1. Introduction
One of the major challenges in the clinical management of cancer is resistance to chemotherapeutics. Multidrug resistance (MDR) has been intensively studied, and overexpression of ATP-binding cassette (ABC) transporters has been considered to be the most prominent underlying mechanism for MDR. Despite research efforts to develop compounds that inhibit the efflux activity of ABC transporters and increase classical chemotherapy efficacy, to date, the Food and Drug Administration has not approved the use of any ABC transporter inhibitor due to toxicity issues [
1]. Therefore, it is necessary to find other targets.
The Hedgehog (Hh) signaling pathway controls cell differentiation and proliferation. It plays a crucial role during embryonic development and, in adulthood, it is involved in stem cell homeostasis and tissue regeneration. However, Hh signaling is also involved in cancer development, progression, and metastasis. Aberrant activation of Hh signaling has been observed in many aggressive cancers [
2], in particular, in cells exhibiting resistance to chemotherapy such as cancer stem cells or tumor-initiating cells [
3]. The Hh receptor, Patched (Ptch1), whose expression is induced upon activation of the Hh pathway, is overexpressed in many cancers, including breast, prostate, ovary, colon, brain, melanoma [
4,
5,
6], and myeloid leukemia [
7,
8] (see the Human Protein Atlas website
http://www.proteinatlas.org/ENSG00000185920-PTCH1/cancer). Studies have even suggested Ptch1 as an early marker of gastric and thyroid cancers [
9,
10]. We previously showed, for the first time, that Ptch1 has a drug efflux activity and contributes to the resistance of cancer cells to chemotherapy [
11]. Remarkably, Ptch1 is not an ABC transporter but uses the proton motive force to efflux drugs. This allows Ptch1 to efflux drugs, at the expense of proton consumption, from the extracellular medium of cancer cells where the extracellular pH is acidic due to the strong glucose consumption (Warburg effect) [
12]. This metabolic feature makes Ptch1 drug efflux activity specific to cancer cells. Hence, Ptch1 is a particularly relevant and highly specific therapeutic target for resistant cancers that express Ptch1. This breakthrough allowed us to propose Ptch1 as a new target to enhance the efficiency of classical or targeted chemotherapeutic treatments and decrease the risk of recurrence and metastasis [
13]. We then developed a test using Ptch1-overexpressing yeast to identify molecules that were able to inhibit the drug efflux activity of Ptch1 [
14]. A first screening of natural compounds purified from marine sponges led to the identification of panicein A hydroquinone (PAH). We showed that this compound strongly inhibited the resistance of Ptch1-overexpressing yeast to doxorubicin (dxr), a chemotherapeutic agent used to treat many cancers, as well as increased the cytotoxic effect of dxr in melanoma cells and strongly inhibited in vitro dxr efflux [
15]. The screening of a chemical library allowed us to identify a second inhibitor, methiothepin, which increases the efficacy of dxr against adrenocortical carcinoma cells, in vitro and in vivo [
16]. These discoveries suggest that the use of inhibitors of Ptch1 drug efflux activity in combination with classical chemotherapy, such as doxorubicin, could be a novel way to circumvent drug resistance, recurrence and metastasis of tumors expressing Ptch1.
Around 45–50% of cutaneous melanomas have mutations in the BRAF serine/threonine kinase. These patients are treated with vemurafenib. This targeted chemotherapy presents heterogeneous clinical responses, and almost all patients who experience an initial response to vemurafenib ultimately acquire resistance and relapse [
17]. In the present study, we have performed the chemical synthesis of the Ptch1 drug efflux inhibitor, PAH, and some analogues, and conducted a preliminary structure activity relationship (SAR) study to enable the identification of a key pharmacophore. We showed that PAH enhances the efficacy of vemurafenib against BRAF
V600E melanoma cells, in vitro and in vivo, by directly interacting with Ptch1 and inhibiting vemurafenib efflux. Our results suggest that the use of this inhibitor of Ptch1 drug efflux in combination with vemurafenib could be a promising therapeutic option to improve vemurafenib efficacy against resistant BRAF
V600E melanomas.
3. Discussion
Cutaneous melanoma is a complex disorder characterized by elevated heterogeneity. It is one of the most aggressive types of cancer and one of the leading causes of cancer-related mortality due its metastatic power. Its therapeutic management is a real challenge as it is amongst the solid malignancies most refractory to conventional cancer therapies [
27]. Up to 90% of melanomas exhibit aberrant MAPK pathway activation that induces cell cycle deregulation and apoptosis inhibition, and marked improvements in cutaneous melanoma treatment have been achieved by targeting the MAPK signaling pathway. Improved overall survival outcomes were observed with targeted therapies in patients with
BRAFV600 mutant unresectable stage III or stage IV melanoma. Nearly half of patients with metastatic melanomas harbor a valine–glutamine substitution in codon 600 of the serine/threonine kinase BRAF [
28]. Vemurafenib, dabrafenib, and encorafenib are BRAF inhibitors (BRAFi) approved by the US Food and Drug Administration (FDA) to treat patients with
BRAFV600E mutated metastatic melanomas [
29]. BRAFi have relatively high response rates; however, patients almost invariably develop disease progression after about 5 months. The addition of a MEK inhibitor to BRAFi extends the median duration of response from 5.6 months to 9.5 months [
30,
31]. However, some patients develop resistance to BRAF (±MEK) inhibitors [
31,
32]. Both intrinsic and acquired resistances can be driven by genetic and epigenetic alterations that drive gene expression changes and intratumor heterogeneity which, in turn, enable tumor regrowth and disease relapse [
33]. Although significant progress has been made in therapeutic approaches, cutaneous melanoma still represents a major problem worldwide due to its high incidence and the lack of a curative treatment for advanced stages. The discovery of therapeutic compounds for treating advanced melanomas that are resistant to existing therapies is paramount to further improve patient outcomes.
Amongst the mechanisms used by cancer cells to become resistant to treatment, multidrug resistance (MDR) has been intensively studied [
34]. A key mechanism underlying MDR is overexpression of ATP-binding cassette (ABC) transporters [
35]. However, we recently discovered that the Hedgehog receptor Ptch1, which is overexpressed in many cancers, also pumps chemotherapeutic agents such as doxorubicin out of cancer cell lines that were derived from melanoma and adrenocortical carcinoma (ACC), thereby conferring resistance to chemotherapy [
11,
16].
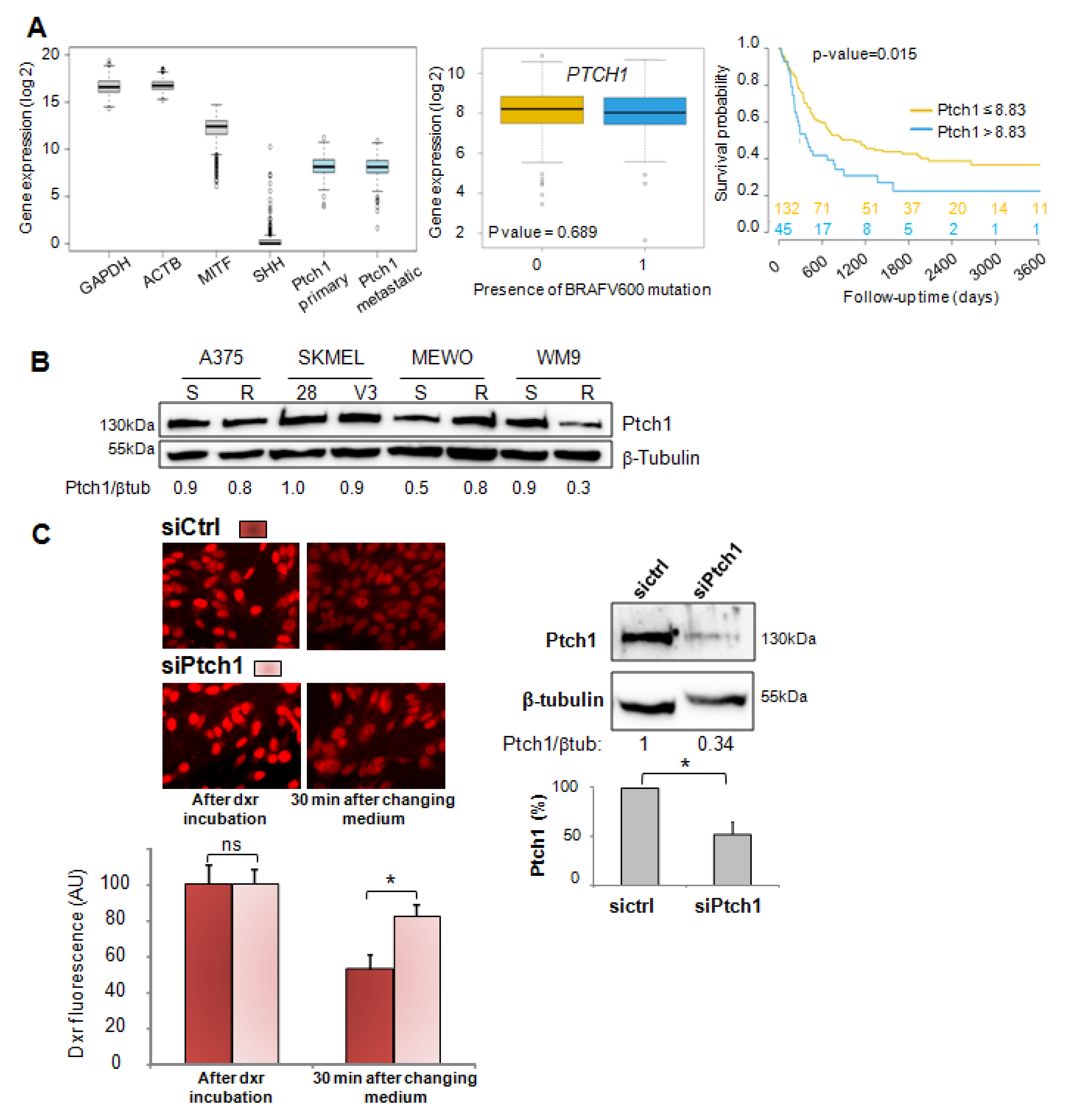
In the present study, we report that Ptch1 is strongly expressed in primary and metastatic specimens from a cohort of 471 cutaneous melanoma patients from The Cancer Genome Atlas (TCGA), and that a high expression level of Ptch1 in patient-derived metastatic samples significantly correlated with a lower overall survival time (
Figure 1A). Ptch1 is endogenously expressed in various melanoma cell lines, and we found that decreased Ptch1 expression strongly inhibited the efflux of doxorubicin, indicating that Ptch1 is involved in doxorubicin efflux in melanoma cells with or without the BRAF mutation (
Figure 1C). Interestingly, we observed that the presence of the BRAF
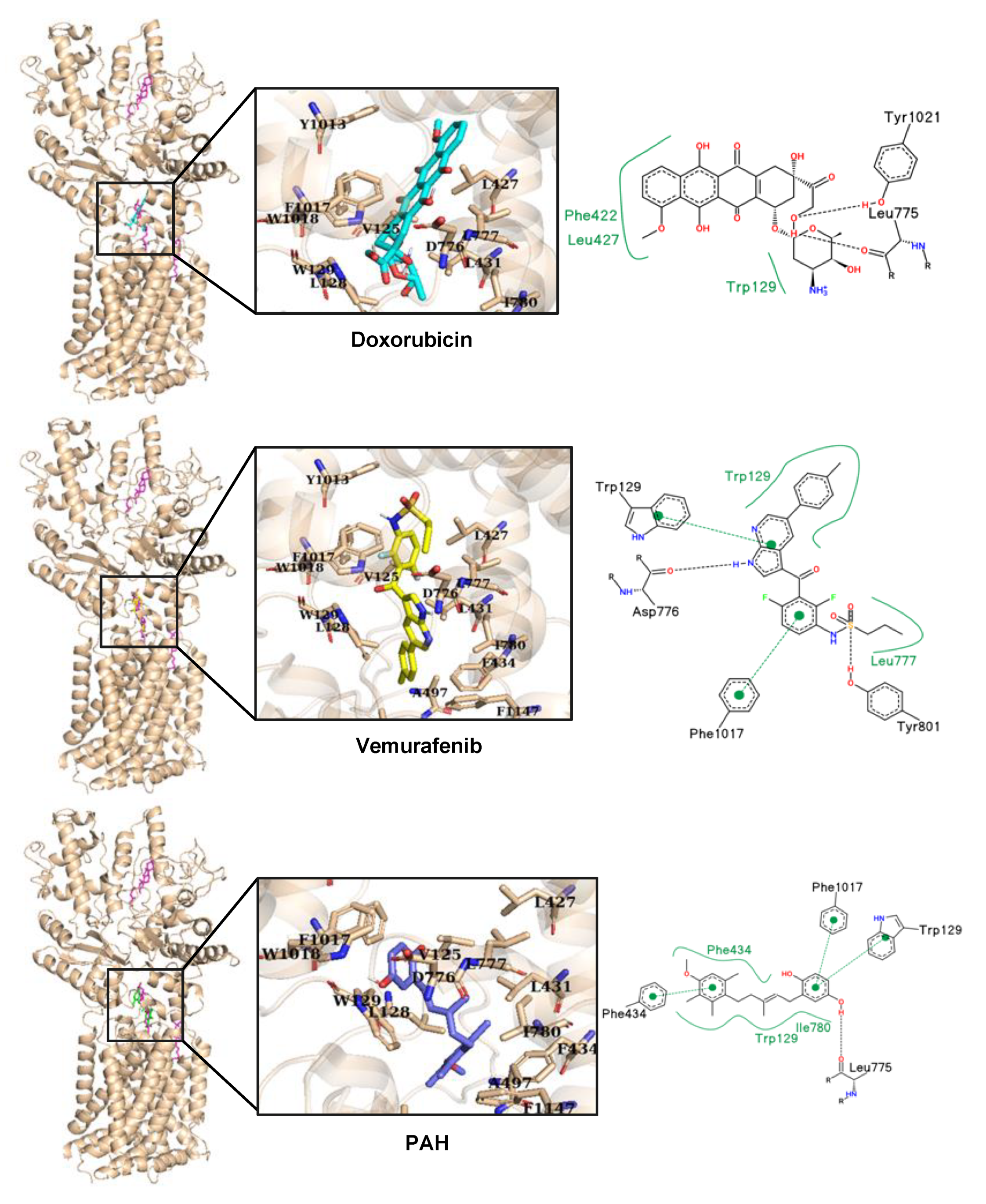
V600E inhibitor, vemurafenib, strongly inhibited the accumulation of doxorubicin in melanoma cells, and in silico docking studies suggested that doxorubicin and vemurafenib bind to Ptch1 at the cholesterol binding site (
Figure 7). These observations suggest that vemurafenib could also be transported by Ptch1 and that Ptch1 could contribute to melanoma cell resistance to vemurafenib, and also that the drugs are exported through the same mechanism as cholesterol.
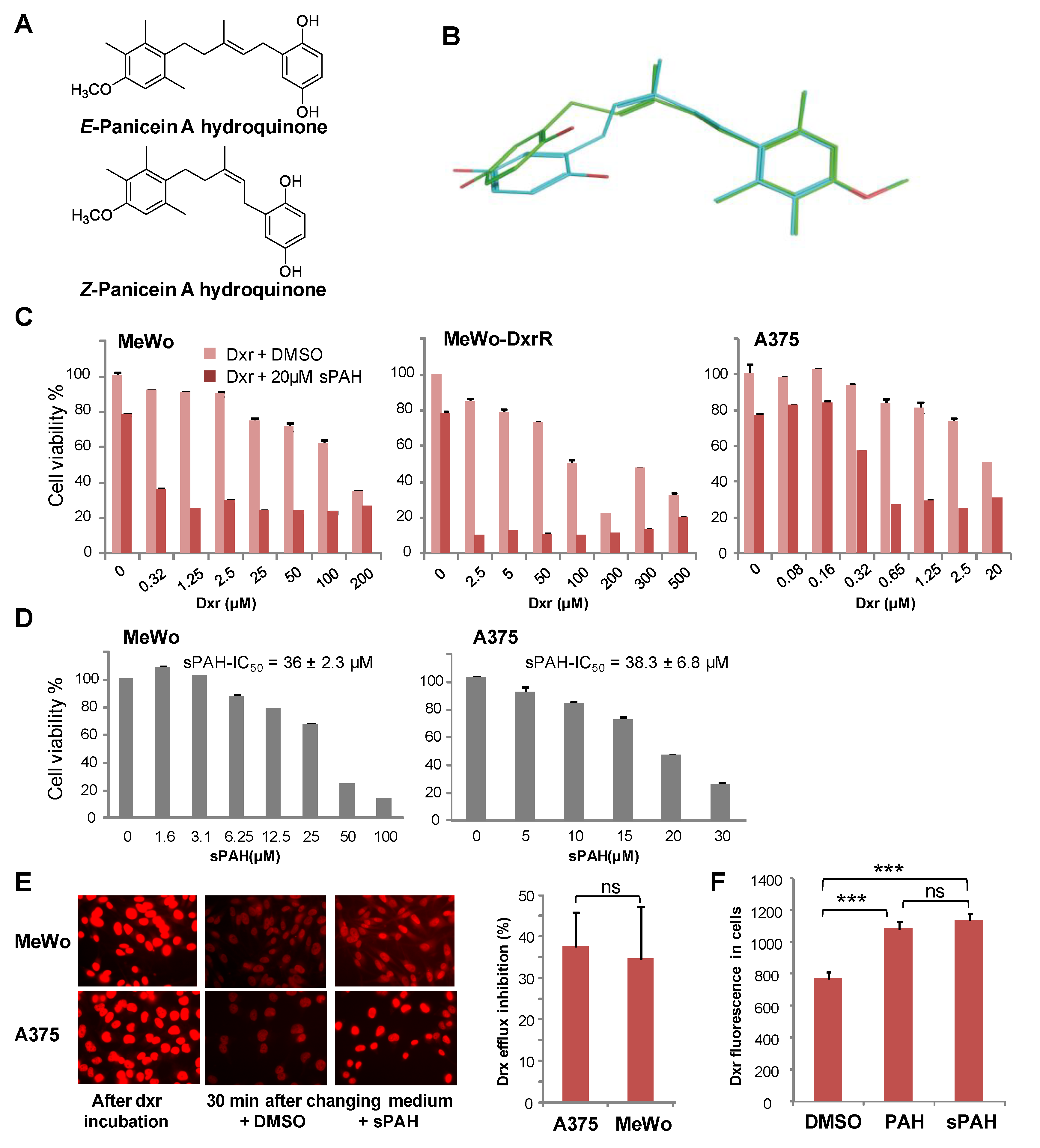
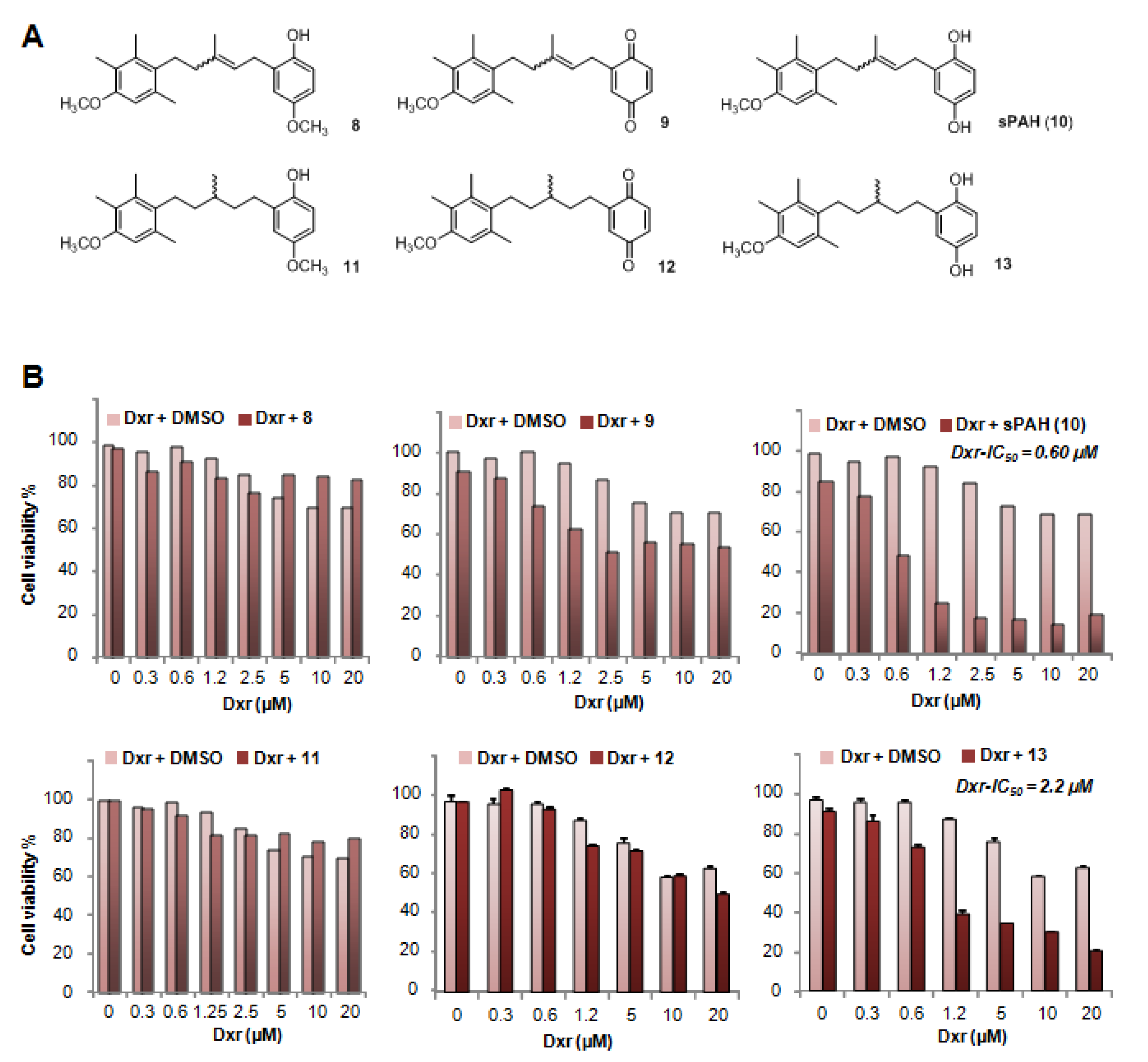
In a previous study, we identified panicein A hydroquinone (PAH) as an inhibitor of the doxorubicin efflux activity of Ptch1 [
15]. Due to the limited availability of PAH that is naturally produced by marine sponges, production of synthetic PAH was necessary. To the best of our knowledge, PAH synthesis has not previously been reported; thus, we carried out the first chemical synthesis of PAH in order to further characterize its activity in vitro and in vivo. While natural PAH is exclusively observed in the E configuration, the chemical synthesis led to a mixture of E and Z stereoisomers that could not be separated. However, the mixture of both stereoisomers proved to be as effective as the natural compound in increasing the cytotoxicity of doxorubicin and inhibiting its efflux in melanoma cells (
Figure 2 and
Table 2). This suggests that the configuration of the double bond does not have a strong influence on the activity. As revealed by the energy minimization study, this may be due to its sufficiently flexible backbone that allows both stereoisomers to superimpose for a large part of the structure of the molecule, coming into proximity with the functional group in such a way that the same interactions can be considered. However, the hydrogenated form of PAH (13) showed a significant loss of activity, indicating that too much flexibility is detrimental. More interestingly, the absence of compound 8 activity highlights the importance of the hydroxyl function of the hydroquinone. This data is perfectly in accordance with the docking study where this function is engaged in a hydrogen bond with Leu775, which could be a key interaction for the activity. This is also confirmed by the fact that the quinone form of PAH (9) is not active, maybe due to the inability to interact as a hydrogen bond donor.
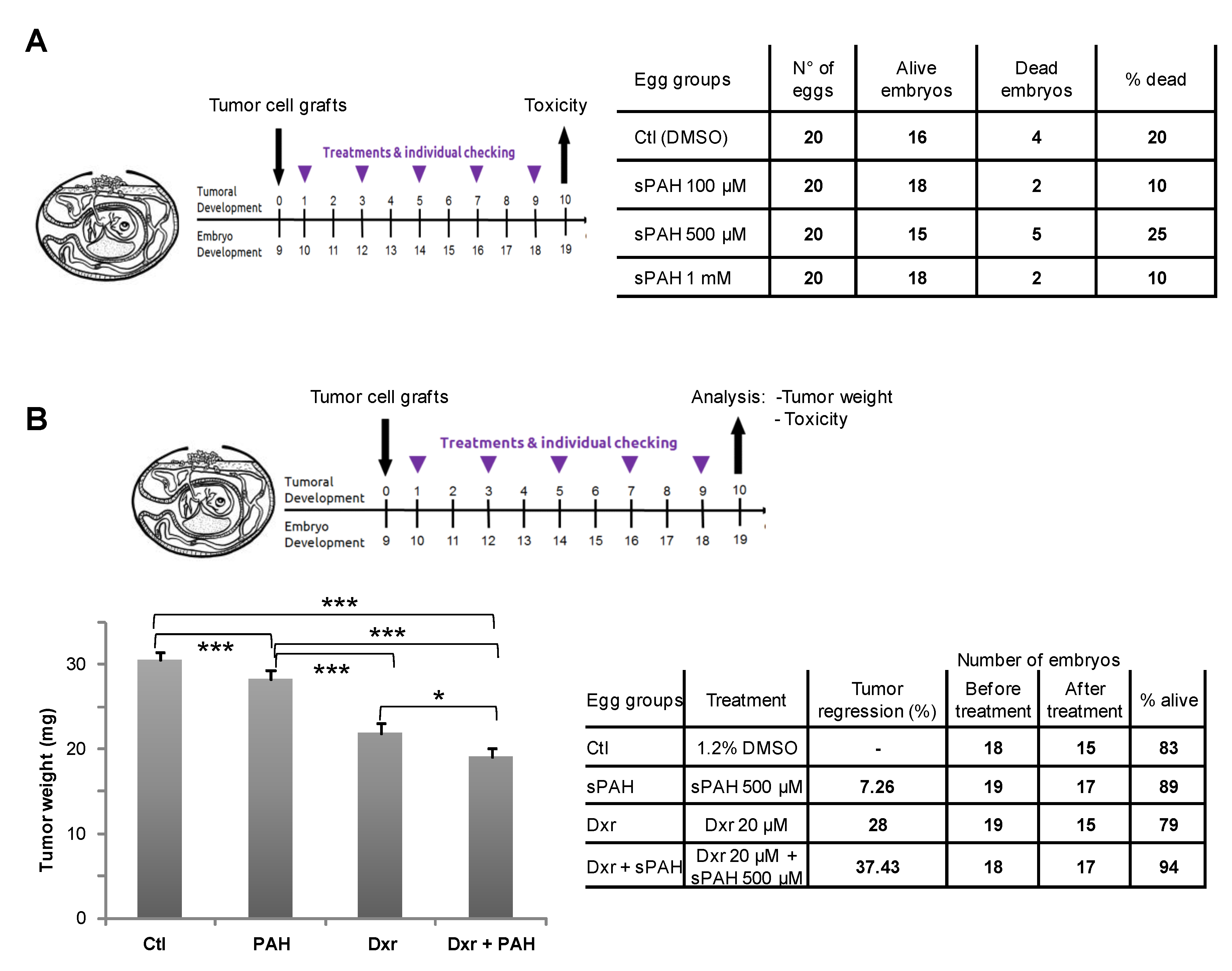
Experiments carried out in embryonated eggs have shown that this synthetic PAH mixture (sPAH) is not toxic to the chicken embryos and, when added in combination with doxorubicin, can inhibit melanoma growth more effectively than doxorubicin alone (
Figure 4), which is a very encouraging in vivo proof of concept.
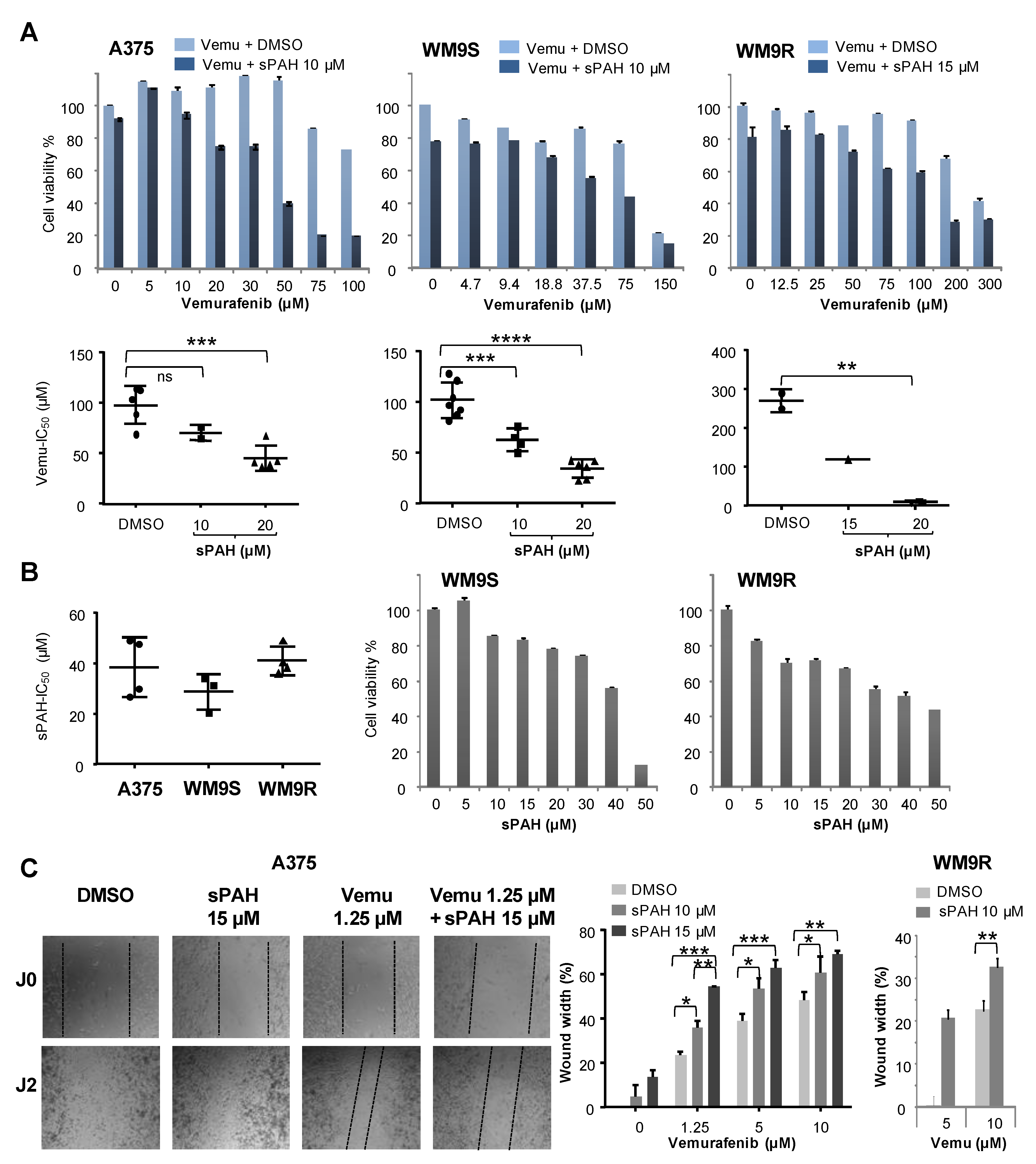
We also observed that sPAH was able to increase the cytotoxicity of cisplatin, another chemotherapeutic agent that we previously identified as a substrate of Ptch1 [
16], against melanoma cells in vitro (
Figure S2). We then wanted to know if sPAH could also enhance the efficiency of targeted chemotherapy such as vemurafenib against BRAF
V600E melanoma cells, and found that sPAH strongly increased the cytotoxicity of vemurafenib, even in resistant BRAF
V600E melanoma cells (
Figure 5). We observed that sPAH itself is slightly cytotoxic for melanoma cells; however, the concentration of sPAH used was not sufficient to explain the strong increase of vemurafenib cytotoxicity induced by sPAH, implying that the combination vemurafenib/sPAH is synergistic, as is the case for combinations of sPAH with doxorubicin or cisplatin. Moreover, wound-healing assays showed that addition of sPAH to vemurafenib significantly reduced the reclosure of wounds compared to vemurafenib alone (
Figure 5), suggesting that a sPAH/vemurafenib combination could be more effective against the migration of BRAF
V600E melanoma cells than vemurafenib alone.
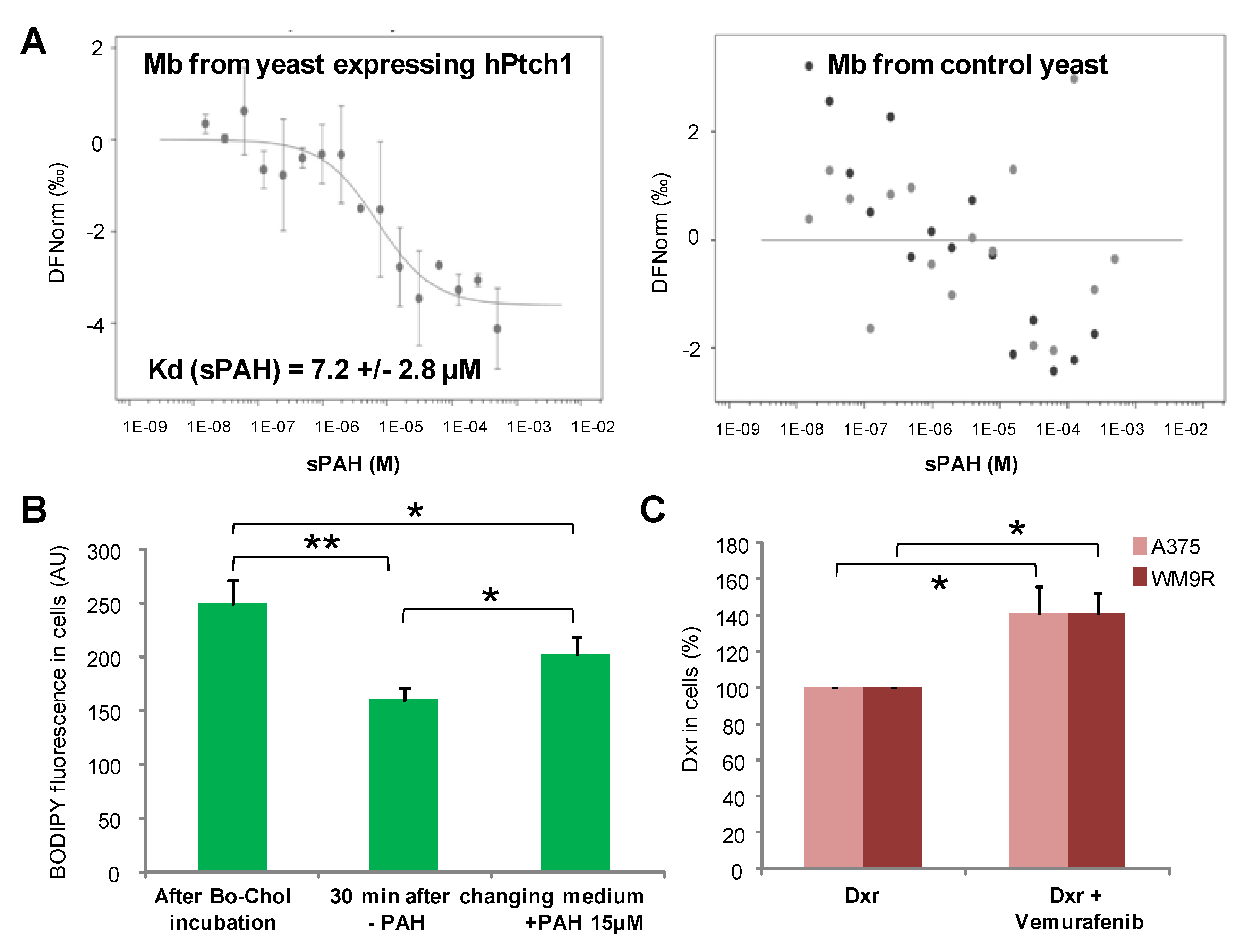
A microscale thermophoresis study allowed us to demonstrate the direct binding of sPAH to Ptch1, and in silico docking of PAH with the Ptch1 structure revealed that the best poses of PAH are located in the cholesterol central binding cavity, where vemurafenib and doxorubicin also present the best docking score (
Figure 6 and
Figure 7).
Altogether, our in vitro results suggest that sPAH increases doxorubicin and vemurafenib efficacy by binding to the same pocket as these chemotherapeutic agents on Ptch1 and inhibiting their efflux by Ptch1. The fact that sPAH also inhibited cholesterol efflux strengthens this interpretation.
Before testing sPAH in mice, we studied its metabolic stability and found that sPAH is very rapidly oxidized on contact with mice liver microsomes. The resulting quinone derivative is unfortunately inactive (
Figure 3,
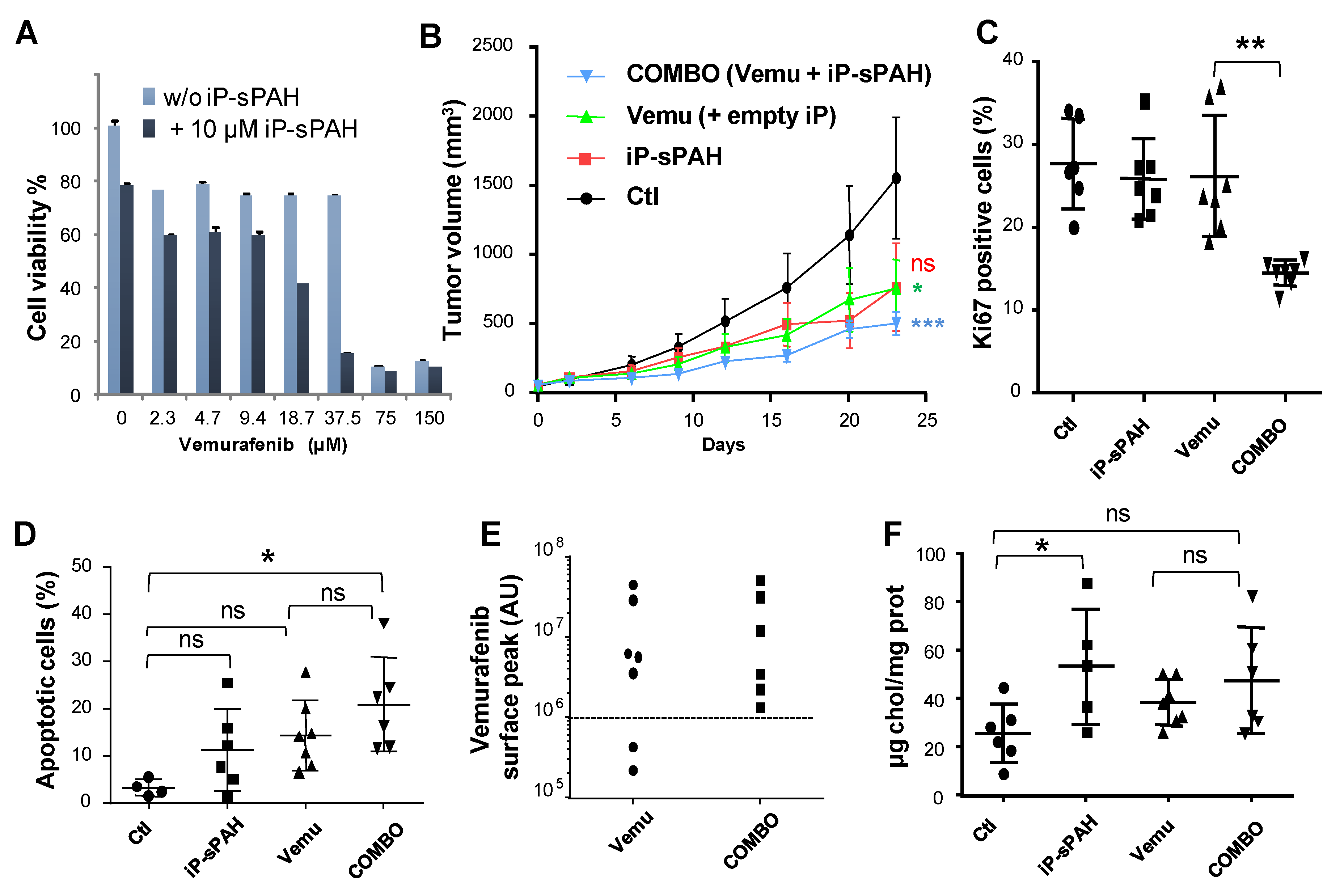
Figures S1 and S4). Therefore, we decided to encapsulate sPAH in particles made of poly(lactic acid) (iP-sPAH). This delivery system is highly effective in protecting sPAH from metabolic degradation and iP-sPAH proved to be as active as free sPAH in increasing the cytotoxicity of vemurafenib in vitro (
Figure 8A). Moreover, experiments performed on mice have shown that iP-sPAH is well tolerated up to high doses by these animals. Based on these results, we subsequently tested the effect of the iP-sPAH/vemurafenib combination on melanoma xenografts in mice. We injected immune-compromised mice with BRAF
V600E A375 melanoma, which is a cell line typically used as a model of xenograft melanoma for testing novel anti-melanoma compounds [
36,
37]. Experiments performed on these mice showed that the addition of iP-sPAH to the vemurafenib treatment inhibited tumor growth more significantly than vemurafenib alone (
Figure 8B). This was accompanied by a decrease in proliferation and an increase in apoptosis of tumor cells, indicating that the treatment is more cytotoxic against melanoma cells, which is supported by the in vitro results. In melanoma cells, BRAF
V600E causes constitutive activation of the BRAF tyrosine kinase, which phosphorylates and activates the MEK1/2 dual kinase, which in turn phosphorylates and activates its only target, the receptor tyrosine kinase ERK1/2. Vemurafenib inhibits BRAF activation and ERK1/2 phosphorylation [
20,
24,
26]. Analyses of tumor extracts revealed that the addition of iP-sPAH to vemurafenib treatment more significantly inhibited the phosphorylation of ERK1/2 than vemurafenib alone, indicating an increase in the effectiveness of vemurafenib. This effect was due to the increased concentration of vemurafenib in tumors (
Figure 8E). Indeed, in the absence of iP-sPAH, we found that some tumors contained little vemurafenib, suggesting that these tumors contained cells that were able to efflux vemurafenib. In the presence of iP-sPAH, all tumors accumulated sufficient vemurafenib to inhibit BRAF and to induce cell apoptosis. All these results strongly suggest that iP-sPAH inhibited the efflux of vemurafenib in melanoma xenografts. Notably, these effects were achieved without obvious undesirable side effects for mice.
We noticed that iP-sPAH alone induced an insignificant decrease in tumor growth (
Figure 8B). Given that sPAH inhibited the cholesterol efflux activity of Ptch1 on A375 cells in vitro (
Figure 6), we wondered if this effect was not due to an increase of cholesterol in melanoma cells. We therefore quantified the amount of cholesterol in tumor extracts and, indeed, found that tumors treated with iP-sPAH contained significantly more cholesterol than other tumors (
Figure 8F), indicating that sPAH inhibited cholesterol efflux mediated by Ptch1 in this system. Previous studies have shown that cholesterol synthesis increases in cancer cells, which helps cancer cell proliferation [
38]. However, Lim and co-workers reported, in 2014, that addition of cholesterol to culture medium led to markedly reduced viability of stomach cancer cells [
39]. Indeed, in healthy cells, accumulation of free cholesterol has been shown to induce many mechanisms of cellular toxicity, including disrupted function of the integral membrane proteins and signaling proteins that reside in membrane domains, intracellular cholesterol crystallization, oxysterol formation, and the triggering of apoptotic signaling pathways [
40]. Thus, we cannot exclude that the increase in cholesterol accumulation caused by the inhibition of Ptch1 cholesterol efflux by sPAH also contributes to toxicity in melanoma cells. In such a hypothesis, the inhibition of Ptch1 efflux activity by sPAH would have dual benefits: to keep the intracellular antineoplastic concentration high and to specifically increase the intracellular cholesterol concentration in cancer cells.
Our data provide strong evidence that PAH is a highly promising lead for the treatment of vemurafenib resistant BRAFV600E melanoma. However, one of the key pharmacophores of sPAH is not stable under biological conditions in living animals and, therefore, in human, since the hydroquinone is metabolized in the inactive quinone form. As the rest of the scaffold is stable and no other modifications have been observed in metabolic experiments, we first plan to replace the hydroquinone part by another group, mainly aromatic, with substitutes that maintain the hydrogen bond interactions. In parallel, an in-depth SAR study will be conducted on other parts of PAH, in particular, on the methoxytrimethylphenyl part, to optimize the activity.
4. Materials and Methods
4.1. Chemical and Biological Material
Reagents and solvents were purchased from Merck and Carlo Erba Reagents and used without further purification. All reactions involving air- or moisture-sensitive reagents or intermediates were performed under an argon atmosphere. Flash column chromatography was carried out on silica gel columns (Interchim Puriflash silica HP 15 μm) on a Puriflash XS420 system (Interchim). Analytical thin-layer chromatography (TLC) was conducted on Sigma Aldrich precoated silica gel and compounds were visualized by irradiation (254 nm) and/or by staining with ninhydrin and phosphomolybdic acid. 1H and 13C NMR spectra were recorded on a Bruker AC 200 MHz or a Bruker AC 400 MHz spectrometer. Chemical shifts are reported in parts per million (ppm, δ) referenced to the residual 1H resonance of the solvent (CDCl3, δ 7.26; CD3OD δ 3.31; DMSO-d6 δ 2.50). Doxorubicin hydrochloride and vemurafenib were purchased from Sigma-Aldrich and Selleckchem, respectively. BODIPY-cholesterol was purchased from Avanti (Topfluor, Avanti). Empty i-Particles® made of poly(d,l-lactic acid) only were purchased from Adjuvatis (Lyon, France).
Human melanoma cell lines A375 and MeWo were purchased from ATCC, and cultured in DMEM medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (Invitrogen) at 37 °C in a 5% CO2/95% water-saturated air atmosphere. Melanoma cell lines SKMEL 28, SKMEL V3, WM9S, and WM9R were provided by Robert Ballotti (C3M, Nice, France). MeWo-dxrR cells were obtained by adding increasing concentrations of doxorubicin, up to 0.2 µM, in the culture medium over 6 months. These cells were grown in medium supplemented with 0.2 µM dxr. WM9R cells were obtained by adding increasing concentrations of vemurafenib, up to 5 µM, in the culture medium over 6 months. These cells were grown in medium supplemented with 5 µM vemurafenib. HEK cells were grown in 75 mm tissue culture dishes (Falcon, Franklin Lakes, NJ, USA) in Dulbecco’s modified Eagle’s medium (Gibco, Life Technologies, Saint Aubin, France) supplemented with 10% fetal calf serum (Hyclone, Thermo Fisher Scientific GMBH, Ulm, Germany) and 1% penicillin/streptomycin (Gibco, Life Technologies, Saint Aubin, France) in a humidified incubator at 37 °C (5% CO2).
4.2. Chemical Synthesis of Panicein A Hydroquinone and Analogues
The first steps of the synthesis were conducted as reported [
18] with minor modifications, as described in
Supplementary Material.
Panicein A hydroquinone (10): A solvent mixture of acetone and water in a ratio of 1:1 was prepared. Panicein A (20 mg) was dissolved in 0.3 mL solvent. A suspension of Na2S2O5 (3 eq) in water (0.1 mL) was added dropwise to the starting material. The reaction was stirred at room temperature for 6 h max (see TLC). An extraction with ethyl acetate followed. The organic fractions were washed with saturated NaCl (aq.) solution, dried over Na2SO4, and evaporated. The crude residue was purified by flash chromatography on silica gel using a gradient of PE/EA (5:1) as the eluent to give 10 as a white solid. Yield = 86%. Rf = 0.105 (Cy/EA 5:1). 1H NMR (400 MHz, CDCl3) δ 6.68 (m, 1H), 6.62–6.47 (m, 3H), 5.34 (m, 1H), 3.80 (s, 3H), 3.34 (d, J = 7.1 Hz) and 3.25 (d, J = 7.1 Hz) (1H, E and Z), 2.78–2.66 (m, 2H), 2.35 (s) and 2.32 (s) (3H, E and Z), 2.26 (s) and 2.22 (s) (3H E and Z), 2.20–2.15 (m, 2H), 2.19 (s) and 2.15 (s) (3H, E and Z), 1.81 (s) and 1.78 (s) (3H, E and Z).13C NMR (101 MHz, CDCl3) δ 155.5, 155.4, 149.6, 149.5, 148.2, 148.1, 138.6, 138.2, 136.1, 136.1, 133.9, 133.8, 131.0, 131.0, 128.4, 123.1, 123.0, 122.6, 121.6, 116.7, 116.7, 116.6, 116.5, 113.9, 113.8, 110.7, 110.6, 55.8, 55.8, 39.7, 32.2, 31.0, 29.5, 29.3, 28.9, 28.2, 23.7, 20.6, 20.5, 16.6, 15.9, 15.9, 12.1. ESI: m/z 341.4 (M + H)+ (theoretical 341.2).
Compound 11: Compound 8 was dissolved in ethyl acetate (10 mL solvent for 1 mmol of starting material). Around 1–2 mol% Pd/C (10 mol%) was added, and the reaction mixture was stirred overnight under hydrogen atmosphere. The crude residue was purified by flash chromatography on silica gel using a gradient of PE/EA (5:1) as the eluent to give 11 as a white solid. Yield = 96%. Rf = 0.36 (Cy/EA 5:1). 1H NMR (400 MHz, CDCl3) δ 6.65–6.46 (m, 4H), 3.70 (s, 3H), 3.67 (s, 3H), 2.68–2.36 (m, 4H), 2.23 (s, 3H), 2.14 (s, 3H), 2.06 (s, 3H), 1.70–1.49 (m, 2H), 1.49–1.35 (m, 2H), 1.22 (m, 1H), 0.99 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.3, 153.9, 147.6, 135.9, 133.5, 131.9, 130.3, 122.9, 116.0, 115.9, 111.7, 110.5, 55.9, 55.7, 37.1, 36.8, 33.7, 28.1, 27.3, 20.5, 19.7, 15.8, 12.1. ESI: m/z 357.1 (M + H)+ (theoretical 357.2).
Compound 12: A solvent mixture of acetonitrile and water in ration of 2:1 was prepared. Around 100 mg of 11 was dissolved in 2 mL of solvent and cooled to 0 °C. Under stirring, CAN (2.2 eq) dissolved in 4 mL solvent was added dropwise to the mixture. The mixture was stirred for maximum 4h. The reaction was extracted with ethyl acetate and the crude residue was purified by flash chromatography on silica gel using a gradient of PE/EA (15:1) as the eluent to give 12 as an orange solid. Yield = 79%. 1H NMR (400 MHz, CDCl3) δ 6.80–6.68 (m, 2H), 6.60–6.53 (m, 2H), 3.79 (s, 3H), 2.76–2.38 (m, 4H), 2.31 (s, 3H), 2.22 (s, 3H), 2.15 (s, 3H), 1.66–1.54 (m, 2H), 1.53–1.37 (m, 3H), 1.07 (d, J = 6.3 Hz, 3H, 9-H). ESI: m/z 341.5 (M + H)+ (theoretical 341.2).
Compound 13: Compound 10 was dissolved in ethyl acetate (for 1 mmol of starting material 10 mL solvent). Around 1–2 mol% Pd/C (10 mol%) was added, and the reaction mixture was stirred overnight under hydrogen atmosphere. The crude residue was purified by flash chromatography on silica gel using a gradient of PE/EA (5:1) as the eluent to give 13 as light yellow oil. Yield = 97%. 1H NMR (200 MHz, CDCl3) δ 6.61–6.36 (m, 4H), 3.69 (s, 3H), 2.65–2.45 (m, 4H), 2.23 (s, 3H), 2.14 (s, 3H), 2.07 (s, 3H), 1.76–1.07 (m, 5H), 0.99 (d, J = 6.0 Hz, 3H). 13C NMR (50 MHz, CDCl3) δ 155.3, 149.5, 147.5, 135.9, 133.6, 131.9, 130.4, 122.9, 116.9, 116.2, 113.4, 110.5, 55.8, 37.0, 36.8, 33.6, 27.8, 27.2, 20.5, 19.7, 15.9, 12.1. ESI: m/z 343.6 (M + H)+ (theoretical 343.2).
4.3. Alignment of E and Z Stereoisomers of PAH
The RDKit Open-Source Cheminformatics Software [
41] was used to generate up to 100 conformers for each molecule by using the e-LEA3D web server (
https://chemoinfo.ipmc.cnrs.fr). This resulted in 60 and 62 conformers for PAH E and Z, respectively. The shape-based alignment program SENSAAS (
https://arxiv.org/abs/1908.11267) was used to align and compare the two ensembles of conformers. The alignment with the best shape score (gfit = 0.761) is displayed in
Figure 2B. A
gfit score ranges from 0 (dissimilar) to 1 (perfect similarity).
4.4. TCGA Data Analysis
Normalized gene expression data and matching clinical information for cutaneous melanoma (SKCM) tumors were downloaded from TCGA using the R package curatedTCGAData, then separated into primary tumor samples (
n = 103) and metastatic tumor samples (
n = 368). Gene expression data was transformed into log2 scale prior to analysis. A count of 1 was added to all expression values to avoid zeros prior to transformation. BRAF
V600 mutation status was downloaded using the R package GenomicDataCommons (MuTect2 Variant Aggregation and Masking workflow). Survival data was further processed as described in [
42]. Statistical significance of the differences in gene expression distribution was performed using the Wilcoxon rank sum test. A Kaplan–Meier analysis was performed using the R package rms (Harrel). Optimal cut-offs were determined using positional scanning [
43].
4.5. Ptch1 Knock-Down
MeWo cells were transfected with 400 pmol of human Ptch1 Silencer® Select pre-designed siRNA (Ambion, #4392420, s11441 (sense: 5′GCACUUACUUUACGACCUAtt3′; as: 5′UAGGUCGUAAAGUAAGUGCtg3′) or control (medium GC) siRNA oligos (Invitrogen) using Lipofectamine RNAiMAX reagent (Invitrogen) following the manufacturer’s protocol, then seeded in 24-well plates and incubated at 37 °C and 5% CO2 for 16 h before Western blotting and dxr efflux measurements.
4.6. Cytotoxicity Assays
Cells were seeded in 96-well plates in triplicate and grown in medium to achieve 70 to 80% confluence. Medium was then removed and replaced with 100 µL/well of complete medium containing PAH or DMSO as a control. After 2 h, 100 µL of complete medium containing serial dilutions of dxr, cisplatin or vemurafenib were added. Plates were incubated at 37 °C and 5% CO2. After 24 or 48 h, cells were incubated for 3 h at 37 °C with 100 µL/well neutral red (NR) solution (50 µg/mL in medium) following the manufacturer’s protocol. Measurements were made in microplate readers (Multiskan Go Microplate Spectrophotometer from Thermo Scientific). IC50 was defined as the concentration that resulted in a 50% decrease in the number of live cells, and IC50 values were calculated using GraphPad Prism 6 software.
4.7. Wound-Healing Assay
Once cells were confluent in 24-well plates, a wound was created using a p200 tip. Two pictures were taken at two different points of each well immediately after wounding, and 48 h after wounding. Images were taken with Leica DM IRB (5×). The width of the wound was measured using ImageJ software and reported as percentage final wound width/initial wound width.
4.8. Efflux Measurements
Dxr efflux measurements were carried out as previously described [
16]. Cells were seeded on coverslips in 24-well plates and allowed to grow to 80% confluence. Coverslips were incubated at 37 °C and 5% CO
2 with 10 μM dxr in physiological buffer (140 mM NaCl, 5 mM KCl, 1 mM CaCl
2, 1 mM MgSO
4, 5 mM glucose, 20 mM HEPES, pH 7.4). After 2 h, three coverslips were immediately fixed with 4% PFA for the dxr loading control, rapidly washed with PBS and mounted in SlowFade Gold antifade reagent with DAPI (Invitrogen). The other coverslips (triplicated per condition) were incubated with physiological buffer supplemented with DMSO or 10 µM of PAH under gentle shaking at room temperature and protected from light. After 30 min, coverslips were fixed with 4% PFA, washed, and mounted as described above. For competition on dxr loading, A375 and WM9R cells seeded on coverslips were incubated for 1 h at 37 °C and 5% CO
2 with 10 μM dxr in physiological buffer in the presence or the absence of 100 µM vemurafenib. Images were acquired with a Zeiss Axioplan 2 fluorescence microscope coupled to a digital charge-coupled device camera using a 40×/1.3 Plan NeoFluar objective and filters for Alexa 594. Dxr fluorescence was quantified using ImageJ software. Sampling of cells was performed randomly. About 100 cells (from three wells) were scored per condition per experiment.
Cholesterol efflux measurements were carried out as described for dxr efflux except that cells were incubated with 10 µM BODIPY-cholesterol, a fluorescent derivative of cholesterol, and images were acquired using a 40×/1.3 Plan NeoFluar objective and filters for FITC.
4.9. Microscale Thermophoresis
Microscale thermophoresis (MST) is a biophysical technique that measures the strength of the interaction between two molecules by detecting a variation in the fluorescence signal of a fluorescently labeled target as a result of an IR-laser induced temperature change. The range of the variation in the fluorescence signal correlates with the binding of a ligand to the fluorescent target.
Membranes from yeast expressing human Ptch1 or human Smoothened were incubated at 30 µg/mL with 20 nM of the fluorescent dye NT-647 2nd gen (NanoTemper Technologies, München, Germany) to label the His-tag present at the c-terminus of both proteins. In this MST experiment, we kept the concentration of labeled membranes constant, while the concentration of non-labeled sPAH was varied between 250 µM and 15 nM. The assay was performed in PBS containing 0.5% DMSO. After a short incubation, the samples were loaded into Monolith™ NT.115 standard treated capillaries from NanoTemper Technologies and the MST analysis was performed using the NanoTemper Technologies Monolith NT.115 (LED: 30%; MST: medium). The fluorescence within the capillary is excited and detected through the same objective. A focused infrared laser is used to locally heat a defined sample volume. The MST response of fluorescent proteins within the temperature gradient is detected. After activation of the IR laser, a decrease in fluorescence is observed which corresponds to temperature-related intensity change (TRIC) triggered by the fast temperature change and thermophoretic movement of the fluorescent proteins out of the heated sample volume. The MST signal of fluorescent proteins changes or not upon binding to sPAH resulting in different MST traces. For analysis, the change in MST signal is expressed as the change in the normalized fluorescence (Fnorm), which is defined as F1/F0. Titration of sPAH results in a gradual change in MST signal, which is plotted as Fnorm against the sPAH concentration to yield a dose–response curve, which has been fitted to derive binding constants (Kd). The sPAH Kd was determined for 3 independent experiments.
4.10. In Silico Docking
Docking of vemurafenib, doxorubicin, and PAH on Ptch1 structure were performed using Vina toolkit [
44] in USCF Chimera [
45]. The structure of Ptch1 was obtained from RCSB Protein Data Bank (PDB ID: 6N7H, chain A) [
23] and prepared using USCF Chimera Predock Toolkit. For the missing side chains, the Dunbrack rotamer 2010 library [
46] was used and charges were assigned with ANTECHAMBER Amber ff14SB force field [
47]. The docking was first done on the whole protein structure. Observing that the poses with lowest scores were in the previously predicted biologically relevant binding sites for cholesterol, docking was then performed by targeting the central cholesterol cavity.
The analysis was performed on the 10 best poses of the docking (ranked by score). Every interaction was compared to the cholesterol equivalents. Amino acids within a radius of 6 Å from the cholesterol were selected and listed in
Table 2. We highlighted amino acid mutations due to single nucleotide variation associated to disease (with a probability above 0.8) which were reported in BioMuta database [
48].
For the docked ligands, interactions were further analyzed using PoseView [
49], a function of the ProteinsPlus web server [
50]. In order to assess conserved amino acids among the proteins from Patched family, sequence alignment of Patched family members was performed on 30 sequences using T-COFFEE server for transmembrane proteins [
51].
4.11. Quantification of Metabolic Stability
A 10 µL aliquot of the sPAH stock solution (10 mM in DMSO) was diluted in 990 µL of a mixture of acetonitrile and water. This solution was then diluted 100-fold in phosphate buffer containing mice liver microsomes (0.5 mg/mL), 1 mM NADPH, and 3 mM MgCl2, and incubated at 37 °C. After 2, 10, 20, 40, and 60 min, 70 µL aliquots were collected and mixed with 70 µL acetonitrile at 0 °C. Equivalent experiments were performed without NADPH in order to identify chemical instability or enzymatic process not depending on NADPH, and with testosterone as a positive control. The enzymatic reaction was stopped by addition of acetonitrile. Samples were analyzed by LC–MS/MS on an UHPLC LC–MS 8030 (Shimazu, Kyoto, Japan).
For metabolite identification, 50 µM of sPAH were incubated with mice liver microsomes and NADPH. Two samples were prepared: one in which acetonitrile was added immediately (t0) and one in which acetonitrile was added after 30 min. After centrifugation for 5 min at 15,000× g, 2 samples of each supernatant were analyzed by LC–MS/MS on a LC–MS 8030 (Shimazu, Kyoto, Japan) and detected by selected ion monitoring (SIM).
4.12. sPAH Toxicity on Chick Embryos
These experiments were performed by the company INOVOTION (La Tronche, France). Fertilized White Leghorn eggs were incubated at 37.5 °C with 50% relative humidity for 9 days. At this time (E9), an access point to the chorioallantoic membrane (CAM) was made by drilling a small hole through the eggshell into the air sac and a 1 cm2 window was cut in the eggshell above the CAM. Twenty-one eggs were used for each condition. Because of some instances of early death just after the opening of the shell (surgical act), data could be collected in fewer than 21 eggs per group. MeWo cells cultivated in DMEM medium with 10% FBS (and 1% penicillin/streptomycin) were detached with trypsin, washed with complete medium, labeled, and suspended in PBS. An inoculum of 3 × 106 cells was added onto the CAM of each egg (E9). Eggs were then randomized in 4 groups. The chick embryos were then treated every two days (E11, E13, E15, E17) for 10 days in total, by adding 100 µL of vehicle (1% DMSO in PBS) as a negative control or 100 µL of sPAH at 3 different doses onto the CAM. The number of dead embryos evaluates the toxicity after 10 days of the treatment, as does the observation of 22 abnormality checkpoints in surviving embryos.
4.13. Effect of sPAH and Doxorubicin on Melanoma Cells Grafted in Chick Eggs
These experiments were performed by the company INOVOTION (La Tronche, France). Fertilized White Leghorn eggs were randomized in 4 groups 9 days after inoculation of 3.106 MeWo cells onto the CAM of each egg. At day 10 (E10), tumors began to be detectable and they were treated by adding 100 µL of vehicle (1.2% DMSO in PBS), doxorubicin, and sPAH alone or with doxorubicin. Eggs were treated every two days (E10, E12, E14, E16, E18) for 10 days in the same way. At day 19 (E19), the upper portion of the CAM was removed, washed in PBS, and directly transferred in PFA for 48 h, and the tumors were then carefully cut away from normal CAM tissue. Tumors were weighed, and one-way ANOVA analysis with post-tests was performed on these data.
4.14. Preparation and Characterization of i-Particles Loaded with sPAH
iP-sPAH synthesis was custom-developed by Adjuvatis (Lyon, France) based on i-Particles
® preparation by nanoprecipitation method [
52], using poly(
d,
l-lactic acid) (PLA) as polymer. No surfactant or stabilizer was required to stabilize the colloid solution. The resulted 180 nm size iP-sPAH were precisely characterized for their physicochemical parameters (hydrodynamic diameter, size distribution, and zeta potential), using a zetasizer NanoZS from Malvern-Panalytical (UK) and after high dilution in 0.22 µm-filtered 1 mM NaCl solution. sPAH loading (%DL) and entrapment efficiency (%EE) within i-Particles were also precisely quantified by fluorimetry after PLA and sPAH solubilization in acetonitrile, using a standard curve of sPAH ranging from 5 to 55 µg/mL in the same solvent. The measurements were made using a fluorimeter equipped with a 96-well microplate reader (infinite M1000, Tecan, Mannedorf, Switzerland) and black microplates (Greiner Bio-One, Courtaboeuf, FranceCity) at 288 nm excitation and 324 nm emission. The DL and EE were calculated by following equations:
4.15. Evaluation of the Toxicity of sPAH in Mice
sPAH encapsulated in i-Particles was diluted in saline solution and injected intraperitoneally at 40, 13.3, 4, and 1.33 mg/kg (10 mL/kg) in Swiss (CD-1) mice (2 mice per dose). Monitoring and evaluation of toxicity was carried out for 72 h based on the behavioral signs and appearance of the animals. All experiments were carried out following protocols approved by the ethics committee from the University of Strasbourg (APAFIS#3671-2016012012046243) and in accordance with the council of the European communities’ guidelines for animal studies (86/609/CEE).
4.16. Melanoma Xenograft Study in Mice
Forty-eight male athymic Nu/NU NMRI mice (4–5 weeks) were purchased from Charles River and housed under pathogen-free conditions. A 200 µL aliquot of 3 Millions A375 BRAFV600E melanoma cells were injected subcutaneously per mouse. Tumor-bearing mice were treated with 4 therapeutic cycles, which consisted of either vemurafenib (8 mg/kg body weight (5 µL/g in olive oil) intraperitoneally 5 days a week) plus empty i-Particles (lot 180509-DF-CP02, 4 µL/g intraperitoneally every other day), iP-sPAH (Lot: 180821-DF-PAH, 5 mg/kg body weight (4 µL/g) intraperitoneally every other day), or a combination of vemurafenib and iP-sPAH (vemurafenib 8 mg/kg body weight intraperitoneally every other day plus iP-sPAH 5 mg/kg body weight intraperitoneally 5 days a week). Mice from the control group were injected with olive oil (5 µL/g) intraperitoneally 5 days a week plus empty i-Particles (lot 180509-DF-CP02, 4 µL/g) intraperitoneally every other day. Animals were monitored daily for symptoms of disease (weight loss >20%, ruffled coat, hunched back, weakness, reduced motility) and tumor sizes were measured every 3 to 4 days. At day 23 after the start of treatment, when the first tumors reached the longest tumor diameter of 1.5 cm, the study was terminated, and animals sacrificed. Tumors were excised and subsequently snap frozen in isopentane using OCT (optimal cutting temperature) compound. This study was conducted in agreement with the French Guidelines for animal handling and approved by local ethics committee (APAFIS#18382-201901081114131v3), and in accordance with the council of the European communities’ guidelines for animal studies.
A part of each tumor was cut in 8 µm thick tissue sections using a microtome-cryostat for quantification of the number of apoptotic tumor cells and of proliferative cells performed using the colorimetric DeadEND TUNEL System (Promega, Mannheim, Germany) and immunofluorescence analysis of Ki67 (BD pharmigen 556003), respectively. OCT was removed from the remainder of each tumor, and tumors were crushed in 50 mM Tris-HCl pH 6.8, 10% glycerol, and 2% SDS), heated at 95 °C, and sonicated for Western blot analyses as well as vemurafenib and cholesterol quantification. Protein concentration in each homogenate was evaluated using a Bio-Rad protein assay based on the Bradford dye-binding method.
4.17. Quantification of Vemurafenib in Tumors
Metabolites were extracted from tumor homogenates using methanol and resuspended in 40% acetonitrile before mass spectrometry analysis. Briefly, the metabolites were separated with UPLC system (ThermoFisher, Illkirch, France on a C18 column in an appropriate gradient. Mass spectrometry data were acquired with a Q-Exactive plus mass spectrometer (ThermoFisher) operating in Parallel Reaction Monitoring (PRM) mode. Finally, vemurafenib was identified using Xcalibur Quan-Browser software version 4.1.31.9 (ThermoFisher).
4.18. Quantification of Cholesterol in Tumors
Total lipids were extracted from the tumor homogenates according to the method developed by Folch [
53]. The total lipids were submitted to alkaline hydrolysis in 5 mL of 0.35 M KOH for 2 h at ambient temperature. The solution was neutralized with 65 µL of phosphoric acid, and the sterols were extracted with 9 mL chloroform in the presence of 3 mL 0.9% sodium chloride. The organic phase was removed, and the solvent was evaporated to dryness. For the quantification of sterols, 5 α-cholestane was added as an internal standard. The samples were derivatized to trimethylsilyl ethers by heating at 60 °C for 30 min after the addition of 100 µL of pyridine and 100 µL of BSTFA (Supelco, Bellafonte, PA, USA). The derivatives were analyzed on a gas chromatograph coupled to a flame ionization detector (GC–FID Hewlett-Packard HP5890A). A 1 µL aliquot was introduced by automated injection in splitless mode at 290 °C on a DB-5MS fused silica capillary column (30 m × 0.25 mm id, 0.25 µm film thickness; J&W Scientific, Agilent Technologies, Massy, France). The initial oven temperature was kept at 60 °C for 1 min, then increased at a rate of 20 °C/min to 290 °C, and then 2 °C/min to a final temperature of 300 °C.
4.19. SDS-PAGE and Western Blotting
Total RIPA extracts from cells or tumor homogenates were prepared. Protein concentrations were determined by the DC Protein Assay (Bio-Rad, Marnes-la-Coquette, France). Samples (50 to 80 µg) were separated on SDS-PAGE and transferred to nitrocellulose membranes (Amersham, Bath, UK) using standard techniques. After 1 h at room temperature in blocking buffer (20 mmol/L Tris-HCl pH 7.5, 45 mmol/L NaCl, 0.1% Tween-20, and 5% non-fat milk), nitrocellulose membranes were incubated overnight at 4 °C with rabbit anti-Patched antibody (Abcam ab53715; 1/1000), rabbit anti-Phospho-Erk1/2 antibody (Cell Signaling Technology (Leiden, The Netherlands); 1/1000), rabbit anti-Erk1/2 antibody (Cell Signaling Technology; 1/1000), or mouse anti-β-tubulin antibody (Sigma; 1/1000). After 3 washes, membranes were incubated for 45 min with anti-rabbit (1:2000) or anti-mouse (1:5000) immunoglobulin coupled to horseradish peroxidase (Dako-Agilent, Santa Clara, CA, USA). Detection was carried out with an ECL Prime Western blotting detection reagent (Amersham) on a Fusion FX imager (Vilber Lourmat, Collegien, France), and analyses were performed using ImageJ software.
4.20. Statistical Analysis
All results represent at least three independent replications. Data are shown as mean value ± SEM. Prism 6 (GraphPad) was used to determine IC50 values and other statistical analyses using one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison tests.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}