Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds

 ,
,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Pyridinyl Imidazole p38 MAPK Inhibitors Disrupt BRAF V600E-Driven ERK Signaling in Human Melanoma Cells
2.2. SB202190-Induced Vacuoles in Melanoma Cells Have an Endocytic Origin
2.3. Pyridinyl Imidazole Compounds Partly Mimic PIKfyve Inhibition in Melanoma Cells
2.4. Pyridinyl Imidazole Compounds Induce Changes in mTOR Subcellular Localization in A375 Melanoma Cells
2.5. Pyridinyl Imidazole Compounds Induce Dynamic Changes in mTORC1 Activity in Melanoma Cells
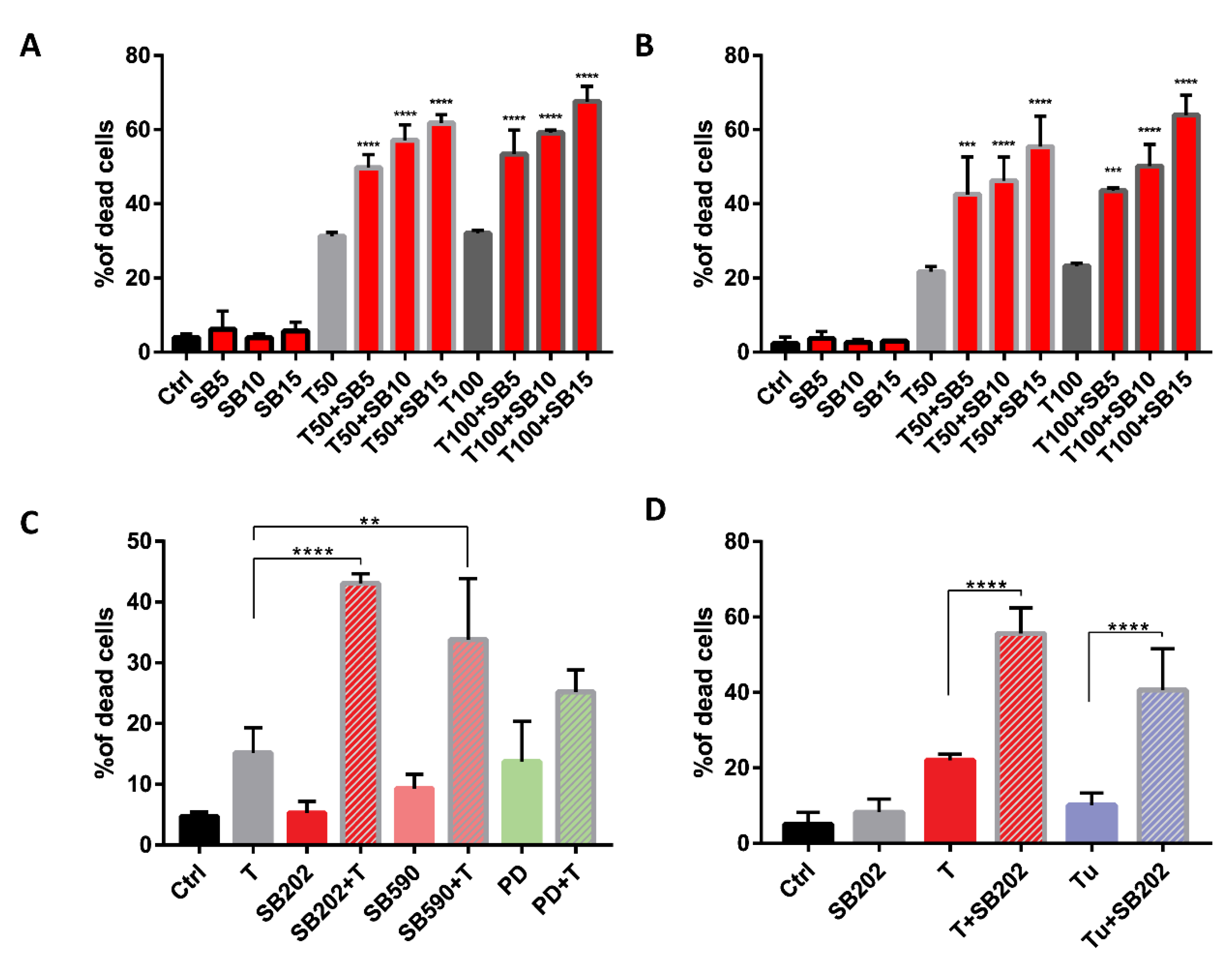
2.6. Pyridinyl Imidazole Drugs Sensitize A375 Melanoma Cells to ER Stress
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Western Blotting
4.3. In Vitro Kinase Assays
4.4. Transient Transfections and Fluorescence Microscopy
4.5. Quantitative Phase Imaging Analysis Using Coherence-Controlled Holographic Microscopy
4.6. Electron Microscopy
4.7. Quantification of Intracellular Metabolites Using NMR Spectroscopy
4.8. Flow Cytometry and MTT Proliferation Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma Genome Sequencing Reveals Frequent PREX2 Mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Holman, D.M.; Maguire-Eisen, M. Ultraviolet Radiation Exposure and Its Impact on Skin Cancer Risk. Semin. Oncol. Nurs. 2016, 32, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Dahl, C.; Guldberg, P. The Genome and Epigenome of Malignant Melanoma. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Fecher, L.A.; Amaravadi, R.K.; Flaherty, K.T. The MAPK Pathway in Melanoma. Curr. Opin. Oncol. 2008, 20, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Agianian, B.; Gavathiotis, E. Current Insights of BRAF Inhibitors in Cancer. J. Med. Chem. 2018, 61, 5775–5793. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Hernández, I.; Baquero, P.; Calleros, L.; Chiloeches, A. Dual Inhibition of V600EBRAF and the PI3K/AKT/MTOR Pathway Cooperates to Induce Apoptosis in Melanoma Cells through a MEK-Independent Mechanism. Cancer Lett. 2012, 314, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS–ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/MTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.-R.; Michael, H.T.; Halsey, C.H.C.; Peer, C.J.; Adhikari, A.; Dwyer, J.E.; Hoover, S.B.; El Meskini, R.; Kozlov, S.; Weaver Ohler, Z.; et al. Synergistic Targeted Inhibition of MEK and Dual PI3K/MTOR Diminishes Viability and Inhibits Tumor Growth of Canine Melanoma Underscoring Its Utility as a Preclinical Model for Human Mucosal Melanoma. Pigment Cell Melanoma Res. 2016, 29, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Deuker, M.M.; McMahon, M. Rational Targeting of BRAF and PI3-Kinase Signaling for Melanoma Therapy. Mol. Cell. Oncol. 2016, 3, e1033095. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of MTORC1 by PI3K Signaling. Trends Cell. Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.-L. MTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; DeVries, B.; Stambolic, V. Localization of Rheb to the Endomembrane Is Critical for Its Signaling Function. Biochem. Biophys. Res. Commun. 2006, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control MTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase Is a Direct Target of TSC2 GAP Activity and Regulates MTOR Signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal Recruitment of TSC2 Is a Universal Response to Cellular Stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator Is a GEF for the Rag GTPases That Signal Amino Acid Levels to MTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets MTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Sekiguchi, T.; Hirose, E.; Nakashima, N.; Ii, M.; Nishimoto, T. Novel G Proteins, Rag C and Rag D, Interact with GTP-Binding Proteins, Rag A and Rag, B. J. Biol. Chem. 2001, 276, 7246–7257. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to MTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. MTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of MTORC1 by Amino Acids. Trends Cell. Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the MTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/MTOR and Raf/MEK/ERK Pathways in the Treatment of Breast Cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Argast, G.M.; Fausto, N.; Campbell, J.S. Inhibition of RIP2/RICK/CARDIAK Activity by Pyridinyl Imidazole Inhibitors of P38 MAPK. Mol. Cell. Biochem. 2005, 268, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; Mclauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The Selectivity of Protein Kinase Inhibitors: A Further Update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Bellei, B.; Pitisci, A.; Migliano, E.; Cardinali, G.; Picardo, M. Pyridinyl Imidazole Compounds Interfere with Melanosomes Sorting through the Inhibition of Cyclin G-Associated Kinase, a Regulator of Cathepsins Maturation. Cell. Signal. 2014, 26, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Dhamija, S.; Kotlyarov, A.; Gaestel, M. The Problem of Pyridinyl Imidazole Class Inhibitors of MAPK14/P38α and MAPK11/P38β in Autophagy Research. Autophagy 2015, 11, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Kalmes, A.; Deou, J.; Clowes, A.W.; Daum, G. Raf-1 Is Activated by the P38 Mitogen-Activated Protein Kinase Inhibitor, SB203580. FEBS Lett. 1998, 444, 71–74. [Google Scholar] [CrossRef]
- Henklova, P.; Vrzal, R.; Papouskova, B.; Bednar, P.; Jancova, P.; Anzenbacherova, E.; Ulrichova, J.; Maurel, P.; Pavek, P.; Dvorak, Z. SB203580, a Pharmacological Inhibitor of P38 MAP Kinase Transduction Pathway Activates ERK and JNK MAP Kinases in Primary Cultures of Human Hepatocytes. Eur. J. Pharmacol. 2008, 593, 16–23. [Google Scholar] [CrossRef] [PubMed]
- New, L.; Li, Y.; Ge, B.; Zhong, H.; Mansbridge, J.; Liu, K.; Han, J. SB203580 Promote EGF-Stimulated Early Morphological Differentiation in PC12 Cell through Activating ERK Pathway. J. Cell. Biochem. 2001, 83, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Thevakumaran, N.; Gavory, G.; Li, J.J.; Padeganeh, A.; Guiral, S.; Duchaine, J.; Mao, D.Y.L.; Bouvier, M.; Sicheri, F.; et al. Inhibitors That Stabilize a Closed RAF Kinase Domain Conformation Induce Dimerization. Nat. Chem. Biol. 2013, 9, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Gudernova, I.; Foldynova-Trantirkova, S.; El Ghannamova, B.; Fafilek, B.; Varecha, M.; Balek, L.; Hruba, E.; Jonatova, L.; Jelinkova, I.; Bosakova, M.K.; et al. One Reporter for In-Cell Activity Profiling of Majority of Protein Kinase Oncogenes. Elife 2017, 6, e21536. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Patrick, D.R.; Batorsky, R.S.; Ho, M.L.; Do, H.T.; Zhang, S.Y.; Kumar, R.; Rusnak, D.W.; Takle, A.K.; Wilson, D.M.; et al. Demonstration of a Genetic Therapeutic Index for Tumors Expressing Oncogenic BRAF by the Kinase Inhibitor SB-590885. Cancer Res. 2006, 66, 11100–11105. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Kotlyarov, A.; Gaestel, M. SB202190-Induced Cell Type-Specific Vacuole Formation and Defective Autophagy Do Not Depend on P38 MAP Kinase Inhibition. PLoS ONE 2011, 6, e23054. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Jefferies, H.B.J.; Cooke, F.T.; Jat, P.; Boucheron, C.; Koizumi, T.; Hayakawa, M.; Kaizawa, H.; Ohishi, T.; Workman, P.; Waterfield, M.D.; et al. A Selective PIKfyve Inhibitor Blocks PtdIns(3,5)P2 Production and Disrupts Endomembrane Transport and Retroviral Budding. EMBO Rep. 2008, 9, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Bridges, D.; Ma, J.-T.; Park, S.; Inoki, K.; Weisman, L.S.; Saltiel, A.R. Phosphatidylinositol 3,5-Bisphosphate Plays a Role in the Activation and Subcellular Localization of Mechanistic Target of Rapamycin 1. Mol. Biol. Cell 2012, 23, 2955–2962. [Google Scholar] [CrossRef] [PubMed]
- Compton, L.M.; Ikonomov, O.C.; Sbrissa, D.; Garg, P.; Shisheva, A. Active Vacuolar H+ ATPase and Functional Cycle of Rab5 Are Required for the Vacuolation Defect Triggered by PtdIns(3,5)P2 Loss under PIKfyve or Vps34 Deficiency. Am. J. Physiol. Cell Physiol. 2016, 311, C366–C377. [Google Scholar] [CrossRef] [PubMed]
- Shisheva, A. PIKfyve: Partners, Significance, Debates and Paradoxes. Cell Biol. Int. 2008, 32, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Palm, W.; Lee, Y.; Yang, W.; Bandyopadhyay, U.; Xu, H.; Florey, O.; Thompson, C.B.; Overholtzer, M. PIKfyve Regulates Vacuole Maturation and Nutrient Recovery Following Engulfment. Dev. Cell 2016, 38, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal V-ATPase-Ragulator Complex Is a Common Activator for AMPK and MTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Flinn, R.J.; Yan, Y.; Goswami, S.; Parker, P.J.; Backer, J.M.; Margolis, B. The Late Endosome Is Essential for MTORC1 Signaling. Mol. Biol. Cell 2010, 21, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Sabatini, D.M. Ragulator and SLC38A9 Activate the Rag GTPases through Noncanonical GEF Mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.; Wang, L.; Deng, W.; Wang, J.; Wu, G. Structural Insight into the Ragulator Complex Which Anchors MTORC1 to the Lysosomal Membrane. Cell Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A Lysosome-to-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef]
- Gayle, S.; Landrette, S.; Beeharry, N.; Conrad, C.; Hernandez, M.; Beckett, P.; Ferguson, S.M.; Mandelkern, T.; Zheng, M.; Xu, T.; et al. Identification of Apilimod as a First-in-Class PIKfyve Kinase Inhibitor for Treatment of B-Cell Non-Hodgkin Lymphoma. Blood 2017, 129, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-Regulation of Lysosomal TRPML1 Channels Is Essential for Lysosomal Adaptation to Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016, 26, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K. Regulation of TORC1 by Rag GTPases in Nutrient Response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of Protein Is an Amino Acid Supply Route in Ras-Transformed Cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Pacitto, R.; Inoki, K.; Swanson, J. Macropinocytosis, MTORC1 and Cellular Growth Control. Cell. Mol. Life Sci. 2018, 75, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Reis, R.C.M.; Sorgine, M.H.F.; Coelho-Sampaio, T. A Novel Methodology for the Investigation of Intracellular Proteolytic Processing in Intact Cells. Eur. J. Cell Biol. 1998, 75, 192–197. [Google Scholar] [CrossRef]
- Mishra, R.; Bhowmick, N.A. Visualization of Macropinocytosis in Prostate Fibroblasts. Bio. Protoc. 2019, 9, e3235. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Suzuki, A.; Weaver, L.; Guzman, J.; Chung, Y.; Jin, H.; Gonzalez, F.; Trasorras, C.; Zhao, L.; Spruce, L.A.; et al. PIKfyve Deficiency in Myeloid Cells Impairs Lysosomal Homeostasis in Macrophages and Promotes Systemic Inflammation in Mice. Mol. Cell. Biol. 2019, 39, e00158-19. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Proud, C.G. The MTOR Pathway in the Control of Protein Synthesis. Physiology 2006, 21, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.-I.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF Inhibitors Prime Wild-Type RAF to Activate the MAPK Pathway and Enhance Growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The Clinical Effect of the Dual-Targeting Strategy Involving PI3K/AKT/MTOR and RAS/MEK/ERK Pathways in Patients with Advanced Cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Penna, I.; Molla, A.; Grazia, G.; Cleris, L.; Nicolini, G.; Perrone, F.; Picciani, B.; del Vecchio, M.; de Braud, F.; Mortarini, R.; et al. Primary Cross-Resistance to BRAFV600E-, MEK1/2- and PI3K/MTOR-Specific Inhibitors in BRAF-Mutant Melanoma Cells Counteracted by Dual Pathway Blockade. Oncotarget 2016, 7, 3947–3965. [Google Scholar] [CrossRef] [PubMed]
- Aramburu, J.; Ortells, M.C.; Tejedor, S.; Buxadé, M.; López-Rodríguez, C. Transcriptional Regulation of the Stress Response by MTOR. Sci. Signal. 2014, 7, re2. [Google Scholar] [CrossRef] [PubMed]
- Heberle, A.M.; Prentzell, M.T.; van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular Mechanisms of MTOR Regulation by Stress. Mol. Cell. Oncol. 2015, 2, e970489. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-O.; Li, R.; Montell, C.; Venkatachalam, K. Drosophila TRPML Is Required for TORC1 Activation. Curr. Biol. 2012, 22, 1616–1621. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) Controls Membrane Trafficking by Direct Activation of Mucolipin Ca(2+) Release Channels in the Endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.H.; Saffi, G.; Gray, M.A.; Wallace, C.; Dayam, R.M.; Ou, Z.Y.A.; Lenk, G.; Puertollano, R.; Watkins, S.C.; Botelho, R.J. Lysosome Enlargement during Inhibition of the Lipid Kinase PIKfyve Proceeds through Lysosome Coalescence. J. Cell Sci. 2018, 131, jcs213587. [Google Scholar] [CrossRef] [PubMed]
- Nada, S.; Hondo, A.; Kasai, A.; Koike, M.; Saito, K.; Uchiyama, Y.; Okada, M. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 2009, 28, 477–489. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nada, S.; Mori, S.; Soma-Nagae, T.; Oneyama, C.; Okada, M. The Late Endosome/Lysosome-Anchored P18-MTORC1 Pathway Controls Terminal Maturation of Lysosomes. Biochem. Biophys. Res. Commun. 2012, 417, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Leli, N.M.; Onorati, A.; Piao, S.; Verginadis, I.I.; Tameire, F.; Rebecca, V.W.; Chude, C.I.; Murugan, S.; Fennelly, C.; et al. ER Translocation of the MAPK Pathway Drives Therapy Resistance in BRAF-Mutant Melanoma. Cancer Discov. 2019, 9, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER Stress: Autophagy Induction, Inhibition and Selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER Stress: Mutual Crosstalk between Autophagy, Oxidative Stress and Inflammatory Response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy Pathway: Cellular and Molecular Mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Hasegawa, J.; Yoshimori, T. Regulation of Lysosomal Phosphoinositide Balance by INPP5E Is Essential for Autophagosome–Lysosome Fusion. Autophagy 2016, 12, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Dossou, A.S.; Basu, A. The Emerging Roles of MTORC1 in Macromanaging Autophagy. Cancers (Basel) 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. MTORC1 as the Main Gateway to Autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [PubMed]
- Martina, J.A.; Diab, H.I.; Brady, O.A.; Puertollano, R. TFEB and TFE3 Are Novel Components of the Integrated Stress Response. EMBO J. 2016, 35, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, H.; Jiang, C.C.; Fang, L.; Chen, C.; Zhang, X.D.; Jiang, Z.W. Connecting Endoplasmic Reticulum Stress to Autophagy through IRE1/JNK/Beclin-1 in Breast Cancer Cells. Int. J. Mol. Med. 2014, 34, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Bondzi, C.; Grant, S.; Krystal, G.W. A Novel Assay for the Measurement of Raf-1 Kinase Activity. Oncogene 2000, 19, 5030–5033. [Google Scholar] [CrossRef] [PubMed]
- Verlande, A.; Krafčíková, M.; Potěšil, D.; Trantírek, L.; Zdráhal, Z.; Elkalaf, M.; Trnka, J.; Souček, K.; Rauch, N.; Rauch, J.; et al. Metabolic stress regulates ERK activity by controlling KSR-RAF heterodimerization. EMBO Rep. 2018, 19, 320–336. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Slabý, T.; Kolman, P.; Dostál, Z.; Antoš, M.; Lošťák, M.; Chmelík, R. Off-Axis Setup Taking Full Advantage of Incoherent Illumination in Coherence-Controlled Holographic Microscope. Opt. Express 2013, 21, 14747–14762. [Google Scholar] [CrossRef] [PubMed]
- Zangle, T.A.; Teitell, M.A. Live-Cell Mass Profiling: An Emerging Approach in Quantitative Biophysics. Nat. Methods 2014, 11, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Dietmair, S.; Timmins, N.E.; Gray, P.P.; Nielsen, L.K.; Krömer, J.O. Towards Quantitative Metabolomics of Mammalian Cells: Development of a Metabolite Extraction Protocol. Anal. Biochem. 2010, 404, 155–164. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palušová, V.; Renzová, T.; Verlande, A.; Vaclová, T.; Medková, M.; Cetlová, L.; Sedláčková, M.; Hříbková, H.; Slaninová, I.; Krutá, M.; et al. Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds. Cancers 2020, 12, 1516. https://doi.org/10.3390/cancers12061516
Palušová V, Renzová T, Verlande A, Vaclová T, Medková M, Cetlová L, Sedláčková M, Hříbková H, Slaninová I, Krutá M, et al. Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds. Cancers. 2020; 12(6):1516. https://doi.org/10.3390/cancers12061516
Chicago/Turabian StylePalušová, Veronika, Tereza Renzová, Amandine Verlande, Tereza Vaclová, Michaela Medková, Linda Cetlová, Miroslava Sedláčková, Hana Hříbková, Iva Slaninová, Miriama Krutá, and et al. 2020. "Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds" Cancers 12, no. 6: 1516. https://doi.org/10.3390/cancers12061516
APA StylePalušová, V., Renzová, T., Verlande, A., Vaclová, T., Medková, M., Cetlová, L., Sedláčková, M., Hříbková, H., Slaninová, I., Krutá, M., Rotrekl, V., Uhlířová, H., Křížová, A., Chmelík, R., Veselý, P., Krafčíková, M., Trantírek, L., Schink, K. O., & Uldrijan, S. (2020). Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds. Cancers, 12(6), 1516. https://doi.org/10.3390/cancers12061516