Vogt, P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA 2005, 102, 802–807. [Google Scholar] [CrossRef]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting oncogenic BRAF: Past, present, and future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Mocellin, S.; Pooley, K.A.; Nitti, D. Telomerase and the search for the end of cancer. Trends Mol. Med. 2013, 19, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.P.; Holt, S.E.; Ouellette, M.; Kaur, K.J.; Yan, Y.; Wilson, K.S.; White, M.A.; Wright, W.E.; Shay, J.W. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat. Genet. 1999, 21, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The longest telomeres: A general signature of adult stem cell compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Casillas, M.A.; Brotherton, S.L.; Andrews, L.G.; Ruppert, J.M.; Tollefsbol, T.O. Induction of endogenous telomerase (hTERT) by c-Myc in WI-38 fibroblasts transformed with specific genetic elements. Gene 2003, 316, 57–65. [Google Scholar] [CrossRef]
- Gil, J.; Kerai, P.; Lleonart, M.; Bernard, D.; Cigudosa, J.C.; Peters, G.; Carnero, A.; Beach, D. Immortalization of primary human prostate epithelial cells by c-Myc. Cancer Res. 2005, 65, 2179–2185. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, L.; Li, Q.; Li, W.; Bjorkholm, M.; Jia, J.; Xu, D. FoxM1 is up-regulated in gastric cancer and its inhibition leads to cellular senescence, partially dependent on p27 kip1. J. Pathol. 2009, 218, 419–427. [Google Scholar] [CrossRef]
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.P. Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J. Biol. Chem. 2008, 283, 23567–23580. [Google Scholar] [CrossRef]
- Liu, C.; Fang, X.; Ge, Z.; Jalink, M.; Kyo, S.; Bjorkholm, M.; Gruber, A.; Sjoberg, J.; Xu, D. The telomerase reverse transcriptase (hTERT) gene is a direct target of the histone methyltransferase SMYD3. Cancer Res. 2007, 67, 2626–2631. [Google Scholar] [CrossRef]
- Liu, T.; Yuan, X.; Xu, D. Cancer-specific telomerase reverse transcriptase (TERT) promoter mutations: Biological and clinical implications. Genes 2016, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Blasco, M.A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11, 161–176. [Google Scholar] [CrossRef]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Van Houten, B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum. Mol. Genet. 2006, 15, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2017, 14, 115–128. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Pickett, H.A. Alternative lengthening of telomeres: DNA repair pathways converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef]
- Chen, L.; Madura, K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005, 65, 5599–5606. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Pathogenesis of colorectal carcinoma and therapeutic implications: The roles of the ubiquitin-proteasome system and Cox-2. J. Cell. Mol. Med. 2007, 11, 252–285. [Google Scholar] [CrossRef]
- Spirina, L.V.; Kondakova, I.V.; Choynzonov, E.L.; Chigevskaya, S.Y.; Shishkin, D.A.; Kulbakin, D.Y. Expression of vascular endothelial growth factor and transcription factors HIF-1, NF-kB expression in squamous cell carcinoma of head and neck; association with proteasome and calpain activities. J. Cancer Res. Clin. Oncol. 2013, 139, 625–633. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Y.; Guo, X. Proteasome dysregulation in human cancer: Implications for clinical therapies. Cancer Metastasis Rev. 2017, 36, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Edkins, A.L. CHIP: A co-chaperone for degradation by the proteasome. Subcell Biochem. 2015, 78, 219–242. [Google Scholar]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.L.; Schreiber, S.; Schafer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Vangala, J.R.; Dudem, S.; Jain, N.; Kalivendi, S.V. Regulation of PSMB5 protein and beta subunits of mammalian proteasome by constitutively activated signal transducer and activator of transcription 3 (STAT3): Potential role in bortezomib-mediated anticancer therapy. J. Biol. Chem. 2014, 289, 12612–12622. [Google Scholar] [CrossRef]
- Xu, H.; Fu, J.; Ha, S.W.; Ju, D.; Zheng, J.; Li, L.; Xie, Y. The CCAAT box-binding transcription factor NF-Y regulates basal expression of human proteasome genes. Biochim. Et Biophys. Acta 2012, 1823, 818–825. [Google Scholar] [CrossRef][Green Version]
- Tsvetkov, P.; Adler, J.; Myers, N.; Biran, A.; Reuven, N.; Shaul, Y. Oncogenic addiction to high 26S proteasome level. Cell Death Dis. 2018, 9, 773. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010, 17, 103–108. [Google Scholar] [CrossRef]
- He, J.; Cui, L.; Zeng, Y.; Wang, G.; Zhou, P.; Yang, Y.; Ji, L.; Zhao, Y.; Chen, J.; Wang, Z.; et al. REGgamma is associated with multiple oncogenic pathways in human cancers. BMC Cancer 2012, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Wang, Z.; Fu, J.; Ji, L.; Liu, J.; Li, L.; Wang, H.; Chen, J.; Caulin, C.; Myers, J.N.; et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat. Commun. 2013, 4, 2667. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, X.; Yu, T.; Yuan, L.; Dai, J.; Wang, W.; Chen, G.; Jiao, C.; Zhou, W.; Huang, Q.; et al. REGgamma controls hippo signaling and reciprocal NF-kappaB-YAP regulation to promote colon cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dang, Y.; Zhang, J.; Yan, W.; Zhai, W.; Chen, H.; Li, K.; Tong, L.; Gao, X.; Amjad, A.; et al. REGgamma is critical for skin carcinogenesis by modulating the Wnt/beta-catenin pathway. Nat. Commun. 2015, 6, 6875. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Hao, J.; Jiang, H.; Jin, J.; Wu, H.; Jin, Z.; Li, Z. Proteasome activator subunit 3 promotes pancreatic cancer growth via c-Myc-glycolysis signaling axis. Cancer Lett. 2017, 386, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, C.; Pan, J.; Wang, X.; Jin, J.; Zhao, L.; Pan, W.; Liao, G.; Cai, X.; Li, X.; et al. Regulation of c-Myc protein stability by proteasome activator REGgamma. Cell Death Differ. 2015, 22, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Murray, M.Y.; Auger, M.J.; Bowles, K.M. Overcoming bortezomib resistance in multiple myeloma. Biochem. Soc. Trans. 2014, 42, 804–808. [Google Scholar] [CrossRef]
- Belalcazar, A.; Shaib, W.L.; Farren, M.R.; Zhang, C.; Chen, Z.; Yang, L.; Lesinski, G.B.; El-Rayes, B.F.; Nagaraju, G.P. Inhibiting heat shock protein 90 and the ubiquitin-proteasome pathway impairs metabolic homeostasis and leads to cell death in human pancreatic cancer cells. Cancer 2017, 123, 4924–4933. [Google Scholar] [CrossRef]
- Li, X.; Liang, M.; Jiang, J.; He, R.; Wang, M.; Guo, X.; Shen, M.; Qin, R. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int. J. Biol. Sci. 2018, 14, 1291–1305. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 synergistically enhances bortezomib-induced cytotoxicity in multiple myeloma. Cell Stress Chaperones 2020, 25, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.J.; Guerrero-Gimenez, M.E.; Prince, T.L.; Ackerman, A.; Bonorino, C.; Calderwood, S.K. Heat shock proteins are essential components in transformation and tumor progression: Cancer cell intrinsic pathways and beyond. Int. J. Mol. Sci. 2019, 20, 4507. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef]
- Liu, X.; Yan, Z.; Huang, L.; Guo, M.; Zhang, Z.; Guo, C. Cell surface heat shock protein 90 modulates prostate cancer cell adhesion and invasion through the integrin-beta1/focal adhesion kinase/c-Src signaling pathway. Oncol. Rep. 2011, 25, 1343–1351. [Google Scholar]
- Shimamura, T.; Li, D.; Ji, H.; Haringsma, H.J.; Liniker, E.; Borgman, C.L.; Lowell, A.M.; Minami, Y.; McNamara, K.; Perera, S.A.; et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008, 68, 5827–5838. [Google Scholar] [CrossRef]
- Toogun, O.A.; Dezwaan, D.C.; Freeman, B.C. The hsp90 molecular chaperone modulates multiple telomerase activities. Mol. Cell. Biol. 2008, 28, 457–467. [Google Scholar] [CrossRef]
- Azoitei, N.; Hoffmann, C.M.; Ellegast, J.M.; Ball, C.R.; Obermayer, K.; Gossele, U.; Koch, B.; Faber, K.; Genze, F.; Schrader, M.; et al. Targeting of KRAS mutant tumors by HSP90 inhibitors involves degradation of STK33. J. Exp. Med. 2012, 209, 697–711. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Huang, S.; Zhang, H.; Alikhan, M.; et al. Activation of MYC, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef]
- Poole, C.J.; Zheng, W.; Lee, H.; Young, D.; Lodh, A.; Chadli, A.; van Riggelen, J. Targeting the MYC oncogene in burkitt lymphoma through HSP90 inhibition. Cancers 2018, 10, 448. [Google Scholar] [CrossRef] [PubMed]
- Tatokoro, M.; Koga, F.; Yoshida, S.; Kihara, K. Heat shock protein 90 targeting therapy: State of the art and future perspective. Excli. J. 2015, 14, 48–58. [Google Scholar] [PubMed]
- Kumar, S.; Stokes, J.; Singh, U.P.; Scissum Gunn, K.; Acharya, A.; Manne, U.; Mishra, M. Targeting Hsp70: A possible therapy for cancer. Cancer Lett. 2016, 374, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kijima, T.; Prince, T.; Neckers, L.; Koga, F.; Fujii, Y. Heat shock factor 1 (HSF1)-targeted anticancer therapeutics: Overview of current preclinical progress. Expert. Opin. Ther. Targets 2019, 23, 369–377. [Google Scholar] [CrossRef]
- McConnell, J.R.; McAlpine, S.R. Heat shock proteins 27, 40, and 70 as combinational and dual therapeutic cancer targets. Bioorg Med. Chem. Lett. 2013, 23, 1923–1928. [Google Scholar] [CrossRef]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 chaperone axis as a novel strategy to treat castration-resistant prostate cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene 2015, 34, 3305–3314. [Google Scholar] [CrossRef]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-kappaB addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef]
- Zavrski, I.; Jakob, C.; Kaiser, M.; Fleissner, C.; Heider, U.; Sezer, O. Molecular and clinical aspects of proteasome inhibition in the treatment of cancer. Recent Results Cancer Res. 2007, 176, 165–176. [Google Scholar]
- Moorthy, A.K.; Savinova, O.V.; Ho, J.Q.; Wang, V.Y.; Vu, D.; Ghosh, G. The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J. 2006, 25, 1945–1956. [Google Scholar] [CrossRef]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, Y.; Li, Y.; Zhou, J.; Wu, Y.; Cui, Y.; Yang, G.; Hong, Y. Mutations of p53 and KRAS activate NF-kappaB to promote chemoresistance and tumorigenesis via dysregulation of cell cycle and suppression of apoptosis in lung cancer cells. Cancer Lett. 2015, 357, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Arnaud, N.; Fradet, M.; Lascaux, H.; Ouk-Martin, C.; Gachard, N.; Zimber-Strobl, U.; Feuillard, J.; Faumont, N. c-Myc dysregulation is a co-transforming event for nuclear factor-kappaB activated B cells. Haematologica 2017, 102, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, V.; Athanassiou, E.; Dubos, S.; Dimou, I.; Papathanasiou, I.; Kitsiou-Tzeli, S.; Kappas, C.; Tsezou, A. HTERT regulation by NF-kappaB and c-myc in irradiated HER2-positive breast cancer cells. Int. J. Radiat Biol. 2011, 87, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K. Fortuitous convergences: The beginnings of JUN. Nat. Rev. Cancer 2002, 2, 465–469. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim.Et Biophys. Acta Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Grigoriadis, A.E.; Schellander, K.; Wang, Z.Q.; Wagner, E.F. Osteoblasts are target cells for transformation in c-fos transgenic mice. J. Cell Biol. 1993, 122, 685–701. [Google Scholar] [CrossRef]
- Young, M.R.; Li, J.J.; Rincon, M.; Flavell, R.A.; Sathyanarayana, B.K.; Hunziker, R.; Colburn, N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc. Natl. Acad. Sci. USA 1999, 96, 9827–9832. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Binetruy, B.; Smeal, T.; Karin, M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature 1991, 351, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Mechta, F.; Lallemand, D.; Pfarr, C.M.; Yaniv, M. Transformation by ras modifies AP1 composition and activity. Oncogene 1997, 14, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Treinies, I.; Paterson, H.F.; Hooper, S.; Wilson, R.; Marshall, C.J. Activated MEK stimulates expression of AP-1 components independently of phosphatidylinositol 3-kinase (PI3-kinase) but requires a PI3-kinase signal to stimulate DNA synthesis. Mol. Cell. Biol. 1999, 19, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Behrens, A.; Sibilia, M.; Wagner, E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999, 21, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dorken, B.; et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef]
- Senapati, P.; Dey, S.; Sudarshan, D.; Das, S.; Kumar, M.; Kaypee, S.; Mohiyuddin, A.; Kodaganur, G.S.; Kundu, T.K. Oncogene c-fos and mutant R175H p53 regulate expression of nucleophosmin implicating cancer manifestation. FEBS J. 2018, 285, 3503–3524. [Google Scholar] [CrossRef]
- Hennigan, R.F.; Hawker, K.L.; Ozanne, B.W. Fos-transformation activates genes associated with invasion. Oncogene 1994, 9, 3591–3600. [Google Scholar]
- Zenz, R.; Scheuch, H.; Martin, P.; Frank, C.; Eferl, R.; Kenner, L.; Sibilia, M.; Wagner, E.F. C-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev. Cell 2003, 4, 879–889. [Google Scholar] [CrossRef]
- Talotta, F.; Cimmino, A.; Matarazzo, M.R.; Casalino, L.; De Vita, G.; D’Esposito, M.; Di Lauro, R.; Verde, P. An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in RAS transformation. Oncogene 2009, 28, 73–84. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; Ferris, M.W.; Hull, M.A.; Chae, H.; Kim, S.; Graves, B.J. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011, 25, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Kamide, D.; Yamashita, T.; Araki, K.; Tomifuji, M.; Tanaka, Y.; Tanaka, S.; Shiozawa, S.; Shiotani, A. Selective activator protein-1 inhibitor T-5224 prevents lymph node metastasis in an oral cancer model. Cancer Sci. 2016, 107, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.K.; Kim, M.P. SR 11302, an AP-1 inhibitor, reduces metastatic lesion formation in Ex Vivo 4D lung cancer model. Cancer Microenviron 2017, 10, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Grainger, S.; Traver, D.; Willert, K. Wnt signaling in hematological malignancies. Prog. Mol. Biol. Transl. Sci. 2018, 153, 321–341. [Google Scholar] [PubMed]
- Kaur, A.; Webster, M.R.; Weeraratna, A.T. In the Wnt-er of life: Wnt signalling in melanoma and ageing. Br. J. Cancer 2016, 115, 1273–1279. [Google Scholar] [CrossRef]
- Yin, P.; Wang, W.; Zhang, Z.; Bai, Y.; Gao, J.; Zhao, C. Wnt signaling in human and mouse breast cancer: Focusing on Wnt ligands, receptors and antagonists. Cancer Sci. 2018, 109, 3368–3375. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Hu, T.; Li, C. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol. Cancer 2010, 9, 236. [Google Scholar] [CrossRef]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef]
- Lien, W.H.; Fuchs, E. Wnt some lose some: Transcriptional governance of stem cells by Wnt/beta-catenin signaling. Genes Dev. 2014, 28, 1517–1532. [Google Scholar] [CrossRef]
- Jung, H.Y.; Jun, S.; Lee, M.; Kim, H.C.; Wang, X.; Ji, H.; McCrea, P.D.; Park, J.I. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol. Cell 2013, 52, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Alaee, M.; Nool, K.; Pasdar, M. Plakoglobin restores tumor suppressor activity of p53(R175H) mutant by sequestering the oncogenic potential of beta-catenin. Cancer Sci. 2018, 109, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, E.; Cagnol, S.; Beaudry, K.; Carrier, J.; Rivard, N. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene 2015, 34, 4914–4927. [Google Scholar] [CrossRef]
- Prowald, A.; Cronauer, M.V.; von Klot, C.; Eilers, T.; Rinnab, L.; Herrmann, T.; Spindler, K.D.; Montenarh, M.; Jonas, U.; Burchardt, M. Modulation of beta-catenin-mediated TCF-signalling in prostate cancer cell lines by wild-type and mutant p53. Prostate 2007, 67, 1751–1760. [Google Scholar] [CrossRef]
- Morris, J.P.t.; Cano, D.A.; Sekine, S.; Wang, S.C.; Hebrok, M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Investig. 2010, 120, 508–520. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.; Gelos, M.; Siedow, A.; Hanski, M.L.; Gratchev, A.; Ilyas, M.; Bodmer, W.F.; Moyer, M.P.; Riecken, E.O.; Buhr, H.J.; et al. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc. Natl. Acad. Sci. USA 1999, 96, 1603–1608. [Google Scholar] [CrossRef]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Oishi, I.; Suzuki, H.; Onishi, N.; Takada, R.; Kani, S.; Ohkawara, B.; Koshida, I.; Suzuki, K.; Yamada, G.; Schwabe, G.C.; et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 2003, 8, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Nomachi, A.; Nishita, M.; Inaba, D.; Enomoto, M.; Hamasaki, M.; Minami, Y. Receptor tyrosine kinase Ror2 mediates Wnt5a-induced polarized cell migration by activating c-Jun N-terminal kinase via actin-binding protein filamin A. J. Biol. Chem. 2008, 283, 27973–27981. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.L.; Hughes, C.C. Endothelial cells stimulate T cell NFAT nuclear translocation in the presence of cyclosporin A: Involvement of the wnt/glycogen synthase kinase-3 beta pathway. J. Immunol. 2002, 169, 3717–3725. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, H.Y. Mitogen-activated protein kinase p38 regulates the Wnt/cyclic GMP/Ca2+ non-canonical pathway. J. Biol. Chem. 2007, 282, 28980–28990. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, H.; Wang, S.; Hao, X.; Li, L. A diterpenoid derivative 15-oxospiramilactone inhibits Wnt/beta-catenin signaling and colon cancer cell tumorigenesis. Cell Res. 2011, 21, 730–740. [Google Scholar] [CrossRef]
- Lenz, H.J.; Kahn, M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014, 105, 1087–1092. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the drosophila homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Toyoda, T.; Nakanishi, H.; Yatabe, Y.; Sato, A.; Matsudaira, Y.; Ito, H.; Murakami, H.; Kondo, Y.; Kondo, E.; et al. TGF-beta synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J. Exp. Med. 2012, 209, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef]
- Croci, O.; De Fazio, S.; Biagioni, F.; Donato, E.; Caganova, M.; Curti, L.; Doni, M.; Sberna, S.; Aldeghi, D.; Biancotto, C.; et al. Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 2017, 31, 2017–2022. [Google Scholar] [CrossRef]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The role of non-coding RNAs in oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Javanmardi, S.; Aghamaali, M.R.; Abolmaali, S.S.; Mohammadi, S.; Tamaddon, A.M. mir-21, an oncogenic target miRNA for cancer therapy: Molecular mechanisms and recent advancements in chemo and radio-resistance. Curr. Gene Ther. 2017, 16, 375–389. [Google Scholar] [CrossRef]
- Wu, Y.R.; Qi, H.J.; Deng, D.F.; Luo, Y.Y.; Yang, S.L. MicroRNA-21 promotes cell proliferation, migration, and resistance to apoptosis through PTEN/PI3K/AKT signaling pathway in esophageal cancer. Tumour Biol. 2016, 37, 12061–12070. [Google Scholar] [CrossRef]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Li, F.; Li, X.; Tian, Y.; Zhang, Y.; Sheng, X.; Song, Y.; Meng, Q.; Yuan, S.; Luan, L.; et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 2017, 8, 1036. [Google Scholar] [CrossRef] [PubMed]
- Mazzarella, G.; MacDonald, T.T.; Salvati, V.M.; Mulligan, P.; Pasquale, L.; Stefanile, R.; Lionetti, P.; Auricchio, S.; Pallone, F.; Troncone, R.; et al. Constitutive activation of the signal transducer and activator of transcription pathway in celiac disease lesions. Am. J. Pathol. 2003, 162, 1845–1855. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, L.F.; Zhang, H.W.; Hu, S.; Lu, M.H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.D.; et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef]
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2013, 32, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Ganci, F.; Sacconi, A.; Bossel Ben-Moshe, N.; Manciocco, V.; Sperduti, I.; Strigari, L.; Covello, R.; Benevolo, M.; Pescarmona, E.; Domany, E.; et al. Expression of TP53 mutation-associated microRNAs predicts clinical outcome in head and neck squamous cell carcinoma patients. Ann. Oncol. 2013, 24, 3082–3088. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Masliah-Planchon, J.; Garinet, S.; Pasmant, E. RAS-MAPK pathway epigenetic activation in cancer: miRNAs in action. Oncotarget 2016, 7, 38892–38907. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Matouk, I.J.; Raveh, E.; Abu-lail, R.; Mezan, S.; Gilon, M.; Gershtain, E.; Birman, T.; Gallula, J.; Schneider, T.; Barkali, M.; et al. Oncofetal H19 RNA promotes tumor metastasis. Biochim. Et Biophys. Acta 2014, 1843, 1414–1426. [Google Scholar] [CrossRef] [PubMed]
- Kallen, A.N.; Zhou, X.B.; Xu, J.; Qiao, C.; Ma, J.; Yan, L.; Lu, L.; Liu, C.; Yi, J.S.; Zhang, H.; et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell 2013, 52, 101–112. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhang, G.; Lu, C.; Chu, H.; Yang, R.; Zhao, G. MALAT1/miR-101-3p/MCL1 axis mediates cisplatin resistance in lung cancer. Oncotarget 2018, 9, 7501–7512. [Google Scholar] [CrossRef] [PubMed]
- Pruszko, M.; Milano, E.; Forcato, M.; Donzelli, S.; Ganci, F.; Di Agostino, S.; De Panfilis, S.; Fazi, F.; Bates, D.O.; Bicciato, S.; et al. The mutant p53-ID4 complex controls VEGFA isoforms by recruiting lncRNA MALAT1. EMBO Rep. 2017, 18, 1331–1351. [Google Scholar] [CrossRef]
- Liu, P.; Yang, H.; Zhang, J.; Peng, X.; Lu, Z.; Tong, W.; Chen, J. The lncRNA MALAT1 acts as a competing endogenous RNA to regulate KRAS expression by sponging miR-217 in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 5186. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, S.J.; Zhu, S.; Jin, Y.; Cui, S.P.; Chen, J.Y.; Xiang, C.; Li, Q.Y.; He, C.; Zhao, S.F.; et al. Hypoxia-induced lncRNA-NUTF2P3-001 contributes to tumorigenesis of pancreatic cancer by derepressing the miR-3923/KRAS pathway. Oncotarget 2016, 7, 6000–6014. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, W.; Gu, S.; Wang, H.; Zhang, Y.; Chen, W.; Xu, W.; Lin, C.; Gong, A.; Xu, M. The UCA1/KRAS axis promotes human pancreatic ductal adenocarcinoma stem cell properties and tumor growth. Am. J. Cancer Res. 2019, 9, 496–510. [Google Scholar] [PubMed]
- Ma, Y.; Wang, P.; Xue, Y.; Qu, C.; Zheng, J.; Liu, X.; Ma, J.; Liu, Y. PVT1 affects growth of glioma microvascular endothelial cells by negatively regulating miR-186. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, B.; Zhang, M.; Guo, W.; Wu, Z.; Wang, Y.; Jia, L.; Li, S.; Xie, W.; Yang, D. LncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell 2018, 33, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Shi, H.; Li, H.; Li, L.; Fang, R.; Cai, X.; Liu, B.; Zhang, X.; Ye, L. HBXIP and LSD1 scaffolded by lncRNA hotair mediate transcriptional activation by c-Myc. Cancer Res. 2016, 76, 293–304. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Hu, X.; Feng, Y.; Zhang, D.; Zhao, S.D.; Hu, Z.; Greshock, J.; Zhang, Y.; Yang, L.; Zhong, X.; Wang, L.P.; et al. A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell 2014, 26, 344–357. [Google Scholar] [CrossRef]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafa, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef]
- Cai, Y.; Li, X.; Shen, P.; Zhang, D. CCAT2 is an oncogenic long non-coding RNA in pancreatic ductal adenocarcinoma. Biol Res. 2018, 51, 1. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Jeon, Y.J.; Cui, R.; Lee, J.H.; Peng, Y.; Kim, S.H.; Tili, E.; Alder, H.; Croce, C.M. Role of MYC-regulated long noncoding RNAs in cell cycle regulation and tumorigenesis. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Verduci, L.; Ferraiuolo, M.; Sacconi, A.; Ganci, F.; Vitale, J.; Colombo, T.; Paci, P.; Strano, S.; Macino, G.; Rajewsky, N.; et al. The oncogenic role of circPVT1 in head and neck squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD transcription-competent complex. Genome Biol. 2017, 18, 237. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S. The Impact of Mutant p53 in the Non-Coding RNA World. Biomolecules 2020, 10, 472. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.J.; Yoo, J.Y.; Shu, D.; Li, H.; Zhang, J.; Yu, J.G.; Jaime-Ramirez, A.C.; Acunzo, M.; Romano, G.; Cui, R.; et al. RNA nanoparticle-based targeted therapy for glioblastoma through inhibition of oncogenic miR-21. Mol. Ther. 2017, 25, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Duo, Y.; Bi, J.; Zeng, X.; Mei, L.; Bao, S.; He, L.; Shan, A.; Zhang, Y.; Yu, X. Targeted delivery of anti-miR-155 by functionalized mesoporous silica nanoparticles for colorectal cancer therapy. Int. J. Nanomed. 2018, 13, 1241–1256. [Google Scholar] [CrossRef]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Tian, T.; Ye, J.; Zhou, S. Effect of pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer: A meta-analysis. Int. J. Clin. Pharm. Ther. 2017, 55, 720–727. [Google Scholar] [CrossRef]
- Do, K.; Cao, L.; Kang, Z.; Turkbey, B.; Lindenberg, M.L.; Larkins, E.; Holkova, B.; Steinberg, S.M.; Raffeld, M.; Peer, C.J.; et al. A phase II study of sorafenib combined with cetuximab in EGFR-expressing, KRAS-mutated metastatic colorectal cancer. Clin. Colorectal Cancer 2015, 14, 154–161. [Google Scholar] [CrossRef]
- Gilbert, J.; Schell, M.J.; Zhao, X.; Murphy, B.; Tanvetyanon, T.; Leon, M.E.; Neil Hayes, D.; Haigentz, M., Jr.; Saba, N.; Nieva, J.; et al. A randomized phase II efficacy and correlative studies of cetuximab with or without sorafenib in recurrent and/or metastatic head and neck squamous cell carcinoma. Oral. Oncol. 2015, 51, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharm. 2018, 81, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Haarberg, H.E.; Wood, E.; Rebecca, V.W.; Chen, Y.A.; Xiang, Y.; Ribas, A.; Lo, R.S.; Weber, J.S.; Sondak, V.K.; et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2502–2514. [Google Scholar] [CrossRef]
- Eroglu, Z.; Chen, Y.A.; Gibney, G.T.; Weber, J.S.; Kudchadkar, R.R.; Khushalani, N.I.; Markowitz, J.; Brohl, A.S.; Tetteh, L.F.; Ramadan, H.; et al. Combined BRAF and HSP90 inhibition in patients with unresectable BRAF (V600E)-mutant melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5516–5524. [Google Scholar] [CrossRef]
- Ambrosi, L.; Khan, S.; Carvajal, R.D.; Yang, J. Novel targets for the treatment of melanoma. Curr. Oncol. Rep. 2019, 21, 97. [Google Scholar] [CrossRef]
- Comunanza, V.; Cora, D.; Orso, F.; Consonni, F.M.; Middonti, E.; Di Nicolantonio, F.; Buzdin, A.; Sica, A.; Medico, E.; Sangiolo, D.; et al. VEGF blockade enhances the antitumor effect of BRAFV600E inhibition. EMBO Mol. Med. 2017, 9, 219–237. [Google Scholar] [CrossRef]
- Gutierrez-Martinez, A.; Sew, W.Q.G.; Molano-Fernandez, M.; Carretero-Junquera, M.; Herranz, H. Mechanisms of oncogenic cell competition-paths of victory. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oncogenic Driver Protein Name | Frequency/Mode of Activation in Multi-Neoplasia Studies | Upstream and Partner Oncogenic Factors Crosstalk | Downstream Oncogenic Process/Pathway/Target Examples | Direct Clinical Options in Neoplasia | Examples of Proposed/Tested Combinations with Other Targeted Therapies | Refs. |
|---|---|---|---|---|---|---|
| Mutant p53 | 42–43% mutated (of which >70% are missense—potential gain of function driver mutations) | Molecular chaperones, transcription co-factors: ETS2, SREBP1/2, NRF2, NFY, NF-κB, STAT3, YAP/TAZ; other partners: MRE11, Drosha/Dicer, DAB2IP | Genomic stability, steroid synthesis, epigenetic alterations, nucleotide synthesis, proteostasis, miRNA maturation, cell cycle/proliferation, migration/invasion | APR-246 (PRIMA-1 MET) in clinical trials | HSP90 inhibitors, HDAC inhibitors, proteasome inhibitors, statins | [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,130,132,162,163,176,194,196,231,232,233, 240,253,254] |
| CMYC | 5–20% mutated (of which >70% are amplifications), estimated hyperactive in majority of neoplasias | Transcription co-factor MAX, Pathways: Hedgehog, WNT, NOTCH, KRAS, PI3K, JAK-STAT3, MAPK-HNRP and mTORC1-S6K1, ncRNAs | Cell cycle (cyclins), metabolism (LDHA), apoptosis (BCL-XL), RNA splicing (PRMT5), immortalization (hTERT) | No direct inhibitors in clinics for neoplasia treatment (only preclinical). | CMYC-MAX complex inhibitors, inhibitors of RAS/PI3K/BRAF-related signaling | [7,8,9,10,29,33,34,49, 55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,91,92,106,107,109,135,136,150,151,164,165,199,220,234,245,246,247,252] |
| KRAS | 7–17% mutated (up to 90% of which are missense driver mutations) | Receptor kinases (e.g. EGFR, CMET, HER2) (mutations in RAS provide independence) | metabolism, angiogenesis, proliferation, diffe-rentiation, migration via PI3K-AKT-mTOR, RAF-MEK-ERK, TKB1- NF-κB pathways, CMYC, hTERT | Small molecule KRAS G12C inhibitors (AMG 510, MRTX849) in clinical trials | Inhibitors of up- or downstream modulators (receptor kinases, PI3K, MEK, mTOR, BRAF etc.) | [4,5,6,7,47,48,50,64, 65,71,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,162,171,172,179,181,195,197,219, 226,234,241,242,243,260] |
| PI3 kinase | 10–18% mutated (>80% of which are missense driver mutations) | Receptor tyrosine kinases, RTK, GRB2, KRAS (mutations in PIK gene provide independence), molecular chaperones | Proliferation/metabolism via AKT-mTOR pathway | Idelalisib (in standard protocol); multiple inhibitors tested in clinical trials | Downstream pathway components targeting (mTOR inhibitors), parallel pathway targeting (BRAF pathway inhibitors) | [64,85,96,97,100,173, 226] |
| BRAF | 1.5–7% (majority are missense driver mutations) | Receptor tyrosine kinases, KRAS (mutations in BRAF gene provide independence), molecular chaperones | Proliferation/metabolism via MEK-ERK pathway | BRAF mutant inhibitors in standard protocols (e.g., vemurafenib and dabrafenib) | Upstream and downstream pathway components (EGFR or MEK inhibitors), parallel pathways (PI3K inhibitors), HSP90 inhibitors, immuno-therapy, angiogenic modulators | [98,99,101,258,264,265, 267] |
| Telomerase (hTERT) | 15% mutated (>80% of which are promoter activating mutations); estimated active in 90% of neoplasias | CMYC, FoxM1, ETS family, SYMD3, molecular chaperones | Telomere-mediated cellular immortality; telomere independent activities: β-catenin/WNT, CMYC, mitochondrial apoptosis inhibition | Small molecule inhibitors (e.g., GRN163L) and peptide-based vaccines (e.g., GV1001) in clinical trials | Tested in combinations with immunotherapies and chemotherapy | [4,5,6,7,51,66,86,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117, 148,165,201] |
| Proteasome machinery (26S, 20S proteasome, immunoproteasome) | Mutations rare, estimated hyperactive and addictive in most neoplasias | NRF1, NRF2, STAT3, NFY, mutant p53, ubiquitin ligases and associated enzymes | Protein homeostasis (including inhibition of tumor suppressive pathways); activation of NF-κB and WNT pathways, molecular chaperones | Inhibitors in standard protocols (Bortezomib, Carfilzomib), other inhibitors tested | Autophagy inhibitors, HDACs inhibitors, oncogenic kinases inhibitors, NRF1/NRF2 inhibitors, mutant p53 targeting | [38,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,159,160,262,263] |
| Molecular chaperones (HSP90 and HSP70 families and co-chaperones) | Mutations rare, estimated addictive in most neoplasias | HSF1, HOP, small HSPs and HSP40 family co-chaperones | Client proteins including: mutant p53, kinases (e.g., SRC, CMET, EGFR, HER2), hTERT, CMYC, proteasome machinery activity | Multiple HSP90 and HSP70 inhibitors in clinical trials | HSF-1 inhibitors, co-chaperone inhibitors (e.g., targeting HOP or HSP40 proteins), oncogenic kinase inhibitors | [22,24,25,124,142,157, 263,264] |
| NF-κB | Overexpressed in 1–12% neoplasias; activatory kinases overexpressed in 1.5–18% neoplasias | EGFR, RAS family, HER2, proteasome | CMYC, cyclins, cytokines influencing cell proliferation, survival, angiogenesis and metastasis | No direct inhibitors in clinics for neoplasia treatment (only preclinical) | Targeting of upstream pathways – proteasome, RAS/PI3K/BRAF-related pathways, receptor kinases (e.g., HER2 or EGFR) | [40,120,133,158,159,160,161,162,163,164,165,175] |
| AP-1 (primarily FOS-JUN family protein dimers) | Mutations in FOS and JUN genes below 1%; estimated addictive in a large proportion of neoplasias | RAS family, MEKs, MAPKs, NF-κB, mutant p53, YAP/TAZ, ETS family | Invasiveness (MMP genes), proliferation (EGF pathway genes), oncogenic miRNAs (e.g., miR-21). | No direct inhibitors in clinics for neoplasia treatment (only preclinical) | Preclinical repositioning of AP-1 inhibitors tested in other diseases (e.g., T-5224 or SR 11302), rarely combined with other therapeutics | [166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183] |
| β-catenin and WNT pathway | 7–10% inactivating mutations in APC, <3% mutations in other WNT pathway proteins, estimated hyperactive especially in tumor-generating neoplasias | Receptor-ligand pairs: FZD-WNT, LGR/R-spondin, EGFR-EGF, EG2-PGE2; β-catenin destruction complex and nuclear co-factors: LEF, TCF, PAF, YAP1, hTERT | Cell proliferation and tumor growth via CMYC, CJUN, hTERT, targets of AP-1 or NFAT (in non-canonical signaling) | Repositioning of approved anti-WNT drugs to cancer treatment (e.g., Niclosamide, Sulindac), other inhibitors in trials | Combination with PI3K/AKT/mTOR inhibitors, multikinase inhibitors (e.g., sorafenib) and chemotherapeutics | [113,134,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209] |
| YAP and TAZ | 1–1.5% mutated YAP1 (up to 40% amplifications); estimated hyperactive in most tumor-generating neoplasias | Rho GTPases and cytoskeleton stiffness, GNAQ and GNA11, mevalonate pathway, β-catenin, CMYC, mutant p53, BRD4, AP-1, TEADs | Inhibition of apoptosis (e.g., BIRC5 and BCL2L1), tissue growth/stiffness/cell proliferation (e.g., AREG, CTGF) | No direct inhibitors in clinics for neoplasia treatment (only preclinical) | BET inhibitors, statins | [32,133,180,210,211,212,213,214,215,216,217,218,219,220,221,222,223] |
| non-coding RNAs (micro RNAs and long non-coding RNAs) | Copy number alterations, SNPs and promoter mutations so far found in single studies | miRNA maturation factors (Dicer, Drosha, DGCR8, KSRP) and their regulators e.g., mutant p53, proteasome; upregulators of specific ncRNA transcription or stability—e.g., CMYC | Cell proliferation (e.g. via RAS and PI3K-AKT pathway, CMYC, MCL1), tumor growth (e.g., via WNT pathway), cancer cell metabolic support (e.g., via STAT1, ATG7, Beclin1), chromatin reprogramming | Inhibition of oncogenic miRNAs (e.g., miR-155 by MRG-106) or targeting cells with overexpression of H19 lncRNA (by BC-819) in clinical trials | Next to none tested so far; suggested are downstream pathways and upstream regulators, such as CMYC pathway or mutant p53-proteasome-KSRP axis | [38,44,67,68,69,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzes, M.; Oron, M.; Staszczak, Z.; Jaiswar, A.; Nowak-Niezgoda, M.; Walerych, D. A Driver Never Works Alone—Interplay Networks of Mutant p53, MYC, RAS, and Other Universal Oncogenic Drivers in Human Cancer. Cancers 2020, 12, 1532. https://doi.org/10.3390/cancers12061532
Grzes M, Oron M, Staszczak Z, Jaiswar A, Nowak-Niezgoda M, Walerych D. A Driver Never Works Alone—Interplay Networks of Mutant p53, MYC, RAS, and Other Universal Oncogenic Drivers in Human Cancer. Cancers. 2020; 12(6):1532. https://doi.org/10.3390/cancers12061532
Chicago/Turabian StyleGrzes, Maria, Magdalena Oron, Zuzanna Staszczak, Akanksha Jaiswar, Magdalena Nowak-Niezgoda, and Dawid Walerych. 2020. "A Driver Never Works Alone—Interplay Networks of Mutant p53, MYC, RAS, and Other Universal Oncogenic Drivers in Human Cancer" Cancers 12, no. 6: 1532. https://doi.org/10.3390/cancers12061532
APA StyleGrzes, M., Oron, M., Staszczak, Z., Jaiswar, A., Nowak-Niezgoda, M., & Walerych, D. (2020). A Driver Never Works Alone—Interplay Networks of Mutant p53, MYC, RAS, and Other Universal Oncogenic Drivers in Human Cancer. Cancers, 12(6), 1532. https://doi.org/10.3390/cancers12061532