Capture-Based Next-Generation Sequencing Improves the Identification of Immunoglobulin/T-Cell Receptor Clonal Markers and Gene Mutations in Adult Acute Lymphoblastic Leukemia Patients Lacking Molecular Probes

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Validation of the Capture-Based NGS Panel
2.2. Retrospective Application of the Capture-Based NGS Panel to MRD-Unknown ALL Patients
2.3. Prospective Application of the Capture-Based NGS Panel and Patient-Specific Probe Design
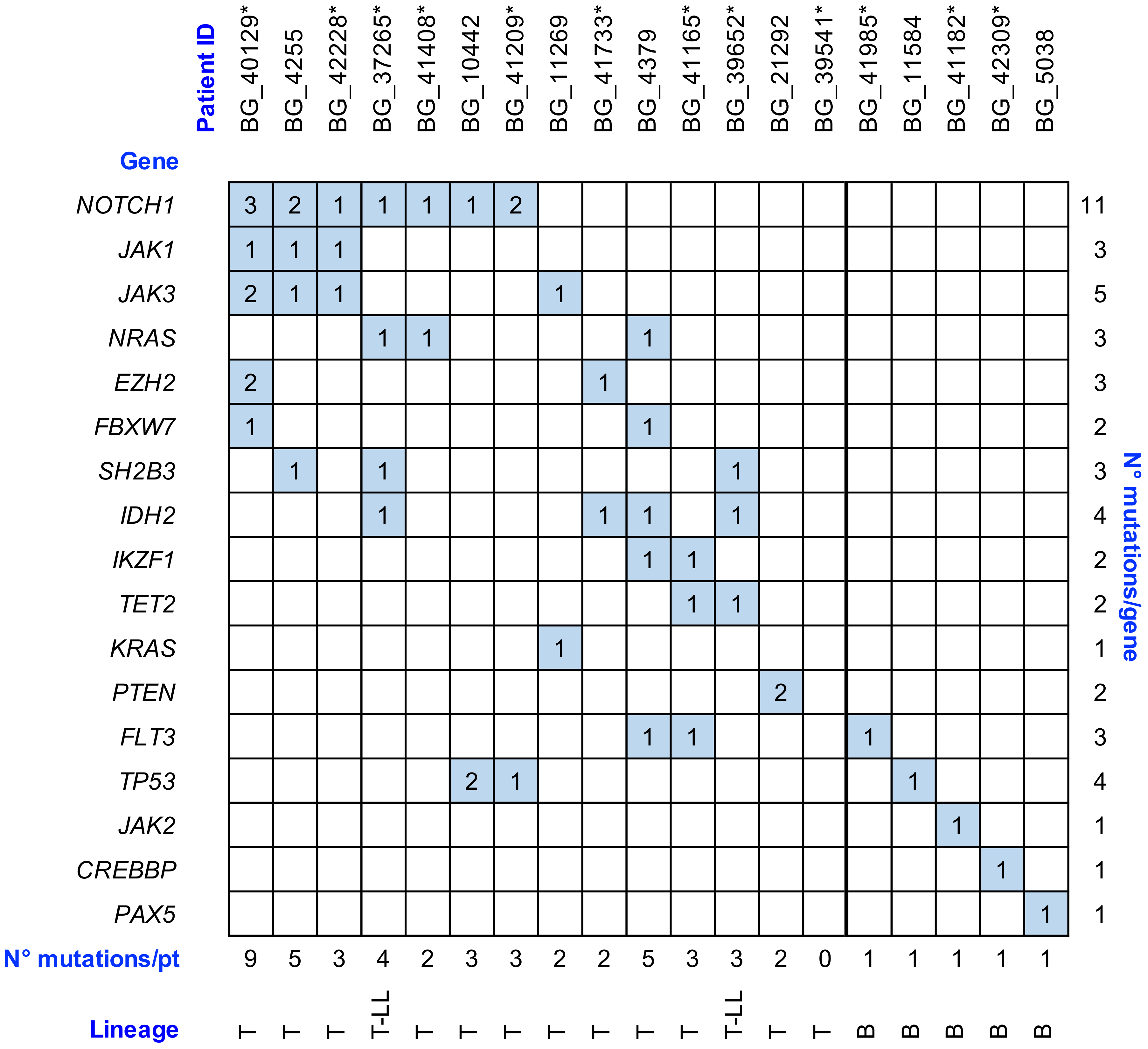
2.4. Gene Variant Analysis by NGS Approach
3. Discussion
4. Materials and Methods
4.1. Capture-Based NGS Panel Design
4.2. Clinical Samples
4.3. DNA Extraction, Library Preparation, and Sequencing
4.4. NGS Data Analysis and Statistical Analysis
4.5. Clonality Assessment Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cavé, H.; van der Werff ten Bosch, J.; Suciu, S.; Guidal, C.; Waterkeyn, C.; Otten, J.; Bakkus, M.; Thielemans, K.; Grandchamp, B.; Vilmer, E. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer--Childhood Leukemia Cooperative Group. N. Engl. J. Med. 1998, 339, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.; Seriu, T.; Panzer-Grümayer, E.R.; Biondi, A.; Pongers-Willemse, M.J.; Corral, L.; Stolz, F.; Schrappe, M.; Masera, G.; Kamps, W.A.; et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet Lond. Engl. 1998, 352, 1731–1738. [Google Scholar] [CrossRef]
- Szczepański, T.; Orfão, A.; van der Velden, V.H.; San Miguel, J.F.; van Dongen, J.J. Minimal residual disease in leukaemia patients. Lancet Oncol. 2001, 2, 409–417. [Google Scholar] [CrossRef]
- Brüggemann, M.; Raff, T.; Flohr, T.; Gökbuget, N.; Nakao, M.; Droese, J.; Lüschen, S.; Pott, C.; Ritgen, M.; Scheuring, U.; et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2006, 107, 1116–1123. [Google Scholar] [CrossRef]
- Bassan, R.; Spinelli, O.; Oldani, E.; Intermesoli, T.; Tosi, M.; Peruta, B.; Rossi, G.; Borlenghi, E.; Pogliani, E.M.; Terruzzi, E.; et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood 2009, 113, 4153–4162. [Google Scholar] [CrossRef]
- Bassan, R.; Hoelzer, D. Modern therapy of acute lymphoblastic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 532–543. [Google Scholar] [CrossRef]
- Lussana, F.; Intermesoli, T.; Gianni, F.; Boschini, C.; Masciulli, A.; Spinelli, O.; Oldani, E.; Tosi, M.; Grassi, A.; Parolini, M.; et al. Achieving Molecular Remission before Allogeneic Stem Cell Transplantation in Adult Patients with Philadelphia Chromosome–Positive Acute Lymphoblastic Leukemia: Impact on Relapse and Long-Term Outcome. Biol. Blood Marrow Transplant. 2016, 22, 1983–1987. [Google Scholar] [CrossRef]
- Van der Velden, V.H.J.; Hochhaus, A.; Cazzaniga, G.; Szczepanski, T.; Gabert, J.; van Dongen, J.J.M. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: Principles, approaches, and laboratory aspects. Leukemia 2003, 17, 1013–1034. [Google Scholar] [CrossRef]
- Pongers-Willemse, M.J.; Seriu, T.; Stolz, F.; d’Aniello, E.; Gameiro, P.; Pisa, P.; Gonzalez, M.; Bartram, C.R.; Panzer-Grümayer, E.R.; Biondi, A.; et al. Primers and protocols for standardized detection of minimal residual disease in acute lymphoblastic leukemia using immunoglobulin and T cell receptor gene rearrangements and TAL1 deletions as PCR targets: Report of the BIOMED-1 CONCERTED ACTION: Investigation of minimal residual disease in acute leukemia. Leukemia 1999, 13, 110–118. [Google Scholar]
- Van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.a.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef]
- Van Dongen, J.J.M.; van der Velden, V.H.J.; Brüggemann, M.; Orfao, A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: Need for sensitive, fast, and standardized technologies. Blood 2015, 125, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, M.; Kotrova, M. Minimal residual disease in adult ALL: Technical aspects and implications for correct clinical interpretation. Blood Adv. 2017, 1, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Langerak, A.W.; Brüggemann, M.; Davi, F.; Darzentas, N.; van Dongen, J.J.M.; Gonzalez, D.; Cazzaniga, G.; Giudicelli, V.; Lefranc, M.-P.; Giraud, M.; et al. High-Throughput Immunogenetics for Clinical and Research Applications in Immunohematology: Potential and Challenges. J. Immunol. 2017, 198, 3765–3774. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, M.; Kotrová, M.; Knecht, H.; Bartram, J.; Boudjogrha, M.; Bystry, V.; Fazio, G.; Froňková, E.; Giraud, M.; Grioni, A.; et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia 2019. [Google Scholar] [CrossRef]
- Knecht, H.; Reigl, T.; Kotrová, M.; Appelt, F.; Stewart, P.; Bystry, V.; Krejci, A.; Grioni, A.; Pal, K.; Stranska, K.; et al. Quality control and quantification in IG/TR next-generation sequencing marker identification: Protocols and bioinformatic functionalities by EuroClonality-NGS. Leukemia 2019. [Google Scholar] [CrossRef]
- Scheijen, B.; Meijers, R.W.J.; Rijntjes, J.; van der Klift, M.Y.; Möbs, M.; Steinhilber, J.; Reigl, T.; van den Brand, M.; Kotrová, M.; Ritter, J.-M.; et al. Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: A technical feasibility study by EuroClonality-NGS. Leukemia 2019, 33, 2227–2240. [Google Scholar] [CrossRef]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.H.; Stow, P.; Coustan-Smith, E.; Pui, C.-H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef]
- Ladetto, M.; Brüggemann, M.; Monitillo, L.; Ferrero, S.; Pepin, F.; Drandi, D.; Barbero, D.; Palumbo, A.; Passera, R.; Boccadoro, M.; et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia 2014, 28, 1299–1307. [Google Scholar] [CrossRef]
- Pulsipher, M.A.; Carlson, C.; Langholz, B.; Wall, D.A.; Schultz, K.R.; Bunin, N.; Kirsch, I.; Gastier-Foster, J.M.; Borowitz, M.; Desmarais, C.; et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood 2015, 125, 3501–3508. [Google Scholar] [CrossRef]
- Wood, B.; Wu, D.; Crossley, B.; Dai, Y.; Williamson, D.; Gawad, C.; Borowitz, M.J.; Devidas, M.; Maloney, K.W.; Larsen, E.; et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 2018, 131, 1350–1359. [Google Scholar] [CrossRef]
- Sánchez, R.; Ayala, R.; Martínez-López, J. Minimal Residual Disease Monitoring with Next-Generation Sequencing Methodologies in Hematological Malignancies. Int. J. Mol. Sci. 2019, 20, 2832. [Google Scholar] [CrossRef] [PubMed]
- Wren, D.; Walker, B.A.; Brüggemann, M.; Catherwood, M.A.; Pott, C.; Stamatopoulos, K.; Langerak, A.W.; Gonzalez, D.; EuroClonality-NGS Consortium. Comprehensive translocation and clonality detection in lymphoproliferative disorders by next-generation sequencing. Haematologica 2017, 102, e57–e60. [Google Scholar] [CrossRef] [PubMed]
- Salmoiraghi, S.; Montalvo, M.L.G.; Ubiali, G.; Tosi, M.; Peruta, B.; Zanghi, P.; Oldani, E.; Boschini, C.; Kohlmann, A.; Bungaro, S.; et al. Mutations of TP53 gene in adult acute lymphoblastic leukemia at diagnosis do not affect the achievement of hematologic response but correlate with early relapse and very poor survival. Haematologica 2016, 101, e245–e248. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 389–396. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.R.; Sulis, M.L.; Duke, V.; Foroni, L.; Jenkinson, S.; Koo, K.; Allen, C.G.; Gale, R.E.; Buck, G.; Richards, S.; et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4352–4356. [Google Scholar] [CrossRef]
- Mansour, M.R.; Duke, V.; Foroni, L.; Patel, B.; Allen, C.G.; Ancliff, P.J.; Gale, R.E.; Linch, D.C. Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 6964–6969. [Google Scholar] [CrossRef]
- Degryse, S.; Bornschein, S.; de Bock, C.E.; Leroy, E.; Vanden Bempt, M.; Demeyer, S.; Jacobs, K.; Geerdens, E.; Gielen, O.; Soulier, J.; et al. Mutant JAK3 signaling is increased by loss of wild-type JAK3 or by acquisition of secondary JAK3 mutations in T-ALL. Blood 2018, 131, 421–425. [Google Scholar] [CrossRef]
- Van der Velden, V.H.J.; Cazzaniga, G.; Schrauder, A.; Hancock, J.; Bader, P.; Panzer-Grumayer, E.R.; Flohr, T.; Sutton, R.; Cave, H.; Madsen, H.O.; et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: Guidelines for interpretation of real-time quantitative PCR data. Leukemia 2007, 21, 604–611. [Google Scholar] [CrossRef]
- Kotrova, M.; Trka, J.; Kneba, M.; Brüggemann, M. Is Next-Generation Sequencing the way to go for Residual Disease Monitoring in Acute Lymphoblastic Leukemia? Mol. Diagn. Ther. 2017, 21, 481–492. [Google Scholar] [CrossRef]
- Giraud, M.; Salson, M.; Duez, M.; Villenet, C.; Quief, S.; Caillault, A.; Grardel, N.; Roumier, C.; Preudhomme, C.; Figeac, M. Fast multiclonal clusterization of V(D)J recombinations from high-throughput sequencing. BMC Genom. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Duez, M.; Giraud, M.; Herbert, R.; Rocher, T.; Salson, M.; Thonier, F. Vidjil: A Web Platform for Analysis of High-Throughput Repertoire Sequencing. PLoS ONE 2016, 11, e0166126. [Google Scholar] [CrossRef] [PubMed]
- Szczepański, T.; Beishuizen, A.; Pongers-Willemse, M.J.; Hählen, K.; Van Wering, E.R.; Wijkhuijs, A.J.; Tibbe, G.J.; De Bruijn, M.A.; Van Dongen, J.J. Cross-lineage T cell receptor gene rearrangements occur in more than ninety percent of childhood precursor-B acute lymphoblastic leukemias: Alternative PCR targets for detection of minimal residual disease. Leukemia 1999, 13, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Szczepański, T.; Pongers-Willemse, M.J.; Langerak, A.W.; van Dongen, J.J. Unusual immunoglobulin and T-cell receptor gene rearrangement patterns in acute lymphoblastic leukemias. Curr. Top. Microbiol. Immunol. 1999, 246, 205–213; discussion 214–215. [Google Scholar] [CrossRef] [PubMed]
- Szczepański, T.; Flohr, T.; van der Velden, V.H.J.; Bartram, C.R.; van Dongen, J.J.M. Molecular monitoring of residual disease using antigen receptor genes in childhood acute lymphoblastic leukaemia. Best Pract. Res. Clin. Haematol. 2002, 15, 37–57. [Google Scholar] [CrossRef]
- Szczepański, T.; van der Velden, V.H.J.; Hoogeveen, P.G.; de Bie, M.; Jacobs, D.C.H.; van Wering, E.R.; van Dongen, J.J.M. Vδ2-Jα rearrangements are frequent in precursor-B–acute lymphoblastic leukemia but rare in normal lymphoid cells. Blood 2004, 103, 3798–3804. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Flohr, T.; Schrauder, A.; Cazzaniga, G.; Panzer-Grümayer, R.; van der Velden, V.; Fischer, S.; Stanulla, M.; Basso, G.; Niggli, F.K.; Schäfer, B.W.; et al. Minimal residual disease-directed risk stratification using real-time quantitative PCR analysis of immunoglobulin and T-cell receptor gene rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for childhood acute lymphoblastic leukemia. Leukemia 2008, 22, 771–782. [Google Scholar] [CrossRef]
- Basso, G.; Veltroni, M.; Valsecchi, M.G.; Dworzak, M.N.; Ratei, R.; Silvestri, D.; Benetello, A.; Buldini, B.; Maglia, O.; Masera, G.; et al. Risk of Relapse of Childhood Acute Lymphoblastic Leukemia Is Predicted By Flow Cytometric Measurement of Residual Disease on Day 15 Bone Marrow. J. Clin. Oncol. 2009, 27, 5168–5174. [Google Scholar] [CrossRef]
- Ribera, J.-M.; Oriol, A.; Morgades, M.; Montesinos, P.; Sarrà, J.; González-Campos, J.; Brunet, S.; Tormo, M.; Fernández-Abellán, P.; Guàrdia, R.; et al. Treatment of High-Risk Philadelphia Chromosome–Negative Acute Lymphoblastic Leukemia in Adolescents and Adults According to Early Cytologic Response and Minimal Residual Disease after Consolidation Assessed by Flow Cytometry: Final Results of the PETHEMA ALL-AR-03 Trial. J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]


{kind=link}
| Patient ID | ALL Lineage | Locus | V Gene | del V | n | del D | D Gene | del D | n | del J | J Gene | Known | Rearrangement Feature |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BG_371 | B | IGH | IGHV3-9*01 | −2 | 15 | −2 | IGHD6-19*01 | −4 | 7 | 0 | IGHJ3*02 | Yes | / |
| TRD+ | TRDV2*02 | −4 | 10 | −28 | TRDD3*01 | NA | NA | NA | NA | Yes | / | ||
| TRG | TRGV2*01 | 0 | 20 | NA | NA | NA | NA | −3 | TRGJ1*01 | Yes | / | ||
| TRG | TRGV3*02 | −4 | 15 | NA | NA | NA | NA | −6 | TRGJ1*02 | Yes | / | ||
| BG_4502 | T | TRB | TRBV11-2*03 | 0 | 9 | −3 | TRBD2*01 | −5 | 1 | −4 | TRBJ2-1*01 | Yes | / |
| TRB | TRBV14*02 | −3 | 4 | −1 | TRBD2*01 | NA | NA | NA | TRBJ2-6*01 | Yes | / | ||
| TRD | TRDV1*01 | 0 | 15 | 0 | TRDD3*01 | −2 | 1 | −3 | TRDJ1*01 | Yes | / | ||
| TRG | TRGV3*02 | −1 | 10 | NA | NA | NA | NA | −4 | TRGJ1*02 | Yes | / | ||
| TRG | TRGV9*01 | −1 | 13 | NA | NA | NA | NA | −6 | TRGJ1*02 | Yes | / | ||
| BG_5038 | B | IGH | IGHV3-49*02 | −15 | 8 | −2 | IGHD2-8*01 | 0 | 2 | −2 | IGHJ6*02 | Yes | / |
| IGK | IGKV1-16*02 | −17 | 9 | −6 | KDE | NA | NA | NA | NA | Yes | / | ||
| TRB | TRBV20-1*05 | 0 | 7 | −3 | TRBD2*01 | −5 | 3 | −2 | TRBJ2-3*01 | Yes | / | ||
| TRD+ | TRDD2*01 | −6 | 5 | NA | NA | NA | NA | 0 | TRDD3*01 | Yes | / | ||
| TRG | TRGV9*01 | 0 | 5 | NA | NA | NA | NA | −1 | TRGJP1*01 | Yes | / | ||
| TRG | TRGV3*02 | 0 | 4 | NA | NA | NA | NA | 0 | TRGJ1*02 | Yes | / | ||
| BG_5418 | B | IGH | IGHV4-30-2*01 | −14 | 34 | 0 | IGHD3-3*01 | −7 | 8 | −11 | IGHJ6*02 | Yes | / |
| IGH | IGHV3-7*02 | −1 | 2 | −10 | IGHD2-2*01 | −5 | 6 | −4 | IGHJ6*02 | Yes | / | ||
| IGK | IGKV3-20*01 | −3 | 3 | NA | NA | NA | NA | 0 | KDE | Yes | / | ||
| IGL | IGLV2-8*01 | −9 | 8 | NA | NA | NA | NA | 0 | IGLJ2*01 | Yes | / | ||
| TRG | TRGV9*01 | −10 | 10 | NA | NA | NA | NA | −5 | TRGJ1*02 | Yes | / | ||
| TRG | TRGV11*01 | −7 | 8 | NA | NA | NA | NA | −3 | TRGJ1*02 | Yes | / | ||
| BG_5452 | T | IGH | IGHV3-11*01 | 0 | 19 | −2 | IGHD6-19*01 | −4 | 4 | −9 | IGHJ4*02 | Yes | / |
| IGH | IGHV3-64D*06 | −2 | 33 | 2 | IGHD6-19*01 | −4 | 4 | 9 | IGHJ4*02 | Yes | / | ||
| IGH | IGHV4-34*01 | 0 | 6 | 1 | IGHD6-6*01 | −7 | 1 | 0 | IGHJ6*02 | Yes | / | ||
| IGH+ | NA | NA | NA | NA | IGHD7-27*01 | −2 | 3 | −3 | IGHJ2*01 | Yes | / | ||
| TRB | NA | NA | NA | NA | TRBD1 | 33 | 0 | 0 | TRBJ2-3 | Yes | / | ||
| TRB | TRBV9*01 | −3 | 10 | NA | TRBD2*01 | NA | NA | 0 | TRBJ2-1*01 | Yes | / | ||
| TRG | TRGV10*02 | −2 | 11 | NA | NA | NA | NA | −6 | TRGJ1*02 | Yes | / | ||
| TRG | TRGV2*01 | 0 | 3 | NA | NA | NA | NA | −1 | TRGJ1*02 | Yes | / | ||
| BG_9574 | T | TRB | TRBV14*01 | −1 | 11 | −4 | NA | NA | NA | NA | TRBJ2-7*01 | Yes | / |
| TRB | TRBV5-3*01 | −2 | 3 | 0 | TRBD1*01 | −3 | 27 | −4 | TRBJ2-7*01 | Yes | / | ||
| TRD | TRDV1*01 | −5 | 5 | 0 | TRDD2*01 | 0 | 11 | 0 | TRDJ1*01 | Yes | / | ||
| TRG | TRGV11*01 | −11 | 3 | −2 | NA | NA | NA | NA | TRGJ2*01 | Yes | / | ||
| TRG | TRGV4*02 | −6 | 2 | −1 | NA | NA | NA | NA | TRGJ1*02 | Yes | / | ||
| TRG | TRGV8*01 | −1 | 3 | −3 | NA | NA | NA | NA | TRGJP2*01 | Yes | / | ||
| BG_9813 | T | TRB+ | NA | NA | NA | NA | TRBD2*01 | −3 | 0 | −5 | TRBJ2-5*01 | Yes | / |
| BG_9813 | T | TRD+ | TRDD2*01 | −10 | 22 | NA | NA | NA | NA | 0 | TRDJ1*01 | Yes | / |
| TRG | TRGV10*02 | −1 | 3 | NA | NA | NA | NA | −6 | TRGJ1*02 | Yes | / | ||
| TRG | TRGV9*01 | −3 | 3 | NA | NA | NA | NA | 0 | TRGJ2*01 | Yes | / | ||
| BG_11360 | B | IGH | IGHV4-4*07 | −2 | 9 | −1 | IGHD2-15*01 | −6 | 12 | −30 | IGHJ6*04 | Yes | / |
| IGH+ | NA | NA | NA | NA | IGHD7-27*01 | 0 | 12 | −7 | IGHJ6*02 | Yes | / | ||
| TRD+ | TRDD2*01 | 0 | 5 | NA | NA | NA | NA | −1 | TRDD3*01 | Yes | / | ||
| TRD+ | TRDV2*02 | 0 | 13 | NA | NA | NA | NA | −3 | TRDD3*01 | Yes | / | ||
| BG_11720 | B | IGH | IGHV6-1*01 | 0 | 5 | −3 | IGHD3-3*01 | −2 | 2 | −6 | IGHJ6*02 | Yes | / |
| TRA+D | TRDV2*03 | 0 | 5 | −2 | TRDD3*01 | 0 | 3 | −4 | TRAJ9*01 | Yes | / | ||
| TRG | TRGV9*01 | −6 | 13 | NA | NA | NA | NA | −1 | TRGJ1*02 | Yes | / | ||
| BG_11806 | B | IGH | IGHV1-2*02 | −1 | 3 | −3 | IGHD2-21*02 | −3 | 24 | −1 | IGHJ5*02 | Yes | / |
| IGK | IGKV2-28*01 | 0 | 6 | NA | NA | NA | NA | −1 | IGKJ4*02 | Yes | / | ||
| IGL | IGLV3-19*01 | −5 | 14 | NA | NA | NA | NA | −4 | IGLJ3*02 | Yes | / | ||
| TRD+ | TRDD2*01 | 0 | 2 | NA | NA | NA | NA | 0 | TRDD3*01 | Yes | / | ||
| TRG | TRGV5*01 | 0 | 3 | NA | NA | NA | NA | −1 | TRGJ1*02 | Yes | / | ||
| BG_11806 | B | TRD+ | TRDD2*01 | 0 | 13 | NA | NA | NA | NA | 0 | TRDJ3*01 | No | Uncommon V/DJ combinations |
| TRA+D | TRDD2*01 | −2 | 3 | NA | NA | NA | NA | −7 | TRAJ30*01 | No | Uncommon V/DJ combinations | ||
| BG_5038 | B | TRA+D | TRDD2*01 | −3 | 53 | NA | NA | NA | NA | −15 | TRAJ48*01 | No | Uncommon V/DJ combinations |
| BG_5418 | B | TRA+D | TRDD2*01 | −1 | 14 | NA | NA | NA | NA | −5 | TRAJ29*01 | No | Uncommon V/DJ combinations |
| TRA+D | TRDD2*01 | −11 | 4 | NA | NA | NA | NA | −1 | TRAJ23*01 | No | Uncommon V/DJ combinations | ||
| TRA | TRAV26-1*01 | 0 | 4 | NA | NA | NA | NA | −2 | TRAJ33*01 | No | Uncommon V/DJ combinations | ||
| TRA | TRAV8-3*01 | 0 | 8 | NA | NA | NA | NA | −7 | TRAJ34*01 | No | Uncommon V/DJ combinations | ||
| BG_5452 | T | TRA | TRAV21*01 | 0 | 1 | NA | NA | NA | NA | 0 | TRAJ48*01 | No | Uncommon V/DJ combinations |
| BG_9574 | T | TRA+D | TRAV29/DV5*01 | 0 | 38 | 0 | TRDD3*01 | −4 | 3 | 0 | TRDJ1*01 | No | Uncommon V/DJ combinations |
| TRA | TRAV21*02 | −7 | 0 | −5 | NA | NA | NA | NA | TRAJ24*02 | No | Uncommon V/DJ combinations | ||
| BG_9813 | T | TRA | TRAV19*01 | −2 | 10 | NA | NA | NA | NA | −4 | TRAJ36*01 | No | Uncommon V/DJ combinations |
| BG_11360 | B | IGH | IGHV4-34*01 | 0 | 9 | −18 | IGHD2-2*01 | 0 | 2 | −4 | IGHJ6*03 | No | Oligoclonality |
| BG_11720 | B | IGH | IGHV4-34*01 | 0 | 6 | −1 | IGHD6-6*01 | −7 | 1 | 0 | IGHJ6*02 | No | Oligoclonality |
| BG_371 | B | IGK | IGKV1-33*01 | −1 | 2 | NA | NA | NA | NA | −2 | IGKJ4*01 | No | Oligoclonality |
| IGK | IGKV1-39*01 | −4 | 8 | NA | NA | NA | NA | −9 | IGKJ2*02 | No | Oligoclonality | ||
| TRG | TRGV4*02 | −4 | 5 | NA | NA | NA | NA | −3 | TRGJ1*01 | No | Oligoclonality | ||
| BG_5418 | B | TRB | TRBV23-1*01 | 0 | 1 | 0 | TRBD2*01 | −7 | 14 | −6 | TRBJ2-7*01 | No | Oligoclonality |
| TRB | TRBV10-3*01 | −8 | 18 | −7 | TRBD2*01 | NA | NA | NA | TRBJ2-3*01 | No | Oligoclonality | ||
| TRB | TRBV24-1*01 | −13 | 5 | −2 | TRBD1*01 | −3 | 5 | 2 | TRBJ2-7*01 | No | Oligoclonality | ||
| BG_5452 | T | IGH | IGHV6-1*01 | 0 | 5 | −3 | IGHD3-3*01 | −2 | 2 | −6 | IGHJ6*02 | No | Oligoclonality |
| BG_9574 | T | TRG | TRGV11*01 | 0 | 1 | −25 | NA | NA | NA | NA | TRGJ1*02 | No | Oligoclonality |
| BG_11360 | B | IGL | IGLV3-10*01 | −2 | 7 | NA | NA | NA | NA | 0 | IGLJ3*02 | No | Low represented clone |
| BG_371 | B | IGH | IGHV1-3*02 | −2 | 4 | −17 | IGHD3-16*01 | −15 | 0 | −18 | IGHJ4*02 | No | Low represented clone |
| BG_9813 | T | TRB | TRBV4-3*01 | −1 | 21 | −6 | TRBD2*02 | NA | NA | NA | TRBJ1-1*01 | No | Low represented clone |
| Patient ID | ALL Lineage | Locus | V Gene | del V | n | del D | D Gene | del D | n | del J | J Gene | Validation | Rearrangement Feature |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BG_11584 | B | IGH | IGHV6-1*01 | −2 | 8 | −6 | IGHD6-6*01 | 0 | 3 | −10 | IGHJ4*02 | Yes | / |
| BG_2097 | B | IGH | NA | NA | NA | NA | IGHD4-23*01 | −2 | 1 | −4 | IGHJ2*01 | Yes | / |
| BG_855 | B | IGH | IGHV6-1*01 | −3 | 2 | −4 | IGHD2-2*01 | −1 | 5 | 0 | IGHJ6*03 | Yes | / |
| IGH | IGHV3-23*01 | 0 | 19 | −2 | IGHD3-9*01 | −8 | 13 | −7 | IGHJ4*02 | Yes | / | ||
| BG_10112 | B | IGH+ | NA | NA | NA | NA | IGHD6-6*01 | −2 | 0 | −7 | IGHJ4*02 | Yes | / |
| IGH+ | NA | NA | NA | NA | IGHD1-7*01 | −6 | 2 | −4 | IGHJ4*02 | Yes | / | ||
| BG_11053 | B | IGH+ | NA | NA | NA | NA | IGHD2-2*01 | −6 | 3 | −4 | IGHJ4*02 | Yes | / |
| BG_1125 | B | IGH+ | NA | NA | NA | NA | IGHD6-25*01 | 0 | −5 | 11 | IGHJ4*02 | Yes | / |
| BG_11584 | B | IGH+ | NA | NA | NA | NA | IGHD1-26*01 | 0 | 3 | −3 | IGHJ3*02 | Yes | / |
| BG_4254 | B | IGH+ | NA | NA | NA | NA | IGHD3-9*01 | −5 | 4 | −11 | IGHJ6*03 | Yes | / |
| IGH+ | NA | NA | NA | NA | IGHD6-6*01 | −4 | 3 | −15 | IGHJ5*02 | Yes | / | ||
| BG_5702 | B | IGH+ | NA | NA | NA | NA | IGHD2-2*02 | −6 | 7 | −5 | IGHJ5*02 | Yes | / |
| BG_8345 | B | IGH+ | NA | NA | NA | NA | IGHD3-22*01 | −2 | 7 | 1 | IGHJ6*02 | Yes | / |
| IGH+ | NA | NA | NA | NA | IGHD1-26*01 | −2 | 0 | 7 | IGHJ4*02 | Yes | / | ||
| BG_9445 | B | IGH+ | NA | NA | NA | NA | IGHD2-2*02 | −3 | 19 | 5 | IGHJ6*03 | Yes | / |
| BG_11269 | T | TRB | TRBV7-8*01 | −3 | 14 | NA | NA | NA | NA | −7 | TRBJ1-4*01 | Yes | / |
| BG_6037 | T | TRB | TRBV4-2*01 | 0 | 25 | NA | NA | NA | NA | −3 | TRBJ2-3*01 | Yes | / |
| TRB | TRBV20-1*02 | −3 | 15 | −2 | TRBD2*02 | −2 | 19 | −6 | TRBJ2-1*01 | Yes | / | ||
| BG_10112 | B | TRG | TRGV11*01 | −4 | 7 | NA | NA | NA | NA | −8 | TRGJP1*01 | Yes | / |
| TRG | TRGV11*01 | −3 | 10 | NA | NA | NA | NA | −1 | TRGJ2*01 | Yes | / | ||
| BG_11269 | T | TRG | TRGV4*02 | −4 | 5 | NA | NA | NA | NA | 0 | TRGJ2*01 | Yes | / |
| TRG | TRGV10*02 | −6 | 0 | NA | NA | NA | NA | −8 | TRGJ1*02 | Yes | / | ||
| BG_11584 | B | TRG | TRGV4*02 | −6 | 2 | NA | NA | NA | NA | 0 | TRGJ2*01 | Yes | / |
| TRG | TRGV10*02 | 0 | NA | NA | NA | NA | 4 | −9 | TRGJP1*01 | Yes | / | ||
| TRG | TRGV11*01 | 0 | 0 | NA | NA | NA | NA | −7 | TRGJP1*01 | Yes | / | ||
| BG_855 | B | TRG | TRGV9*01 | −10 | 3 | NA | NA | NA | NA | −2 | TRGJ1*02 | Yes | / |
| BG_11053 | B | TRD+ | NA | NA | NA | NA | TRDD2*01 | −1 | 22 | −1 | TRDJ1*01 | Yes | / |
| TRD+ | NA | NA | NA | NA | TRDD2*01 | 0 | 19 | 0 | TRDJ4*01 | Yes | / | ||
| BG_4254 | B | TRD+ | NA | NA | NA | NA | TRDD2*01 | 0 | 5 | −2 | TRDD3*01 | Yes | / |
| BG_8345 | B | TRD+ | NA | NA | NA | NA | TRDD2*01 | −3 | 15 | 0 | TRDD3*01 | Yes | / |
| BG_11053 | B | IGH+ | NA | NA | NA | NA | IGHD3-9*01 | −5 | 3 | −4 | IGHJ4*02 | Yes | Oligoclonality |
| IGH+ | NA | NA | NA | NA | IGHD3-9*01 | −5 | 0 | −4 | IGHJ6*02 | Yes | Oligoclonality | ||
| IGH+ | NA | NA | NA | NA | IGHD3-9*01 | −3 | 13 | −6 | IGHJ6*02 | Yes | Oligoclonality | ||
| BG_11345 | T | TRD+ | NA | NA | NA | NA | TRDD2*01 | −8 | 0 | −14 | TRDD3*01 | Yes | Oligoclonality |
| BG_1125 | B | IGK | IGKV1-27*01 | −1 | 0 | NA | NA | NA | NA | 2 | IGKJ1*01 | YesŦ | / |
| BG_11269 | T | IGK | IGKV4-1*01 | −1 | 0 | NA | NA | NA | NA | −4 | IGKJ4*01 | YesŦ | / |
| IGK | IGKV2D-29*01 | −2 | 6 | NA | NA | NA | NA | 0 | IGKJ2*01 | YesŦ | / | ||
| IGK | IGKV1-6*01 | −3 | 0 | NA | NA | NA | NA | 0 | IGKJ1*01 | YesŦ | / | ||
| BG_6037 | T | TRG | TRGV3*01 | 0 | 0 | NA | NA | NA | NA | 0 | TRGJ1*02 | YesŦ | / |
| TRG | TRGV2*01 | 0 | 4 | NA | NA | NA | NA | −2 | TRGJ1*02 | YesŦ | / | ||
| BG_2097 | B | TRA+D | TRDV1*01 | −2 | 8 | NA | NA | NA | NA | −8 | TRAJ29*01 | YesŦ | / |
| BG_855 | B | TRA+D | NA | NA | NA | NA | TRDD2*01 | −4 | 24 | −5 | TRAJ29*01 | YesŦ | / |
| TRA+D | TRDV2*01 | 0 | 6 | 0 | TRDD3*01 | 0 | 5 | −4 | TRAJ58*01 | YesŦ | / | ||
| BG_11269 | T | TRA | TRAV26-1*01 | −3 | 3 | NA | NA | NA | NA | −4 | TRAJ4*01 | YesŦ | / |
| BG_12438 | T | TRA | TRAV19*01 | −8 | 4 | NA | NA | NA | NA | 0 | TRAJ47*02 | YesŦ | / |
| BG_2097 | B | TRA | TRAV13-1*01 | −3 | 0 | NA | NA | NA | NA | −3 | TRAJ35*01 | YesŦ | / |
| BG_5702 | B | TRA | TRAV16*01 | −6 | 2 | NA | NA | NA | NA | −4 | TRAJ9*01 | YesŦ | / |
| BG_10487 | B | IGH+ | NA | NA | NA | NA | IGHD3-16*02 | −5 | 8 | 0 | IGHJ4*02 | YesŦ | Biclonal sequence |
| IGH+ | NA | NA | NA | NA | IGHD3-3*01 | 0 | 11 | −3 | IGHJ5*02 | YesŦ | Biclonal sequence | ||
| BG_6490 | B | IGH+ | NA | NA | NA | NA | IGHD2-21*02 | −1 | 2 | 0 | IGHJ6*03 | YesŦ | Biclonal sequence |
| IGH+ | NA | NA | NA | NA | IGHD2-8*01 | −7 | 8 | −2 | IGHJ3*02 | YesŦ | Biclonal sequence | ||
| BG_8646 | T | TRD+ | NA | NA | NA | NA | TRDD2*01 | −8 | 0 | −14 | TRDD3*01 | YesŦ | Biclonal sequence |
| TRD+ | NA | NA | NA | NA | TRDD2*01 | −8 | 0 | −16 | TRDD3*01 | YesŦ | Biclonal sequence | ||
| BG_1125 | B | IGH | IGHV4-34*02 | 0 | 10 | NA | NA | NA | NA | −4 | IGHJ5*01 | No | Low represented clone |
| BG_5702 | B | IGK | IGKV2-28*01 | −3 | 2 | −7 | KDE | NA | NA | NA | NA | No | Low represented clone |
| BG_12438 | T | TRB | TRBV4-1*02 | −3 | 2 | −1 | TRBD1*01 | −3 | 8 | −2 | TRBJ2-7*01 | No | Low represented clone |
| BG_5702 | B | TRB | TRBV4-1*02 | 0 | 2 | 0 | TRBD1*01 | −4 | 10 | −8 | TRBJ1-1*01 | No | Low represented clone |
| BG_11053 | B | TRG | TRGV9*01 | −1 | 6 | NA | NA | NA | NA | 0 | TRGJ1*02 | No | Low represented clone |
| BG_3895 | T | TRG | TRGV2*01 | 0 | 4 | NA | NA | NA | NA | −10 | TRGJP2*01 | No | Low represented clone |
| BG_11053 | B | TRD+ | NA | NA | NA | NA | TRDD2*01 | 0 | 14 | −10 | TRDJ1*01 | No | Low represented clone |
| BG_12438 | T | TRA | TRAV21*01 | −4 | 7 | NA | NA | NA | NA | 0 | TRAJ27*01 | No | Low represented clone |
| BG_2097 | B | TRA | TRAV19*01 | −10 | 5 | NA | NA | NA | NA | 0 | TRAJ47*01 | No | Low represented clone |
| BG_6490 | B | IGH | IGHV4-34*12 | 0 | 9 | 1 | IGHD2-8*02 | −7 | 8 | −2 | IGHJ3*02 | No | Oligoclonality |
| BG_5702 | B | IGK | IGKV3-20*01 | −4 | 6 | NA | NA | NA | NA | −2 | IGKJ2*01 | No | Oligoclonality |
| BG_2097 | B | TRB | TRBV6-5*01 | 0 | 2 | −4 | TRBD2*01 | −7 | 0 | −4 | TRBJ1-5*01 | No | Oligoclonality |
| BG_11053 | B | TRG | TRGV4*02 | −3 | 0 | NA | NA | NA | NA | −4 | TRGJ2*01 | No | Oligoclonality |
| TRG | TRGV11*01 | −7 | 7 | NA | NA | NA | NA | −1 | TRGJ2*01 | No | Oligoclonality | ||
| BG_2097 | B | TRG | TRGV3*01 | 0 | 15 | NA | NA | NA | NA | −1 | TRGJ2*01 | No | Oligoclonality |
| BG_9445 | B | TRG | TRGV10*02 | 0 | 3 | NA | NA | NA | NA | 4 | TRGJ1*01 | No | Oligoclonality |
| BG_11584 | B | IGH | IGHV4-31*02 | −1 | 1 | −3 | IGHD3-10*01 | −6 | 0 | 0 | IGHJ4*02 | ND | <5% |
| BG_2481 | T | IGH | NA | NA | NA | NA | IGHD7-27*01 | −11 | 0 | 0 | IGHJ1*01 | ND | <5% |
| BG_11054 | B | IGH+ | NA | NA | NA | NA | IGHD6-13*01 | 3 | 0 | 21 | IGHJ1*01 | ND | <5% |
| IGH+ | NA | NA | NA | NA | IGHD7-27*01 | 0 | 2 | 6 | IGHJ4*02 | ND | <5% | ||
| BG_10112 | B | TRG | TRGV5*01 | 0 | 6 | NA | NA | NA | NA | −1 | TRGJ1*02 | ND | <5% |
| TRG | TRGV9*01 | 0 | 9 | NA | NA | NA | NA | 0 | TRGJ1*02 | ND | <5% | ||
| TRG | TRGV11*01 | −9 | 12 | NA | NA | NA | NA | −5 | TRGJ1*02 | ND | <5% | ||
| TRG | TRGV9*01 | −2 | 4 | NA | NA | NA | NA | 0 | TRGJ2*01 | ND | <5% | ||
| TRG | TRGV9*01 | 0 | 4 | NA | NA | NA | NA | −2 | TRGJ2*01 | ND | <5% | ||
| BG_11053 | B | TRG | TRGV10*02 | −2 | 0 | NA | NA | NA | NA | −1 | TRGJ2*01 | ND | <5% |
| TRG | TRGV11*01 | −2 | 15 | NA | NA | NA | NA | 0 | TRGJ1*02 | ND | <5% | ||
| TRG | TRGV11*01 | 0 | 10 | NA | NA | NA | NA | −8 | TRGJ1*01 | ND | <5% | ||
| TRG | TRGV5*01 | 0 | 3 | NA | NA | NA | NA | −5 | TRGJ1*02 | ND | <5% | ||
| BG_11584 | B | TRG | TRGV11*01 | 0 | NA | NA | NA | NA | 5 | −2 | TRGJ1*02 | ND | <5% |
| TRG | TRGV11*01 | −7 | NA | NA | NA | NA | 3 | −7 | TRGJP1*01 | ND | <5% | ||
| TRG | TRGV9*01 | −2 | NA | NA | NA | NA | 11 | −8 | TRGJ1*01 | ND | <5% | ||
| TRG | TRGV11*01 | −3 | NA | NA | NA | NA | 0 | −16 | TRGJ1*02 | ND | <5% | ||
| BG_2097 | B | TRG | TRGV4*02 | 0 | 6 | NA | NA | NA | NA | −5 | TRGJP1*01 | ND | <5% |
| TRG | TRGV4*01 | −5 | 14 | NA | NA | NA | NA | 0 | TRGJ1*02 | ND | <5% | ||
| BG_11053 | B | TRD+ | NA | NA | NA | NA | TRDD2*01 | 0 | 16 | 0 | TRDJ1*01 | ND | <5% |
| BG_12438 | T | TRG | TRGV8*01 | 1 | 0 | NA | NA | NA | NA | −7 | TRGJP2*01 | ND | ND |
| BG_10640 | B | IGH+ | NA | NA | NA | NA | IGHD3-3*01 | −4 | 6 | −5 | IGHJ6*02 | ND• | ND |
| IGH+ | NA | NA | NA | NA | IGHD2-2*02 | −3 | 4 | −10 | IGHJ6*02 | ND• | ND | ||
| BG_4005 | T | Na | Na | Na | Na | Na | Na | Na | Na | Na | Na | ND | / |
| BG_4255 | T | Na | Na | Na | Na | Na | Na | Na | Na | Na | Na | ND | / |
| BG_4379 | T | Na | Na | Na | Na | Na | Na | Na | Na | Na | Na | ND | / |
| Patient ID | ALL lineage | Locus | V Gene | del V | n | del D | D Gene | del D | n | del J | J Gene | Known | Rearrangement feature |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BG_41182 | B | TRG | TRGV9*01 | −1 | 0 | NA | NA | NA | NA | −8 | TRGJ1*02 | Yes | / |
| TRG | TRGV9*01 | −2 | 3 | NA | NA | NA | NA | 0 | TRGJ2*01 | Yes | / | ||
| BG_41408 | T | TRB | TRBV6-1*01 | −10 | 26 | NA | NA | NA | NA | 0 | TRBJ2-1*01 | Yes | / |
| TRG | TRGV11*01 | −2 | 8 | NA | NA | NA | NA | 0 | TRGJP2*01 | Yes | / | ||
| TRG | TRGV2*01 | −25 | 0 | NA | NA | NA | NA | 0 | TRGJP2*01 | Yes | / | ||
| TRD | TRDV2*02 | −4 | 31 | NA | NA | NA | NA | 0 | TRDJ1*01 | Yes | / | ||
| BG_41985 | B | TRG | TRGV2*02 | −4 | 5 | NA | NA | NA | NA | −7 | TRGJ1*02 | Yes | / |
| TRG | TRGV3*01 | −1 | 5 | NA | NA | NA | NA | 0 | TRGJP2*01 | Yes | / | ||
| TRG | TRGV10*02 | −1 | 6 | NA | NA | NA | NA | −8 | TRGJ1*02 | Yes | / | ||
| BG_42228 | T | TRB+ | NA | NA | NA | NA | TRBD2*01 | −2 | 1 | −5 | TRBJ2-7*01 | Yes | / |
| TRG | TRGV2*02 | 0 | 4 | NA | NA | NA | NA | −2 | TRGJ1*02 | Yes | / | ||
| TRG | TRGV2*01 | 0 | 6 | NA | NA | NA | NA | −9 | TRGJ1*02 | Yes | / | ||
| BG_42309 | B | IGH | IGHV3-23*01 | −4 | 19 | −9 | IGHD3/OR15-3a*01 | −9 | 8 | −11 | IGHJ6*02 | Yes | / |
| IGK+ | Intron | 0 | 2 | NA | NA | NA | NA | −5 | KDE | Yes | / | ||
| BG_41985 | B | IGH+ | NA | NA | NA | NA | IGHD1-7*01 | 0 | 1 | −2 | IGHJ6*02 | ND• | / |
| IGH+ | NA | NA | NA | NA | IGHD1-7*01 | −3 | 11 | −17 | IGHJ6*02 | ND• | / | ||
| BG_41165 | T | TRB | TRBV30*02 | −2 | 10 | NA | NA | NA | NA | 0 | TRBJ1-4*01 | Na | Uncommon V/DJ combinations and low represented clone |
| TRA | TRAV19*01 | −2 | 4 | NA | NA | NA | NA | −3 | TRAJ50*01 | Na | Uncommon V/DJ combinations and low represented clone | ||
| BG_41408 | T | TRD | TRDV1*01 | −8 | 20 | 0 | TRDD3*01 | −3 | 5 | −3 | TRDJ4*01 | Na | Uncommon V/DJ combinations |
| BG_42228 | T | TRD | TRDV3*01 | −1 | 9 | 0 | TRDD3*01 | −1 | 0 | −3 | TRDJ1*01 | Na | Uncommon V/DJ combinations |
| TRA | TRAV21*01 | −4 | 1 | NA | NA | NA | NA | −1 | TRAJ29*01 | Na | Uncommon V/DJ combinations | ||
| BG_42309 | B | IGK | GKV3-15*01 | 0 | 3 | NA | NA | NA | NA | −1 | IGKJ2*03 | Na | Uncommon V/DJ combinations |
| IGL | IGLV2-23*01 | 0 | 1 | NA | NA | NA | NA | 0 | IGLJ3*02 | Na | Uncommon V/DJ combinations | ||
| BG_37265 | T-LL | IGK+ | IGKV3-15*01 | −2 | 5 | NA | NA | NA | NA | 6 | KDE | Na | Uncommon and NT in T-lineage |
| IGK+ | IGKV2-28*01 | −3 | 0 | NA | NA | NA | NA | 0 | IGKJ4*01 | Na | Uncommon and NT in T-lineage | ||
| IGK+ | IGKV1-33*01 | −3 | 0 | NA | NA | NA | NA | 0 | IGKJ4*01 | Na | Uncommon and NT in T-lineage | ||
| BG_41182 | B | TRA+D | NA | NA | NA | NA | TRDD2*01 | −3 | 7 | −3 | TRAJ48*01 | No-Na | Uncommon V/DJ combinations |
| IGL | IGLV4-3*01 | 0 | 2 | NA | NA | NA | NA | −2 | IGLJ3*02 | No | Uncommon V/DJ combinations | ||
| IGL | IGLV3-1*01 | −4 | 3 | NA | NA | NA | NA | −5 | IGLJ2*01 | No | Uncommon V/DJ combinations | ||
| TRG | TRGV10*02 | −19 | 8 | NA | NA | NA | NA | −17 | TRGJP1*01 | No | Uncommon V/DJ combinations | ||
| BG_41408 | T | TRB | TRBV9*01 | −4 | 15 | NA | NA | NA | NA | 0 | TRBJ2-1*01 | No | Oligoclonality |
| BG_40129 | T | TRD+ | TRDV2*01 | 0 | 2 | NA | NA | NA | NA | 0 | TRDD3*01 | No | Missed by heteroduplex |
| BG_41182 | B | IGH | IGHV3-38-3*01 | 11 | 9 | NA | NA | NA | NA | 2 | IGHJ6*03 | No | Missed by heteroduplex |
| BG_41209 | T | TRD+ | NA | NA | NA | NA | TRDD2*01 | −7 | 4 | −13 | TRDD3*01 | No | Missed by heteroduplex |
| BG_42228 | T | TRB | TRBV4-1*02 | 0 | 4 | 0 | TRBD2*01 | −2 | 0 | −5 | TRBJ2-1*01 | No | Missed by heteroduplex |
| BG_42309 | B | IGK+ | IGKV1-17*01 | −1 | 0 | NA | NA | NA | NA | 8 | KDE | No | Missed by heteroduplex |
| BG_39652 | T-LL | TRG | TRGV4*01 | −2 | 0 | NA | NA | NA | NA | −5 | TRGJ1*01 | No | Low represented clone |
| BG_41165 | T | TRG | TRGV4*01 | 0 | 4 | NA | NA | NA | NA | −11 | TRGJ1*02 | No | Low represented clone |
| TRG | TRGV11*01 | −11 | 17 | NA | NA | NA | NA | 6 | TRGJP*01 | No | Low represented clone | ||
| BG_41182 | B | TRG | TRGV2*01 | −3 | 2 | NA | NA | NA | NA | −8 | TRGJ1*02 | No | Low represented clone |
| BG_41733 | T | Na | Na | Na | Na | Na | Na | Na | Na | Na | Na | ND | / |
| BG_9541 | T | Na | Na | Na | Na | Na | Na | Na | Na | Na | Na | ND | / |
| Patient ID | Gene | HGVSc | HGVSp | VAF | RD | ARD | Consequence |
|---|---|---|---|---|---|---|---|
| BG_42309 | CREBBP | NM_004380.2:c.4427C>T | NP_004371.2:p.Pro1476Leu | 78.6 | 641 | 504 | missense_variant |
| BG_40129 | EZH2 | NM_004456.4:c.1613C>T | NP_004447.2:p.Ser538Leu | 51.7 | 410 | 212 | missense_variant |
| BG_40129 | EZH2 | NM_004456.4:c.347T>C | NP_004447.2:p.Leu116Pro | 43.3 | 282 | 122 | missense_variant |
| BG_41733 | EZH2 | NM_004456.4:c.1987T>A | NP_004447.2:p.Tyr663Asn | 18.2 | 292 | 53 | missense_variant |
| BG_40129 | FBXW7 | NM_033632.3:c.1513C>T | NP_361014.1:p.Arg505Cys | 7.4 | 542 | 40 | missense_variant |
| BG_4379 | FBXW7 | NM_033632.3:c.62G>A | NP_361014.1:p.Gly21Asp | 47.7 | 2514 | 1200 | missense_variant |
| BG_41165 | FLT3 | NM_004119.2:c.1779_1793dupTTTCAGAGAATATGA | NP_004110.2:p.Asp593_Tyr597dup | 11.8 | 330 | 39 | inframe_insertion |
| BG_41985 | FLT3 | NM_004119.2:c.2503G>A | NP_004110.2:p.Asp835Asn | 27.6 | 181 | 50 | missense_variant |
| BG_4379 | FLT3 | NM_004119.2:c.2864A>G | NP_004110.2:p.Tyr955Cys | 51.7 | 1412 | 730 | missense_variant |
| BG_37265 | IDH2 | NM_002168.2:c.419G>A | NP_002159.2:p.Arg140Gln | 22.7 | 1122 | 255 | missense_variant |
| BG_39652 | IDH2 | NM_002168.2:c.547delGACinsAAG | NP_002159.2:p.Asp183Lys | 5.6 | 144 | 8 | missense_variant |
| BG_41733 | IDH2 | NM_002168.2:c.419G>A | NP_002159.2:p.Arg140Gln | 45.8 | 690 | 316 | missense_variant |
| BG_4379 | IDH2 | NM_002168.2:c.419G>A | NP_002159.2:p.Arg140Gln | 42.9 | 1559 | 668 | missense_variant |
| BG_41165 | IKZF1 | NM_006060.4_dupl12.1:c.849G>T | NP_006051.1_dupl12.1:p.Arg284Leu | 43.6 | 165 | 72 | missense_variant |
| BG_4379 | IKZF1 | NM_006060.4_dupl12.1:c.396G>T | NP_006051.1_dupl12.1:p.Gly133Val | 49.0 | 514 | 252 | missense_variant |
| BG_40129 | JAK1 | NM_002227.2:c.1954T>C | NP_002218.2:p.Tyr652His | 52.5 | 179 | 94 | missense_variant |
| BG_42228 | JAK1 | NM_002227.2:c.2107A>T | NP_002218.2:p.Ser703Cys | 42.8 | 566 | 242 | missense_variant |
| BG_4255 | JAK1 | NM_002227.2:c.2170C>T | NP_002218.2:p.Arg724Cys | 12.2 | 1199 | 146 | missense_variant |
| BG_41182 | JAK2 | NM_004972.3:c.2171T>C | NP_004963.1:p.Ile724Thr | 12.0 | 465 | 56 | missense_variant |
| BG_11269 | JAK3 | NM_000215.3:c.1370G>A | NP_000206.2:p.Cys457Tyr | 49.2 | 177 | 87 | missense_variant |
| BG_40129 | JAK3 | NM_000215.3:c.2570T>C | NP_000206.2:p.Leu857Pro | 47.2 | 301 | 142 | missense_variant |
| BG_40129 | JAK3 | NM_000215.3:c.2536G>A | NP_000206.2:p.Asp846Asn | 13.4 | 307 | 41 | missense_variant |
| BG_42228 | JAK3 | NM_000215.3:c.1533G>A | NP_000206.2:p.Met511Ile | 44.0 | 650 | 286 | missense_variant |
| BG_4255 | JAK3 | NM_000215.3:c.1533G>A | NP_000206.2:p.Met511Ile | 29.1 | 955 | 278 | missense_variant |
| BG_11269 | KRAS | NM_033360.2:c.182A>T | NP_203524.1:p.Gln61Leu | 6.6 | 802 | 53 | missense_variant |
| BG_10442 | NOTCH1 | NM_017617.3:c.7324_7325insTC | NP_060087.3:p.Asp2442ValfsTer36 | 31.3 | 412 | 129 | frameshift_variant |
| BG_37265 | NOTCH1 | NM_017617.3:c.5033T>C | NP_060087.3:p.Leu1678Pro | 5.8 | 291 | 17 | missense_variant |
| BG_40129 | NOTCH1 | NM_017617.3:c.4799T>A | NP_060087.3:p.Leu1600Gln | 43.8 | 105 | 46 | missense_variant |
| BG_40129 | NOTCH1 | NM_017617.3:c.7541C>A | NP_060087.3:p.Pro2514His | 28.6 | 276 | 79 | missense_variant |
| BG_40129 | NOTCH1 | NM_017617.3:c.7387delG | NP_060087.3:p.Ala2463ProfsTer14 | 5.8 | 223 | 13 | frameshift_variant |
| BG_41209 | NOTCH1 | NM_017617.3:c.5165A>C | NP_060087.3:p.Gln1722Pro | 24.7 | 174 | 43 | missense_variant, splice_region_variant |
| BG_41209 | NOTCH1 | NM_017617.3:c.4787T>C | NP_060087.3:p.Leu1596Pro | 6.0 | 117 | 7 | missense_variant |
| BG_41408 | NOTCH1 | NM_017617.3:c.3394C>T | NP_060087.3:p.Arg1132Cys | 20.6 | 194 | 40 | missense_variant |
| BG_42228 | NOTCH1 | NM_017617.3:c.4778T>C | NP_060087.3:p.Leu1593Pro | 48.7 | 189 | 92 | missense_variant |
| BG_4255 | NOTCH1 | NM_017617.3:c.4776_4777insAGAACC | NP_060087.3:p.Phe1592_Leu1593insArgThr | 19.6 | 168 | 33 | inframe_insertion |
| BG_4255 | NOTCH1 | NM_017617.3:c.141-4A>G | 5.2 | 155 | 8 | splice_region_variant, intron_variant | |
| BG_37265 | NRAS | NM_002524.4:c.37G>C | NP_002515.1:p.Gly13Arg | 19.3 | 1464 | 283 | missense_variant |
| BG_41408 | NRAS | NM_002524.4:c.35G>A | NP_002515.1:p.Gly12Asp | 26.8 | 1131 | 303 | missense_variant |
| BG_4379 | NRAS | NM_002524.4:c.35G>A | NP_002515.1:p.Gly12Asp | 43.4 | 2130 | 924 | missense_variant |
| BG_5038 | PAX5 | NM_016734.2:c.780+5G>T | 45.5 | 321 | 146 | splice_region_variant, intron_variant | |
| BG_21292 | PTEN | NM_000314.4:c.493G>T | NP_000305.3:p.Gly165Ter | 32.9 | 347 | 114 | stop_gained, splice_region_variant |
| BG_21292 | PTEN | NM_000314.4:c.736_737insAG | NP_000305.3:p.Pro246GlnfsTer11 | 34.3 | 1130 | 388 | frameshift_variant |
| BG_37265 | SH2B3 | NM_005475.2:c.1345G>A | NP_005466.1:p.Glu449Lys | 5.8 | 924 | 54 | missense_variant |
| BG_39652 | SH2B3 | NM_005475.2:c.927-2delAGinsCT | 5.4 | 112 | 6 | splice_acceptor_variant | |
| BG_4255 | SH2B3 | NM_005475.2:c.1038dupG | NP_005466.1:p.Leu347AlafsTer38 | 50.2 | 944 | 474 | frameshift_variant |
| BG_39652 | TET2 | NM_001127208.2:c.1588delCAinsTG | NP_001120680.1:p.Gln530Trp | 6.2 | 242 | 15 | stop_gained |
| BG_41165 | TET2 | NM_001127208.2:c.5733delA | NP_001120680.1:p.Lys1911AsnfsTer39 | 35.7 | 493 | 176 | frameshift_variant |
| BG_10442 | TP53 | NM_000546.5:c.684_685insGGGGTTTGACC | NP_000537.3:p.Cys229GlyfsTer3 | 5.9 | 236 | 14 | stop_gained,frameshift_variant |
| BG_10442 | TP53 | NM_000546.5:c.651_654dupGGTG | NP_000537.3:p.Pro219GlyfsTer4 | 33.7 | 943 | 318 | frameshift_variant |
| BG_11584 | TP53 | NM_000546.5:c.844C>G | NP_000537.3:p.Arg282Gly | 4.6 | 1034 | 47 | missense_variant |
| BG_41209 | TP53 | NM_000546.5:c.390_426delCAACAAGATGTTTTGCCAACTGGCCAAGACCTGCCCT | NP_000537.3:p.Asn131CysfsTer27 | 35.0 | 117 | 41 | frameshift_variant |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavagna, R.; Guinea Montalvo, M.L.; Tosi, M.; Paris, M.; Pavoni, C.; Intermesoli, T.; Bassan, R.; Mosca, A.; Rambaldi, A.; Spinelli, O. Capture-Based Next-Generation Sequencing Improves the Identification of Immunoglobulin/T-Cell Receptor Clonal Markers and Gene Mutations in Adult Acute Lymphoblastic Leukemia Patients Lacking Molecular Probes. Cancers 2020, 12, 1505. https://doi.org/10.3390/cancers12061505
Cavagna R, Guinea Montalvo ML, Tosi M, Paris M, Pavoni C, Intermesoli T, Bassan R, Mosca A, Rambaldi A, Spinelli O. Capture-Based Next-Generation Sequencing Improves the Identification of Immunoglobulin/T-Cell Receptor Clonal Markers and Gene Mutations in Adult Acute Lymphoblastic Leukemia Patients Lacking Molecular Probes. Cancers. 2020; 12(6):1505. https://doi.org/10.3390/cancers12061505
Chicago/Turabian StyleCavagna, Roberta, Marie L. Guinea Montalvo, Manuela Tosi, Michela Paris, Chiara Pavoni, Tamara Intermesoli, Renato Bassan, Andrea Mosca, Alessandro Rambaldi, and Orietta Spinelli. 2020. "Capture-Based Next-Generation Sequencing Improves the Identification of Immunoglobulin/T-Cell Receptor Clonal Markers and Gene Mutations in Adult Acute Lymphoblastic Leukemia Patients Lacking Molecular Probes" Cancers 12, no. 6: 1505. https://doi.org/10.3390/cancers12061505
APA StyleCavagna, R., Guinea Montalvo, M. L., Tosi, M., Paris, M., Pavoni, C., Intermesoli, T., Bassan, R., Mosca, A., Rambaldi, A., & Spinelli, O. (2020). Capture-Based Next-Generation Sequencing Improves the Identification of Immunoglobulin/T-Cell Receptor Clonal Markers and Gene Mutations in Adult Acute Lymphoblastic Leukemia Patients Lacking Molecular Probes. Cancers, 12(6), 1505. https://doi.org/10.3390/cancers12061505